
Epigenetic Alterations in Alzheimer’s Disease: Impact on Insulin Signaling and Advanced Drug Delivery Systems
 , , ,
, , ,  and
and 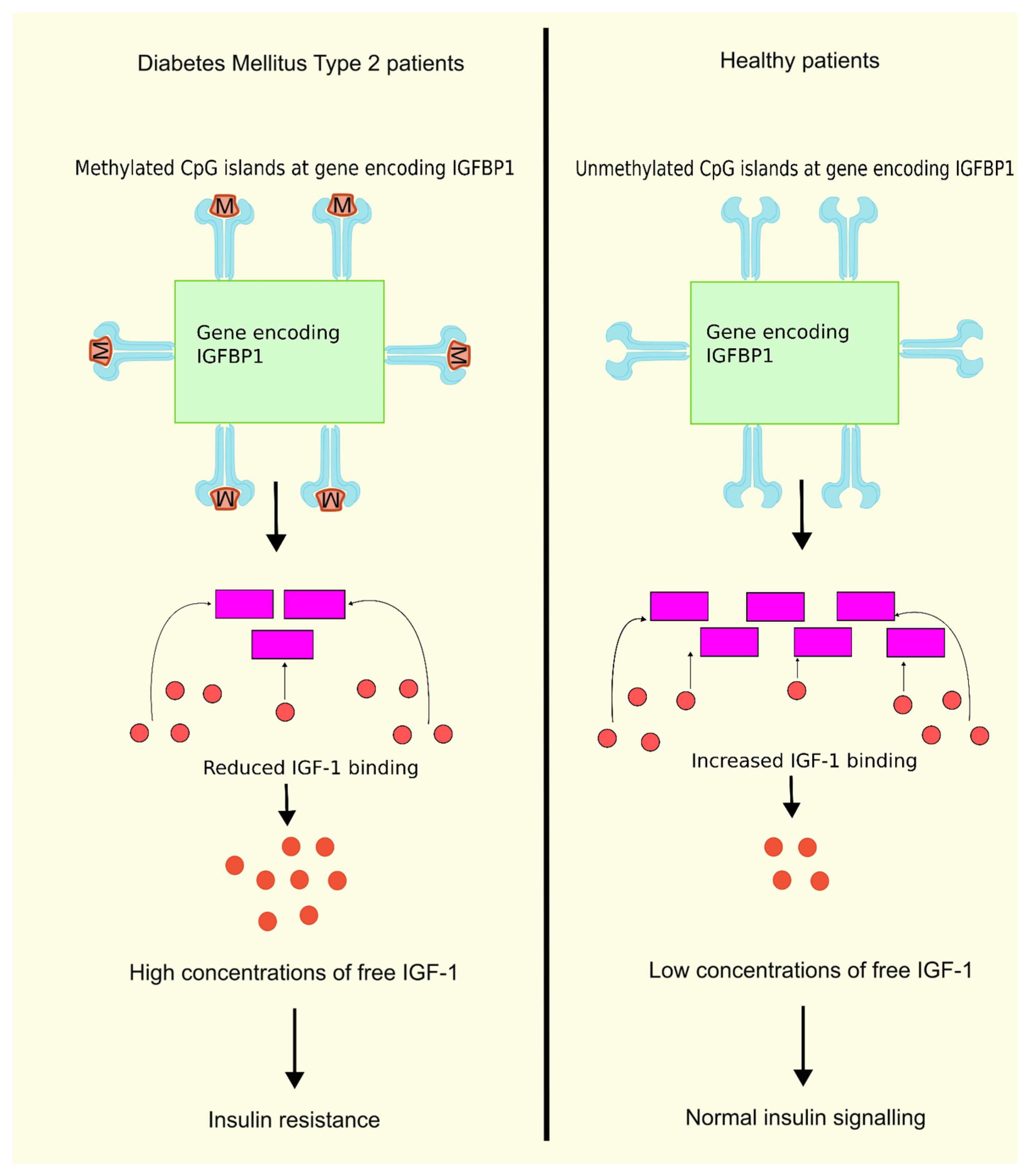{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Epigenetics
2.1. DNA Methylation
2.2. Histone Modifications
2.3. Non-Coding RNA
3. Insulin and Epigenetic Mechanisms
4. DNA Methylation and Insulin Resistance
4.1. DNA Methylation at IRS1/2
4.2. DNA Methylation at PPARGC1A
4.3. FADS2
4.4. DNA Methylation and Insulin-Like Growth Factor Binding Proteins
4.5. DNA Methylation and IGFBP 7
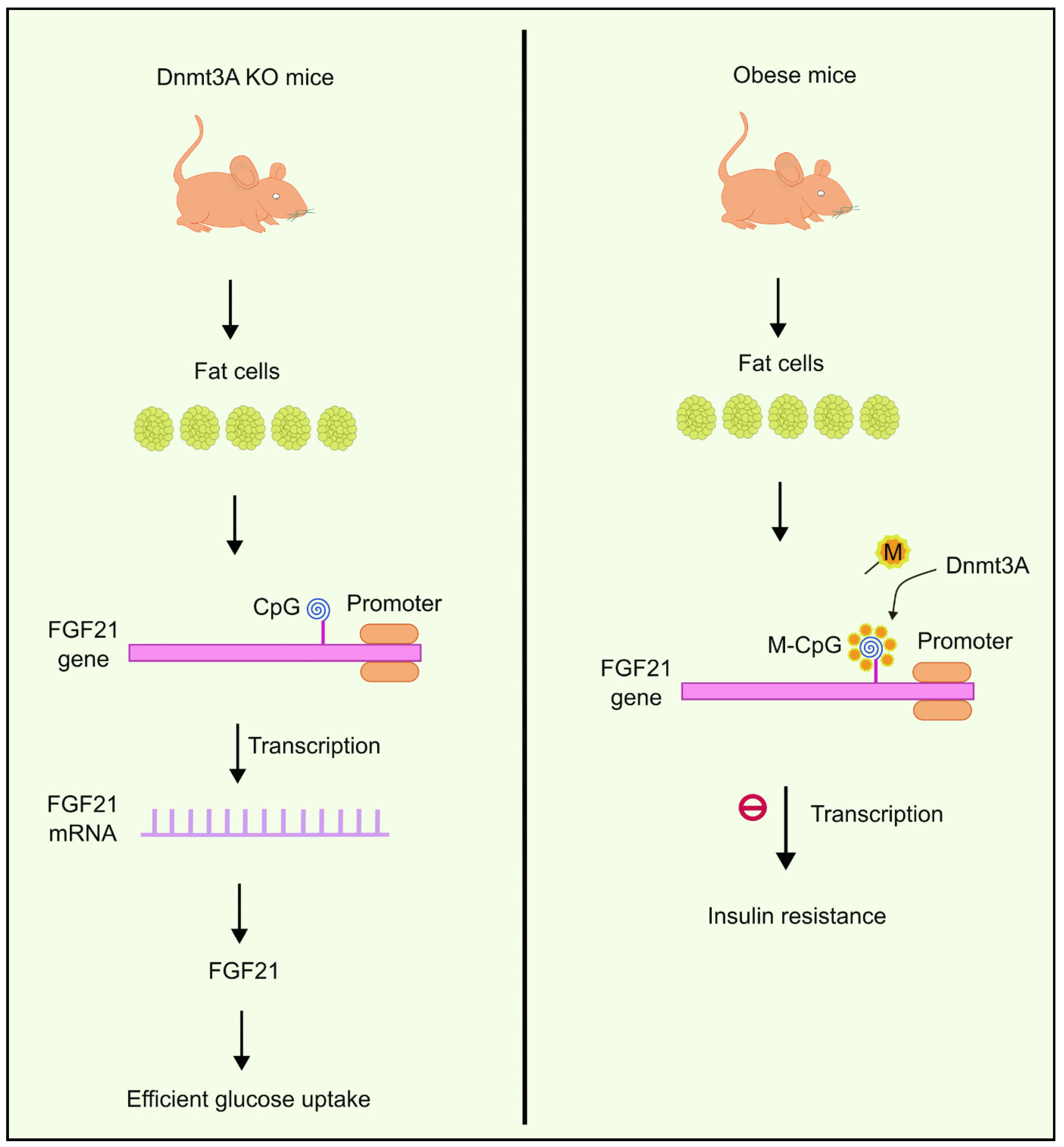
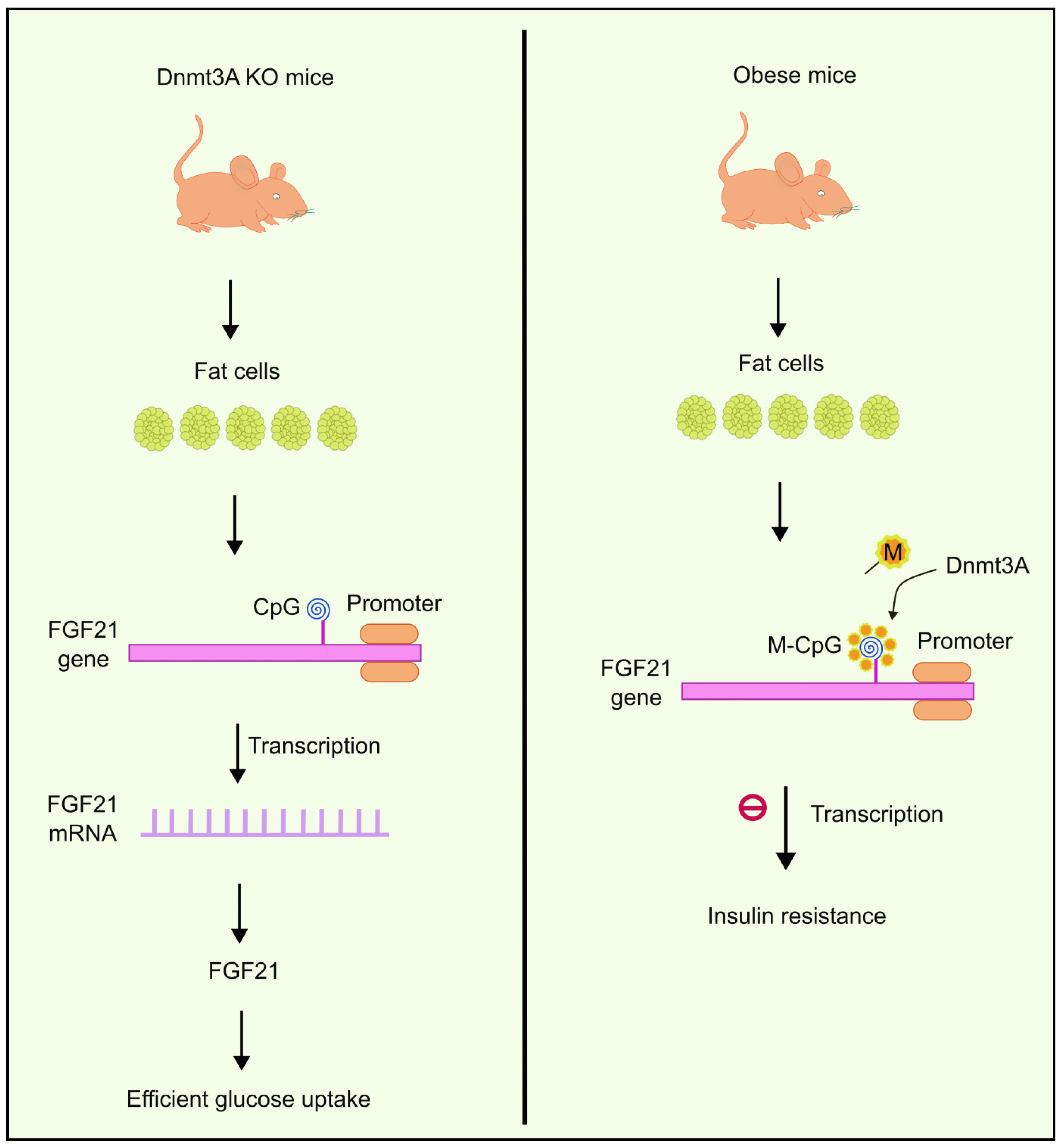
4.6. The Role of Dnmt3a in Insulin Resistance
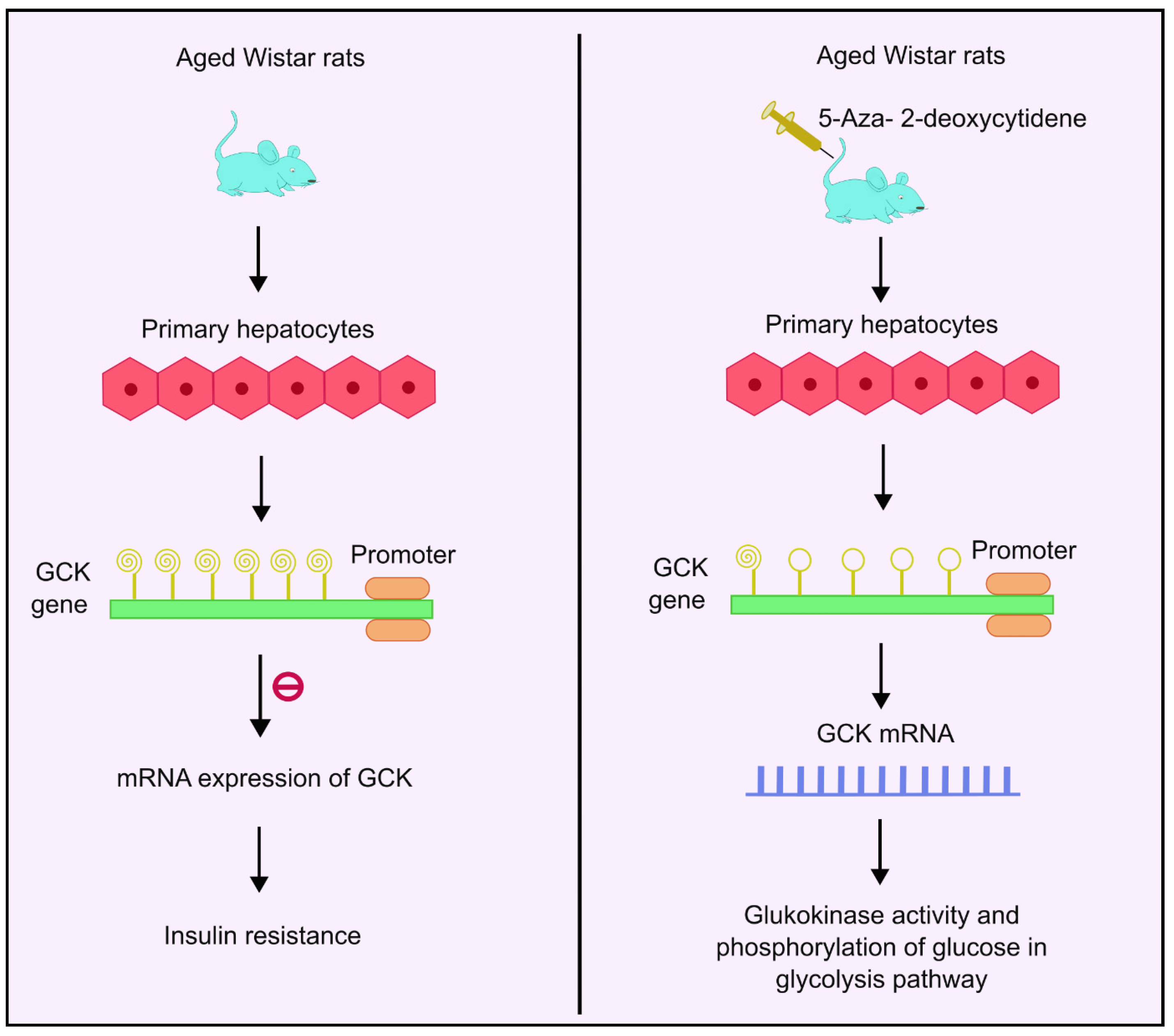
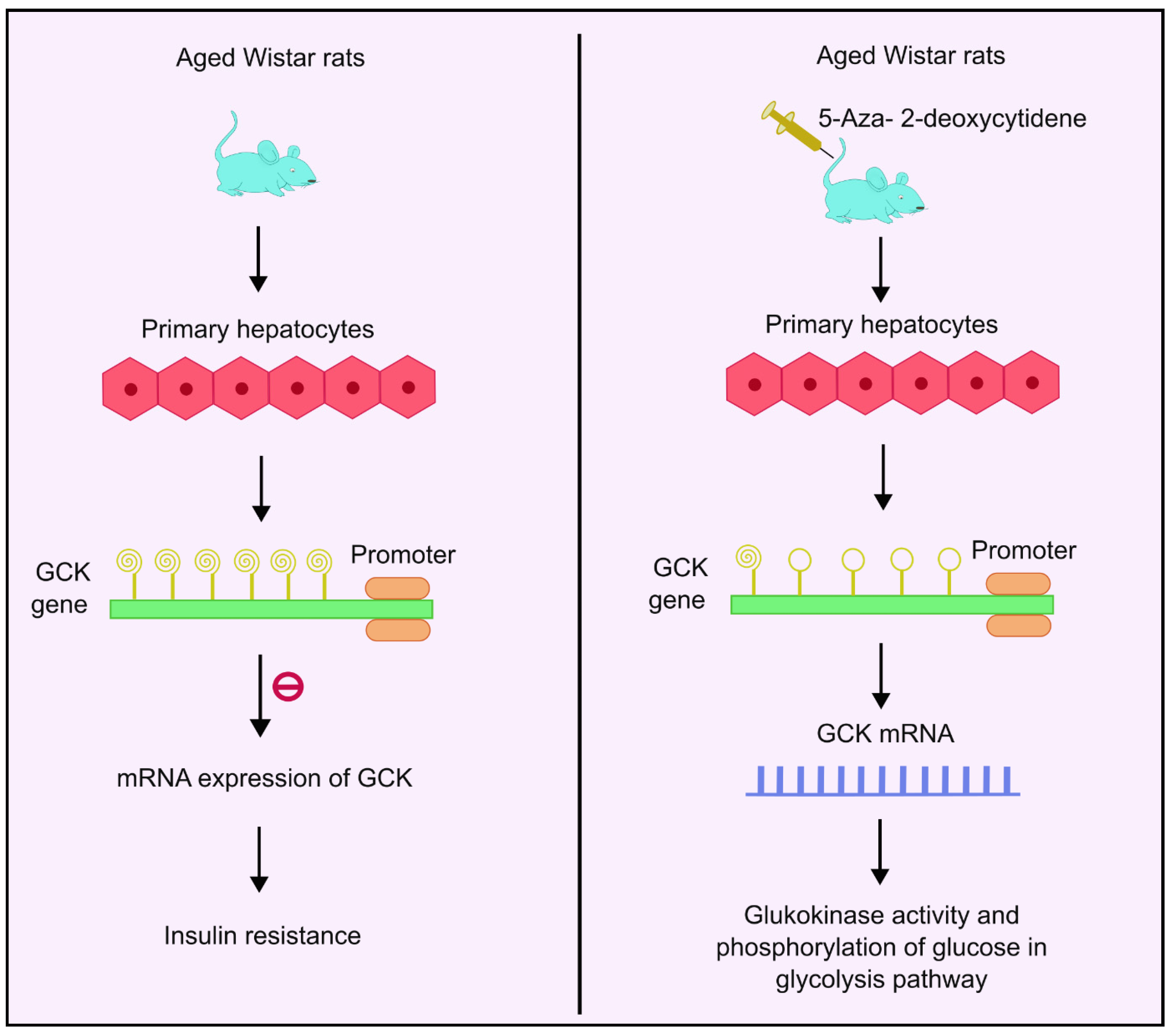
4.7. DNA Methylation and Glucokinase
4.8. DNA Methylation and Glucagon like Peptide 1
4.9. Histone Modifications: Acetylation and Deacetylation
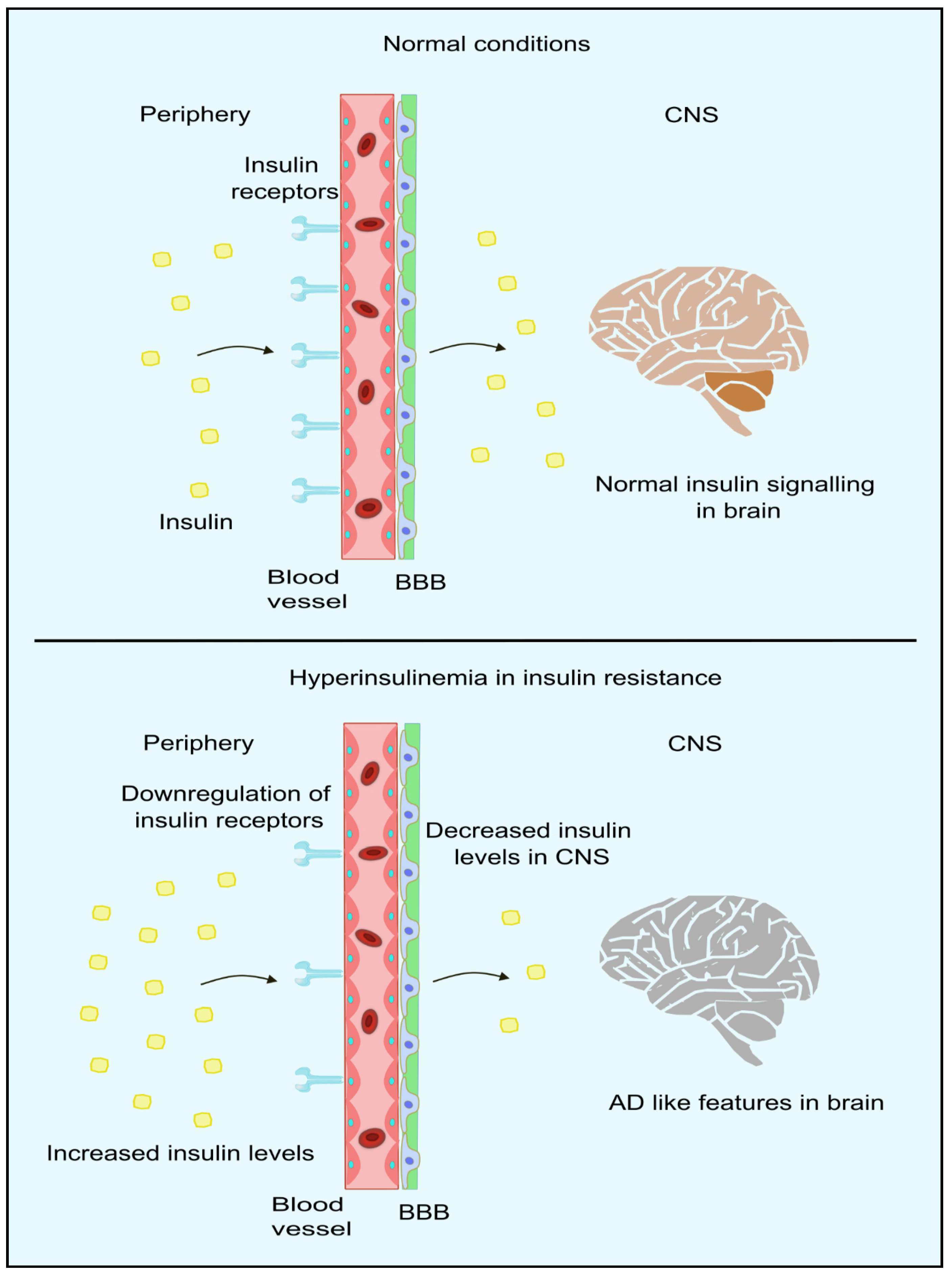
5. Linking Peripheral IR with Impaired Brain Insulin Signaling and AD
6. Advanced Drug Delivery Systems in Epigenetics
6.1. Nanoparticle-Based Systems
6.2. Vesicular Systems
6.3. Network Systems and Dendrimers
6.4. Hydrogel-Based Systems
6.5. Biological Vectors
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β |
| AD | Alzheimer’s Disease |
| ATF-2 | Activator Transcripton Factor-2 |
| BBB | Blood Brain Barrier |
| CRE | cAMP Responsive Element |
| CREB | cAMP Responsive Binding Protein-1 |
| CRB | CREB Binding Protein |
| Dnmts | DNA methyl transferases |
| FADS | Fatty Acid Desaturase |
| FGF | Fibroblast Growth Factor |
| GCK | Glucokinase |
| GLP-1 | Glucagon Like Peptide-1 |
| HAT | Histone Acetyl Transferases |
| HDAC | Histone Deacetylases |
| HFD | High Fat Diet |
| IDE | Insulin Degrading Enzyme |
| IGFBP | Insulin Like Growth Factor Binding Protein |
| IR | Insulin Resistance |
| KO | Knock Out |
| MEF-2 | Monocyte Enhancer Factor-2 |
| ncRNA | non-coding RNA |
| NP | Nano Particles |
| NT | Neurofibrillary Tangles |
| SiRNA | Short Interfering RNA |
| T2DM | Type 2 Diabetes Mellitus |
References
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 16 November 2023).
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s Disease and Some Therapeutic Approaches Targeting Aβ by Using Several Synthetic and Herbal Compounds. Oxid. Med. Cell. Longev. 2016, 2016, 7361613. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756. [Google Scholar] [CrossRef] [PubMed]
- Schachter, A.S.; Davis, K.L. Alzheimer’s Disease. Dialogues Clin. Neurosci. 2000, 2, 91. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, S.; Hafeez, D.A.; Riaz, S.U.; Ali, A.; Ghauri, M.I.; Zehra, M. Low Vitamin B12 Levels: An Underestimated Cause of Minimal Cognitive Impairment and Dementia. Cureus 2020, 12, e6976. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Araujo, A.P.B.; Melo, H.M.; da Silva, G.S.S.; Alves-Leon, S.V.; de Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Aβ Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797. [Google Scholar] [CrossRef] [PubMed]
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s Disease. Subcell. Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s Disease: Definition, Natural History, and Diagnostic Criteria. Alzheimers Dement. 2016, 12, 292. [Google Scholar] [CrossRef]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022; pp. 1–27. [Google Scholar]
- Wattmo, C.; Minthon, L.; Wallin, Å.K. Mild versus Moderate Stages of Alzheimer’s Disease: Three-Year Outcomes in a Routine Clinical Setting of Cholinesterase Inhibitor Therapy. Alzheimer’s Res. Ther. 2016, 8, 7. [Google Scholar] [CrossRef]
- Gao, X.; Chen, Q.; Yao, H.; Tan, J.; Liu, Z.; Zhou, Y.; Zou, Z. Epigenetics in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 911635. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The Epigenotype. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S. The Epigenetic Landscape: An Evolving Concept. Front. Epigenet. Epigenom. 2023, 1, 1176449. [Google Scholar] [CrossRef]
- Fyfe, I. Epigenetics Links Ageing with Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 254. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Nisticò, R.; Seyfried, N.T.; Levey, A.I.; Modeste, E.; Lemercier, P.; Baldacci, F.; Toschi, N.; Garaci, F.; Perry, G.; et al. Omics Sciences for Systems Biology in Alzheimer’s Disease: State-of-the-Art of the Evidence. Ageing Res. Rev. 2021, 69, 101346. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, J.I.; Oliver, S.G. Alzheimer’s as a Systems-Level Disease Involving the Interplay of Multiple Cellular Networks. Methods Mol. Biol. 2016, 1303, 3–48. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F.; Mattick, J.S. Non-coding RNAs and RNA Editing in Brain Development, Functional Diversification, and Neurological Disease. Physiol. Rev. 2007, 87, 799–823. [Google Scholar] [CrossRef]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T.; et al. Histone Deacetylase Inhibitors Enhance Memory and Synaptic Plasticity via CREB: CBP-Dependent Transcriptional Activation. J. Neurosci. 2007, 27, 6128. [Google Scholar] [CrossRef]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 Negatively Regulates Memory Formation and Synaptic Plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Michán, S.; Li, Y.; Chou, M.M.H.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 Is Essential for Normal Cognitive Function and Synaptic Plasticity. J. Neurosci. 2010, 30, 9695. [Google Scholar] [CrossRef]
- Nassar, A.; Satarker, S.; Gurram, P.C.; Upadhya, D.; Fayaz, S.; Nampoothiri, M. Repressor Element-1 Binding Transcription Factor (REST) as a Possible Epigenetic Regulator of Neurodegeneration and MicroRNA-Based Therapeutic Strategies. Mol. Neurobiol. 2023, 60, 5557–5577. [Google Scholar] [CrossRef] [PubMed]
- Nampoothiri, M.; Kumar, N.; Ramalingayya, G.V.; Kutty, N.G.; Krishnadas, N.; Rao, C.M. Effect of Insulin on Spatial Memory in Aluminum Chloride-Induced Dementia in Rats. Neuroreport 2017, 28, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Nampoothiri, M.; Ramalingayya, G.V.; Kutty, N.G.; Krishnadas, N.; Rao, C.M. Insulin Combined with Glucose Improves Spatial Learning and Memory in Aluminum Chloride & minus; Induced Dementia in Rats. J. Environ. Pathol. Toxicol. Oncol. 2017, 36, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Khan, S.; Wani, I.A.; Gupta, R.; Verma, S.; Alam, P.; Alaklabi, A. Unravelling the Role of Epigenetic Modifications in Development and Reproduction of Angiosperms: A Critical Appraisal. Front. Genet. 2022, 13, 819941. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An Operational Definition of Epigenetics. Genes Dev. 2009, 23, 781. [Google Scholar] [CrossRef] [PubMed]
- Al Aboud, N.M.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Jirtle, R.L.; Skinner, M.K. Environmental Epigenomics and Disease Susceptibility. Nat. Rev. Genet. 2007, 8, 253. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a Are Required for the Maintenance of DNA Methylation and Synaptic Function in Adult Forebrain Neurons. Nat. Neurosci. 2010, 13, 423. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Numata, M.; Komura, J.I.; Ono, T.; Bestor, T.H.; Kondo, H. Expression of DNA Methyltransferase Gene in Mature and Immature Neurons as Well as Proliferating Cells in Mice. Differentiation 1994, 56, 39–44. [Google Scholar] [CrossRef]
- Volkov, P.; Bacos, K.; Ofori, J.K.; Esguerra, J.L.S.; Eliasson, L.; Rönn, T.; Ling, C. Whole-Genome Bisulfite Sequencing of Human Pancreatic Islets Reveals Novel Differentially Methylated Regions in Type 2 Diabetes Pathogenesis. Diabetes 2017, 66, 1074–1085. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA Methylomes at Base Resolution Show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Balaji, E.V.; Kumar, N.; Satarker, S.; Nampoothiri, M. Zinc as a Plausible Epigenetic Modulator of Glioblastoma Multiforme. Eur. J. Pharmacol. 2020, 887, 173549. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, M.A. Epigenetic Editing: How Cutting-Edge Targeted Epigenetic Modification Might Provide Novel Avenues for Autoimmune Disease Therapy. Clin. Immunol. 2018, 196, 49. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, E.; Zink, D. Nuclear Architecture and Gene Regulation. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications: Basic Mechanisms and Role in Cardiovascular Disease. Circulation 2011, 123, 2145. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Denu, J.M. Catalysis and Substrate Selection by Histone/Protein Lysine Acetyltransferases. Curr. Opin. Struct. Biol. 2008, 18, 682. [Google Scholar] [CrossRef]
- Frías-Lasserre, D.; Villagra, C.A. The Importance of NcRNAs as Epigenetic Mechanisms in Phenotypic Variation and Organic Evolution. Front. Microbiol. 2017, 8, 298939. [Google Scholar] [CrossRef]
- Parveen, N.; Dhawan, S. DNA Methylation Patterning and the Regulation of Beta Cell Homeostasis. Front. Endocrinol. 2021, 12, 651258. [Google Scholar] [CrossRef]
- Nassar, A.; Kodi, T.; Satarker, S.; Chowdari Gurram, P.; Upadhya, D.; SM, F.; Mudgal, J.; Nampoothiri, M. Astrocytic MicroRNAs and Transcription Factors in Alzheimer’s Disease and Therapeutic Interventions. Cells 2022, 11, 4111. [Google Scholar] [CrossRef]
- Non-Coding RNAs as Regulators in Epigenetics (Review). Available online: https://www.spandidos-publications.com/or/37/1/3 (accessed on 18 November 2023).
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Bogan, J.S. Regulation of Glucose Transporter Translocation in Health and Diabetes. Annu. Rev. Biochem. 2012, 81, 507–532. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Mitron, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin Effects in Muscle and Adipose Tissue. Diabetes Res. Clin. Pract. 2011, 93, S52–S59. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.W.; Ferguson, L.A.; Docherty, K. ATF-2 Stimulates the Human Insulin Promoter through the Conserved CRE2 Sequence. Biochim. Biophys. Acta BBA-Gene Struct. Expr. 2007, 1769, 79–91. [Google Scholar] [CrossRef]
- Hay, C.W.; Docherty, K. Comparative Analysis of Insulin Gene Promoters Implications for Diabetes Research. Diabetes 2006, 55, 3201–3213. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, A.; Rauch, T.A.; Todorov, I.; Ku, H.T.; Al-Abdullah, I.H.; Kandeel, F.; Mullen, Y.; Pfeifer, G.P.; Ferreri, K. Insulin Gene Expression Is Regulated by DNA Methylation. PLoS ONE 2009, 4, e6953. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23. [Google Scholar] [CrossRef]
- Rui, J.; Deng, S.; Lebastchi, J.; Clark, P.L.; Usmani-Brown, S.; Herold, K.C. Methylation of Insulin DNA in Response to Pro-inflammatory Cytokines during the Progression of Autoimmune Diabetes in NOD Mice. Diabetologia 2016, 59, 1021. [Google Scholar] [CrossRef]
- Zhang, K.; Lin, G.; Han, Y.; Xie, J.; Li, J. Circulating Unmethylated Insulin DNA as a Potential Non-Invasive Biomarker of Beta Cell Death in Type 1 Diabetes: A Review and Future Prospect. Clin. Epigenet. 2017, 9, 44. [Google Scholar] [CrossRef]
- Yang, B.T.; Dayeh, T.A.; Kirkpatrick, C.L.; Taneera, J.; Kumar, R.; Groop, L.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Insulin Promoter DNA Methylation Correlates Negatively with Insulin Gene Expression and Positively with HbA1c Levels in Human Pancreatic Islets. Diabetologia 2011, 54, 360. [Google Scholar] [CrossRef]
- Ishikawa, K.; Tsunekawa, S.; Ikeniwa, M.; Izumoto, T.; Iida, A.; Ogata, H.; Uenishi, E.; Seino, Y.; Ozaki, N.; Sugimura, Y.; et al. Long-Term Pancreatic Beta Cell Exposure to High Levels of Glucose but Not Palmitate Induces DNA Methylation within the Insulin Gene Promoter and Represses Transcriptional Activity. PLoS ONE 2015, 10, e0115350. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Pinney, S.E. DNA Methylation and Its Role in the Pathogenesis of Diabetes. Pediatr. Diabetes 2017, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Ponnaluri, V.K.C.; Ehrlich, K.C.; Zhang, G.; Lacey, M.; Johnston, D.; Pradhan, S.; Ehrlich, M. Association of 5-Hydroxymethylation and 5-Methylation of DNA Cytosine with Tissue-Specific Gene Expression. Epigenetics 2017, 12, 123–138. [Google Scholar] [CrossRef] [PubMed]
- De Mello, V.D.F.; Pulkkinen, L.; Lalli, M.; Kolehmainen, M.; Pihlajamäki, J.; Uusitupa, M. DNA Methylation in Obesity and Type 2 Diabetes. Ann. Med. 2014, 46, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Ronald Kahn, C. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Tobe, K.; Terauchi, Y.; Eto, K.; Yamauchi, T.; Suzuki, R.; Tsubamoto, Y.; Komeda, K.; Nakano, R.; Miki, H.; et al. Disruption of Insulin Receptor Substrate 2 Causes Type 2 Diabetes Because of Liver Insulin Resistance and Lack of Compensatory Beta-Cell Hyperplasia. Diabetes 2000, 49, 1880–1889. [Google Scholar] [CrossRef]
- Krause, C.; Geißler, C.; Tackenberg, H.; El Gammal, A.T.; Wolter, S.; Spranger, J.; Mann, O.; Lehnert, H.; Kirchner, H. Multi-Layered Epigenetic Regulation of IRS2 Expression in the Liver of Obese Individuals with Type 2 Diabetes. Diabetologia 2020, 63, 2182. [Google Scholar] [CrossRef]
- Ide, T.; Shimano, H.; Yahagi, N.; Matsuzaka, T.; Nakakuki, M.; Yamamoto, T.; Nakagawa, Y.; Takahashi, A.; Suzuki, H.; Sone, H.; et al. SREBPs Suppress IRS-2-Mediated Insulin Signalling in the Liver. Nat. Cell Biol. 2004, 6, 351–357. [Google Scholar] [CrossRef]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional Regulatory Circuits Controlling Mitochondrial Biogenesis and Function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Sirén, J.; Hamsten, A.; Fisher, R.M.; Yki-Järvinen, H. Genes Involved in Fatty Acid Partitioning and Binding, Lipolysis, Monocyte/Macrophage Recruitment, and Inflammation Are Overexpressed in the Human Fatty Liver of Insulin-Resistant Subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; Von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA Methylation Analysis in Nonalcoholic Fatty Liver Disease Suggests Distinct Disease-Specific and Remodeling Signatures after Bariatric Surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueñ, A.L.; Gianotti, T.F.; Castañ, G.O.; Pirola, C.J. Epigenetic Regulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease: Impact of Liver Methylation of the Peroxisome Proliferator-Activated Receptor c Coactivator 1a Promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Gol, S.; Pena, R.N.; Rothschild, M.F.; Tor, M.; Estany, J. A Polymorphism in the Fatty Acid Desaturase-2 Gene Is Associated with the Arachidonic Acid Metabolism in Pigs. Sci. Rep. 2018, 8, 14336. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, R.; Jiang, F.; Zhang, H.; Zhao, A.; Xu, B.; Jin, L.; Wang, T.; Jia, W.; Jia, W.; et al. FADS1-FADS2 Genetic Polymorphisms Are Associated with Fatty Acid Metabolism through Changes in DNA Methylation and Gene Expression. Clin. Epigenet. 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Glaser, C.; Heinrich, J.; Koletzko, B. Role of FADS1 and FADS2 Polymorphisms in Polyunsaturated Fatty Acid Metabolism. Metabolism 2010, 59, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Walle, P.; Takkunen, M.; Männistö, V.; Vaittinen, M.; Lankinen, M.; Kärjä, V.; Käkelä, P.; Ågren, J.; Tiainen, M.; Schwab, U.; et al. Fatty Acid Metabolism Is Altered in Non-Alcoholic Steatohepatitis Independent of Obesity. Metabolism 2016, 65, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic, M.; Ramdas, S.; Lorenz, K.M.; Guo, X.; Darlay, R.; Cordell, H.J.; He, J.; Gindin, Y.; Chung, C.; Myers, R.P.; et al. A Multiancestry Genome-Wide Association Study of Unexplained Chronic ALT Elevation as a Proxy for Nonalcoholic Fatty Liver Disease with Histological and Radiological Validation. Nat. Genet. 2022, 54, 761. [Google Scholar] [CrossRef]
- Wang, X.; Sato, R.; Brown, M.S.; Hua, X.; Goldstein, J.L. SREBP-1, a Membrane-Bound Transcription Factor Released by Sterol-Regulated Proteolysis. Cell 1994, 77, 53–62. [Google Scholar] [CrossRef]
- Walle, P.; Männistö, V.; De Mello, V.D.; Vaittinen, M.; Perfilyev, A.; Hanhineva, K.; Ling, C.; Pihlajamäki, J. Liver DNA Methylation of FADS2 Associates with FADS2 Genotypex. Clin. Epigenet. 2019, 11, 10. [Google Scholar] [CrossRef]
- Ding, H.; Wu, T. Insulin-Like Growth Factor Binding Proteins in Autoimmune Diseases. Front. Endocrinol. 2018, 9, 499. [Google Scholar] [CrossRef] [PubMed]
- Rajpathak, S.N.; Gunter, M.J.; Wylie-Rosett, J.; Ho, G.Y.F.; Kaplan, R.C.; Muzumdar, R.; Rohan, T.E.; Strickler, H.D. The Role of Insulin-like Growth Factor-I and Its Binding Proteins in Glucose Homeostasis and Type 2 Diabetes. Diabetes Metab. Res. Rev. 2009, 25, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Gu, H.F.; Hilding, A.; Sjöholm, L.K.; Östenson, C.G.; Ekström, T.J.; Brismar, K. Increased DNA Methylation Levels of the Insulin-like Growth Factor Binding Protein 1 Gene Are Associated with Type 2 Diabetes in Swedish Men. Clin. Epigenet. 2013, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Shen, F.; Weinfeld, M.; Sergi, C. Insulin Growth Factor Binding Protein 7 (IGFBP7)-Related Cancer and IGFBP3 and IGFBP7 Crosstalk. Front. Oncol. 2020, 10, 727. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.F.; Gu, T.; Hilding, A.; Zhu, Y.; Kärvestedt, L.; Östenson, C.G.; Lai, M.; Kutsukake, M.; Frystyk, J.; Tamura, K.; et al. Evaluation of IGFBP-7 DNA Methylation Changes and Serum Protein Variation in Swedish Subjects with and without Type 2 Diabetes. Clin. Epigenet. 2013, 5, 20. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA Methylation and Human Disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A.; et al. Obesity-Induced DNA Hypermethylation of the Adiponectin Gene Mediates Insulin Resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef]
- You, D.; Nilsson, E.; Tenen, D.E.; Lyubetskaya, A.; Lo, J.C.; Jiang, R.; Deng, J.; Dawes, B.A.; Vaag, A.; Ling, C.; et al. Dnmt3a Is an Epigenetic Mediator of Adipose Insulin Resistance. Elife 2017, 6, e30766. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a Novel Metabolic Regulator. J. Clin. Investig. 2005, 115, 1627. [Google Scholar] [CrossRef]
- Yang, M.L.; Horstman, S.; Gee, R.; Guyer, P.; Lam, T.K.T.; Kanyo, J.; Perdigoto, A.L.; Speake, C.; Greenbaum, C.J.; Callebaut, A.; et al. Citrullination of Glucokinase Is Linked to Autoimmune Diabetes. Nat. Commun. 2022, 13, 1870. [Google Scholar] [CrossRef]
- Ren, Y.; Li, L.; Wan, L.; Huang, Y.; Cao, S. Glucokinase as an Emerging Anti-Diabetes Target and Recent Progress in the Development of Its Agonists. J. Enzyme Inhib. Med. Chem. 2022, 37, 606. [Google Scholar] [CrossRef] [PubMed]
- Bogdarina, I.; Murphy, H.C.; Burns, S.P.; Clark, A.J.L. Investigation of the Role of Epigenetic Modification of the Rat Glucokinase Gene in Fetal Programming. Life Sci. 2004, 74, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.H.; Fei, J.; Lan, M.S.; Lu, Z.P.; Liu, M.; Fan, W.W.; Gao, X.; Lu, D.R. Hypermethylation of Hepatic Gck Promoter in Ageing Rats Contributes to Diabetogenic Potential. Diabetologia 2008, 51, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, C.; Donnelly, D.; Wootten, D.; Lau, J.; Sexton, P.M.; Miller, L.J.; Ahn, J.M.; Liao, J.; Fletcher, M.M.; Yang, D.; et al. Glucagon-Like Peptide-1 and Its Class B G Protein–Coupled Receptors: A Long March to Therapeutic Successes. Pharmacol. Rev. 2016, 68, 954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 1040. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Dayeh, T.; Kirkpatrick, C.L.; Wollheim, C.B.; Dekker Nitert, M.; Ling, C. DNA Methylation of the Glucagon-like Peptide 1 Receptor (GLP1R) in Human Pancreatic Islets. BMC Med. Genet. 2013, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone Acetyltransferases. Annu. Rev. Biochem. 2003, 70, 81–120. [Google Scholar] [CrossRef]
- Fang, Z.; Wang, X.; Sun, X.; Hu, W.; Miao, Q.R. The Role of Histone Protein Acetylation in Regulating Endothelial Function. Front. Cell Dev. Biol. 2021, 9, 672447. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Kaiser, C.; James, S.R. Acetylation of Insulin Receptor Substrate-1 Is Permissive for Tyrosine Phosphorylation. BMC Biol. 2004, 2, 23. [Google Scholar] [CrossRef]
- Beljanski, V.; Trichostatin, A. xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2009; pp. 1–4. [Google Scholar] [CrossRef]
- Zeng, Z.; Liao, R.; Yao, Z.; Zhou, W.; Ye, P.; Zheng, X.; Li, X.; Huang, Y.; Chen, S.; Chen, Q. Three Single Nucleotide Variants of the HDAC Gene Are Associated with Type 2 Diabetes Mellitus in a Chinese Population: A Community-Based Case–Control Study. Gene 2014, 533, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Miller, R.A.; Patel, R.T.; Chen, J.; Dhir, R.; Wang, H.; Zhang, D.; Graham, M.J.; Unterman, T.G.; Shulman, G.I.; et al. Hepatic Hdac3 Promotes Gluconeogenesis by Repressing Lipid Synthesis and Sequestration. Nat. Med. 2012, 18, 934. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 Promotes Ligand-Independent PPARγ Activation by Protein Acetylation. J. Mol. Endocrinol. 2014, 53, 191. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. Fibroblast Growth Factors. Genome Biol. 2001, 2, reviews3005.1. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, Z.; Zhang, J.; Ye, X.; Xu, A.; Ye, J.; Jia, W. Sodium Butyrate Stimulates Expression of Fibroblast Growth Factor 21 in Liver by Inhibition of Histone Deacetylase 3. Diabetes 2012, 61, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, N.; Guo, X.; Li, J.; Zhang, T.; Ren, G.; Li, D. Fibroblast Growth Factor 21 Regulates Glucose Metabolism in Part by Reducing Renal Glucose Reabsorption. Biomed. Pharmacother. 2018, 108, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.G.; Jornayvaz, F.R.; Petersen, M.C.; Pesta, D.; Guigni, B.A.; Serr, J.; Zhang, D.; Kahn, M.; Samuel, V.T.; Jurczak, M.J.; et al. Cellular Mechanisms by Which FGF21 Improves Insulin Sensitivity in Male Mice. Endocrinology 2013, 154, 3099. [Google Scholar] [CrossRef] [PubMed]
- Szczepańska, E.; Gietka-Czernel, M. FGF21: A Novel Regulator of Glucose and Lipid Metabolism and Whole-Body Energy Balance. Horm. Metab. Res. 2022, 54, 203–211. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Z.; Gu, J.; Jiang, S.; Liu, Q.; Zheng, Y.; Freedman, J.H.; Sun, J.; Cai, L. HDAC3 Inhibition in Diabetic Mice May Activate Nrf2 Preventing Diabetes-Induced Liver Damage and FGF21 Synthesis and Secretion Leading to Aortic Protection. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E150–E162. [Google Scholar] [CrossRef]
- Thai, M.V.; Guruswamy, S.; Cao, K.T.; Pessin, J.E.; Olson, A.L. Myocyte Enhancer Factor 2 (MEF2)-Binding Site Is Required ForGLUT4 Gene Expression in Transgenic Mice. J. Biol. Chem. 1998, 273, 14285–14292. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Miska, E.A.; Langley, E.; Reynaud-Deonauth, S.; Kotecha, S.; Towers, N.; Spohr, G.; Kouzarides, T.; Mohun, T.J. MEF-2 Function Is Modified by a Novel Co-Repressor, MITR. EMBO J. 1999, 18, 5085. [Google Scholar] [CrossRef] [PubMed]
- Nikzamir, A.; Palangi, A.; Kheirollaha, A.; Tabar, H.; Malakaskar, A.; Shahbazian, H.; Fathi, M. Expression of Glucose Transporter 4 (GLUT4) Is Increased by Cinnamaldehyde in C2C12 Mouse Muscle Cells. Iran. Red Crescent Med. J. 2014, 16, 13426. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An Autoregulatory Loop Controls Peroxisome Proliferator-Activated Receptor γ Coactivator 1α Expression in Muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-Dependent Activation of the MEF2 Transcription Factor by Dissociation from Histone Deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-Dependent Nuclear Export of a Histone Deacetylase Regulates Muscle Differentiation. Nature 2000, 408, 106. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Benz, V.; Clemenz, M.; Bloch, M.; Foryst-Ludwig, A.; Wardat, S.; Witte, N.; Trappiel, M.; Namsolleck, P.; Mai, K.; et al. Histone Deacetylase 6 (HDAC6) Is an Essential Modifier of Glucocorticoid-Induced Hepatic Gluconeogenesis. Diabetes 2012, 61, 513. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell 2011, 145, 607. [Google Scholar] [CrossRef]
- Wang, J.; Gong, B.; Zhao, W.; Tang, C.; Varghese, M.; Nguyen, T.; Bi, W.; Bilski, A.; Begum, S.; Vempati, P.; et al. Epigenetic Mechanisms Linking Diabetes and Synaptic Impairments. Diabetes 2014, 63, 645–654. [Google Scholar] [CrossRef]
- Kellar, D.; Craft, S. Brain Insulin Resistance in Alzheimer’s Disease and Related Disorders: Mechanisms and Therapeutic Approaches. Lancet Neurol. 2020, 19, 758. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.R.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis Model Assessment: Insulin Resistance and Fl-Cell Function from Fasting Plasma Glucose and Insulin Concentrations in Man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Stennis Watson, G.; Craft, S. Insulin Resistance Is Associated with Alzheimer-Like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults with Pre-Diabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51. [Google Scholar] [CrossRef]
- Craft, S. Insulin Resistance Syndrome and Alzheimer’s Disease: Age- and Obesity-Related Effects on Memory, Amyloid, and Inflammation. Neurobiol. Aging 2005, 26, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.R.; Astell, A.; Green, C.; Sutherland, C. Molecular Connexions between Dementia and Diabetes. Neurosci. Biobehav. Rev. 2007, 31, 1046–1063. [Google Scholar] [CrossRef] [PubMed]
- Neumann, K.; Rojo, L.; Navarrete, L.; Farias, G.; Reyes, P.; Maccioni, R. Insulin Resistance and Alzheimers Disease: Molecular Links & Clinical Implications. Curr. Alzheimer Res. 2008, 5, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How Does Diabetes Accelerate Alzheimer Disease Pathology? Nat. Rev. Neurol. 2010, 6, 551. [Google Scholar] [CrossRef] [PubMed]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin Resistance and Pathological Brain Ageing. Diabet. Med. 2011, 28, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Townsend, M. Insulin Resistance and Amyloidogenesis as Common Molecular Foundation for Type 2 Diabetes and Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 482–496. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Perry, G.; Zhu, X.; IMoreira, P.I.; Smith, M.A. Insulin-Resistant Brain State: The Culprit in Sporadic Alzheimer’s Disease? Ageing Res. Rev. 2011, 10, 264. [Google Scholar] [CrossRef]
- Hoyer, S. Sporadic Alzheimer Disease: A Challenging Hypothesis Is Sporadic Alzheimer Disease the Brain Type of Non-Insulin Dependent Diabetes Mellitus? A Challenging Hypothesis. J. Neural Transm. 1998, 105, 415–422. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient Brain Insulin Signalling Pathway in Alzheimer’s Disease and Diabetes. J. Pathol. 2011, 225, 54. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; De La Monte, S.M. Insulin and Insulin-like Growth Factor Expression and Function Deteriorate with Progression of Alzheimer’s Disease: Link to Brain Reductions in Acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Han, L.; Schneider, J.A.; Wilson, R.S.; Bennett, D.A.; Arnold, S.E. O3–02–02: Expression of PIRS–1 (S312 and S616) Is Elevated in MCI and AD and Correlates with Cognitive Impairment and Neurofibrillary Pathology. Alzheimer’s Dement. 2006, 2, S54. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 Receptor, Insulin Receptor and IRS-1/2 in Alzheimer’s Disease Indicate Possible Resistance to IGF-1 and Insulin Signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Ping, P.C.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. β-Amyloid Oligomers Induce Phosphorylation of Tau and Inactivation of Insulin Receptor Substrate via c-Jun N-Terminal Kinase Signaling: Suppression by Omega-3 Fatty Acids and Curcumin. J. Neurosci. 2009, 29, 9078. [Google Scholar] [CrossRef] [PubMed]
- Apelt, J.; Mehlhorn, G.; Schliebs, R. Insulin-Sensitive GLUT4 Glucose Transporters Are Colocalized with GLUT3-Expressing Cells and Demonstrate a Chemically Distinct Neuron-Specific Localization in Rat Brain. J. Neurosci. Res. 1999, 57, 693–705. [Google Scholar] [CrossRef]
- Ibberson, M.; Uldry, M.; Thorens, B. GLUTX1, a Novel Mammalian Glucose Transporter Expressed in the Central Nervous System and Insulin-Sensitive Tissues. J. Biol. Chem. 2000, 275, 4607–4612. [Google Scholar] [CrossRef]
- Craft, S.; Peskind, E.; Schwartz, M.W.; Schellenberg, G.D.; Raskind, M.; Porte, D. Cerebrospinal Fluid and Plasma Insulin Levels in Alzheimer’s Disease. Neurology 1998, 50, 164–168. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Molecular Indices of Oxidative Stress and Mitochondrial Dysfunction Occur Early and Often Progress with Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Erol, A. An Integrated and Unifying Hypothesis for the Metabolic Basis of Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 13, 241–253. [Google Scholar] [CrossRef]
- Choeiri, C.; Staines, W.; Messier, C. Immunohistochemical Localization and Quantification of Glucose Transporters in the Mouse Brain. Neuroscience 2002, 111, 19–34. [Google Scholar] [CrossRef]
- Moreira, P.I.; Duarte, A.I.; Santos, M.S.; Rego, A.C.; Oliveira, C.R. An Integrative View of the Role of Oxidative Stress, Mitochondria and Insulin in Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 16, 741–761. [Google Scholar] [CrossRef] [PubMed]
- Tschritter, O.; Preissl, H.; Hennige, A.M.; Stumvoll, M.; Porubska, K.; Frost, R.; Marx, H.; Klösel, B.; Lutzenberger, W.; Birbaumer, N.; et al. The Cerebrocortical Response to Hyperinsulinemia Is Reduced in Overweight Humans: A Magnetoencephalographic Study. Proc. Natl. Acad. Sci. USA 2006, 103, 12103. [Google Scholar] [CrossRef] [PubMed]
- Anthony, K.; Reed, L.J.; Dunn, J.T.; Bingham, E.; Hopkins, D.; Marsden, P.K.; Amiel, S.A. Attenuation of Insulin-Evoked Responses in Brain Networks Controlling Appetite and Reward in Insulin Resistance the Cerebral Basis for Impaired Control of Food Intake in Metabolic Syndrome? Diabetes 2006, 55, 2986–2992. [Google Scholar] [CrossRef] [PubMed]
- Tschritter, O.; Hennige, A.M.; Preissl, H.; Porubska, K.; Schäfer, S.A.; Lutzenberger, W.; Machicao, F.; Birbaumer, N.; Fritsche, A.; Häring, H.U. Cerebrocortical Beta Activity in Overweight Humans Responds to Insulin Detemir. PLoS ONE 2007, 2, 1196. [Google Scholar] [CrossRef] [PubMed]
- Hirvonen, J.; Virtanen, K.A.; Nummenmaa, L.; Hannukainen, J.C.; Honka, M.J.; Bucci, M.; Nesterov, S.V.; Parkkola, R.; Rinne, J.; Iozzo, P.; et al. Effects of Insulin on Brain Glucose Metabolism in Impaired Glucose Tolerance. Diabetes 2011, 60, 443. [Google Scholar] [CrossRef]
- Kullmann, S.; Frank, S.; Heni, M.; Ketterer, C.; Veit, R.; Häring, H.U.; Fritsche, A.; Preissl, H. Intranasal Insulin Modulates Intrinsic Reward and Prefrontal Circuitry of the Human Brain in Lean Women. Neuroendocrinology 2013, 97, 176–182. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Veit, R.; Scheffler, K.; Machann, J.; Häring, H.U.; Fritsche, A.; Preissl, H. Selective Insulin Resistance in Homeostatic and Cognitive Control Brain Areas in Overweight and Obese Adults. Diabetes Care 2015, 38, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.-U. Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef]
- DeFronzo, R.A. The Triumvirate: β-Cell, Muscle, Liver: A Collusion Responsible for NIDDM. Diabetes 1988, 37, 667–687. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular Mechanisms of Insulin Resistance. J. Clin. Investig. 2000, 106, 171. [Google Scholar] [CrossRef]
- Shulman, G.I.; Rothman, D.L.; Jue, T.; Stein, P.; DeFronzo, R.A.; Shulman, R.G. Quantitation of Muscle Glycogen Synthesis in Normal Subjects and Subjects with Non-Insulin-Dependent Diabetes by 13C Nuclear Magnetic Resonance Spectroscopy. N. Engl. J. Med. 2010, 322, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Sesti, G. Pathophysiology of Insulin Resistance. Best. Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain Insulin and Insulin Receptors in Aging and Sporadic Alzheimer’s Disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-Degrading Enzyme as a Downstream Target of Insulin Receptor Signaling Cascade: Implications for Alzheimer’s Disease Intervention. J. Neurosci. 2004, 24, 11120. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.A.; Adjei, I.M.; Labhasetwar, V. Advancements in the Delivery of Epigenetic Drugs. Expert. Opin. Drug Deliv. 2015, 12, 1501. [Google Scholar] [CrossRef] [PubMed]
- Mayersohn, M. Pharmacokinetics in the Elderly. Environ. Health Perspect. 1994, 102, 119. [Google Scholar] [CrossRef] [PubMed]
- El Bahhaj, F.; Dekker, F.J.; Martinet, N.; Bertrand, P. Delivery of Epidrugs. Drug Discov. Today 2014, 19, 1337–1352. [Google Scholar] [CrossRef] [PubMed]
- Narang, A.S.; Boddu, S.H. Excipient Applications in Formulation Design and Drug Delivery. In Excipient Applications in Formulation Design and Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–10. [Google Scholar] [CrossRef]
- Wang, Z.; Tiruppathi, C.; Minshall, R.D.; Malik, A.B. Size and Dynamics of Caveolae Studied Using Nanoparticles in Living Endothelial Cells. ACS Nano 2009, 3, 4110. [Google Scholar] [CrossRef]
- González-Maciel, A.; Reynoso-Robles, R.; Torres-Jardón, R.; Mukherjee, P.S.; Calderón-Garcidueñas, L. Combustion-Derived Nanoparticles in Key Brain Target Cells and Organelles in Young Urbanites: Culprit Hidden in Plain Sight in Alzheimer’s Disease Development. J. Alzheimer’s Dis. 2017, 59, 189–208. [Google Scholar] [CrossRef]
- Sadanandan, P.; Payne, N.L.; Sun, G.; Ashokan, A.; Gowd, S.G.; Lal, A.; Satheesh Kumar, M.K.; Pulakkat, S.; Nair, S.V.; Menon, K.N.; et al. Exploiting the Preferential Phagocytic Uptake of Nanoparticle-Antigen Conjugates for the Effective Treatment of Autoimmunity. Nanomedicine 2022, 40, 102481. [Google Scholar] [CrossRef]
- Yang, X.; He, C.; Li, J.; Chen, H.; Ma, Q.; Sui, X.; Tian, S.; Ying, M.; Zhang, Q.; Luo, Y.; et al. Uptake of Silica Nanoparticles: Neurotoxicity and Alzheimer-like Pathology in Human SK-N-SH and Mouse Neuro2a Neuroblastoma Cells. Toxicol. Lett. 2014, 229, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Fatima, M.T.; Islam, Z.; Khan, R.H.; Uversky, V.N.; Salahuddin, P. Nanoparticle Formulations in the Diagnosis and Therapy of Alzheimer’s Disease. Int. J. Biol. Macromol. 2019, 130, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Del Amo, L.; Cano, A.; Ettcheto, M.; Souto, E.B.; Espina, M.; Camins, A.; García, M.L.; Sánchez-López, E. Surface Functionalization of PLGA Nanoparticles to Increase Transport across the BBB for Alzheimer’s Disease. Appl. Sci. 2021, 11, 4305. [Google Scholar] [CrossRef]
- Mathew, A.; Fukuda, T.; Nagaoka, Y.; Hasumura, T.; Morimoto, H.; Yoshida, Y.; Maekawa, T.; Venugopal, K.; Kumar, D.S. Curcumin Loaded-PLGA Nanoparticles Conjugated with Tet-1 Peptide for Potential Use in Alzheimer’s Disease. PLoS ONE 2012, 7, e32616. [Google Scholar] [CrossRef]
- Jeon, S.G.; Cha, M.Y.; Kim, J.I.; Hwang, T.W.; Kim, K.A.; Kim, T.H.; Song, K.C.; Kim, J.J.; Moon, M. Vitamin D-Binding Protein-Loaded PLGA Nanoparticles Suppress Alzheimer’s Disease-Related Pathology in 5XFAD Mice. Nanomedicine 2019, 17, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Luppi, B.; Bigucci, F.; Corace, G.; Delucca, A.; Cerchiara, T.; Sorrenti, M.; Catenacci, L.; Di Pietra, A.M.; Zecchi, V. Albumin Nanoparticles Carrying Cyclodextrins for Nasal Delivery of the Anti-Alzheimer Drug Tacrine. Eur. J. Pharm. Sci. 2011, 44, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Li, M.; Wu, L.; Chen, C.; Qu, X. Mesoporous Silica Nanoparticle-Based H2O2 Responsive Controlled-Release System Used for Alzheimer’s Disease Treatment. Adv. Healthc. Mater. 2012, 1, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Geetha, K.M. Neurotherapeutic Applications of Nanomedicine for Treating Alzheimer’s Disease. J. Control. Release 2020, 325, 25–37. [Google Scholar] [CrossRef]
- Ashokan, A.; Somasundaram, V.H.; Gowd, G.S.; Anna, I.M.; Malarvizhi, G.L.; Sridharan, B.; Jobanputra, R.B.; Peethambaran, R.; Unni, A.K.K.; Nair, S.; et al. Biomineral Nano-Theranostic Agent for Magnetic Resonance Image Guided, Augmented Radiofrequency Ablation of Liver Tumor. Sci. Rep. 2017, 7, 14481. [Google Scholar] [CrossRef]
- Ramachandran, R.; Junnuthula, V.R.; Gowd, G.S.; Ashokan, A.; Thomas, J.; Peethambaran, R.; Thomas, A.; Unni, A.K.K.; Panikar, D.; Nair, S.V.; et al. Theranostic 3-Dimensional Nano Brain-Implant for Prolonged and Localized Treatment of Recurrent Glioma. Sci. Rep. 2017, 7, 43271. [Google Scholar] [CrossRef]
- Wang, W.; Liu, M.; Gao, W.; Sun, Y.; Dong, X. Coassembled Chitosan-Hyaluronic Acid Nanoparticles as a Theranostic Agent Targeting Alzheimer’s β-Amyloid. ACS Appl. Mater. Interfaces 2021, 13, 55879–55889. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Taylor, M.; Fullwood, N.; Allsop, D. Liposome Delivery Systems for the Treatment of Alzheimer’s Disease. Int. J. Nanomed. 2018, 13, 8507. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Shao, X.; Zhang, C.; Tan, Y.; Liu, Q.; Wan, X.; Zhang, Q.; Xu, S.; Jiang, X. Intranasal H102 Peptide-Loaded Liposomes for Brain Delivery to Treat Alzheimer’s Disease. Pharm. Res. 2015, 32, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Mancini, S.; Minniti, S.; La Vitola, P.; Zotti, M.; Sancini, G.; Mauri, M.; Cagnotto, A.; Colombo, L.; Fiordaliso, F.; et al. Multifunctional Liposomes Reduce Brain β-Amyloid Burden and Ameliorate Memory Impairment in Alzheimer’s Disease Mouse Models. J. Neurosci. 2014, 34, 14022. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.; Lopes, I.; Magalhães, L.; Sárria, M.P.; Machado, R.; Sousa, J.C.; Botelho, C.; Teixeira, J.; Gomes, A.C. Novel Concept of Exosome-like Liposomes for the Treatment of Alzheimer’s Disease. J. Control. Release 2021, 336, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Rawtani, D.; Barot, T. Design, Development and in-Vitro/in-Vivo Evaluation of Intranasally Delivered Rivastigmine and N-Acetyl Cysteine Loaded Bifunctional Niosomes for Applications in Combinative Treatment of Alzheimer’s Disease. Eur. J. Pharm. Biopharm. 2021, 163, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, Y.S.R.; Etman, S.M.; Abdelmonsif, D.A.; Abdallah, O.Y. Novel Piperine-Loaded Tween-Integrated Monoolein Cubosomes as Brain-Targeted Oral Nanomedicine in Alzheimer’s Disease: Pharmaceutical, Biological, and Toxicological Studies. Int. J. Nanomed. 2015, 10, 5459–5473. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, A.M.; Salama, A.A.A.; Asfour, M.H. Cubosome-Based Thermosensitive in Situ Gelling System for Intranasal Administration of Lamotrigine with Enhanced Antiepileptic Efficacy. Pharm. Dev. Technol. 2023, 28, 520–534. [Google Scholar] [CrossRef]
- Raina, N.; Rani, R.; Khan, A.; Nagpal, K.; Gupta, M. Interpenetrating Polymer Network as a Pioneer Drug Delivery System: A Review. Polym. Bull. 2020, 77, 5027–5050. [Google Scholar] [CrossRef]
- Singh, A.; Ansari, V.A.; Mahmood, T.; Ahsan, F.; Wasim, R. Dendrimers: A Neuroprotective Lead in Alzheimer Disease: A Review on Its Synthetic Approach and Applications. Drug Res. 2022, 72, 417–423. [Google Scholar] [CrossRef]
- Singh, A.; Ujjwal, R.R.; Naqvi, S.; Verma, R.K.; Tiwari, S.; Kesharwani, P.; Shukla, R. Formulation Development of Tocopherol Polyethylene Glycol Nanoengineered Polyamidoamine Dendrimer for Neuroprotection and Treatment of Alzheimer Disease. J. Drug Target. 2022, 30, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, T.; Marcinkowska, M.; Pieszynski, I.; Zablocka, M.; Caminade, A.M.; Majoral, J.P.; Klajnert-Maculewicz, B. Cationic Phosphorus Dendrimers and Therapy for Alzheimer’s Disease. New J. Chem. 2015, 39, 4852–4859. [Google Scholar] [CrossRef]
- Zha, L.; Banik, B.; Alexis, F. Stimulus Responsive Nanogels for Drug Delivery. Soft Matter 2011, 7, 5908–5916. [Google Scholar] [CrossRef]
- Fonseca-Santos, B.; Gremião, M.P.D.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for the Treatment of Alzheimer’s Disease. Int. J. Nanomed. 2015, 10, 4981. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) in Cosmetic and Dermatological Preparations. Adv. Drug Deliv. Rev. 2002, 54, S131–S155. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Yeom, S.Y.; Kim, J.; Jung, S.; Jung, S.; Shim, T.S.; Kim, S.K.; Kang, J.Y.; Lee, S.H.; Cho, I.J.; et al. Hydrogel Micropost-Based QPCR for Multiplex Detection of MiRNAs Associated with Alzheimer’s Disease. Biosens. Bioelectron. 2018, 101, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Das, G.; Barman, S.; Mohapatra, S.; Bhunia, D.; Jana, B.; Ghosh, S. Biodegradable Neuro-Compatible Peptide Hydrogel Promotes Neurite Outgrowth, Shows Significant Neuroprotection, and Delivers Anti-Alzheimer Drug. ACS Appl. Mater. Interfaces 2017, 9, 5067–5076. [Google Scholar] [CrossRef]
- Macdiarmid, J.A.; Mugridge, N.B.; Weiss, J.C.; Phillips, L.; Burn, A.L.; Paulin, R.P.; Haasdyk, J.E.; Dickson, K.-A.; Brahmbhatt, V.N.; Pattison, S.T.; et al. Article Bacterially Derived 400 Nm Particles for Encapsulation and Cancer Cell Targeting of Chemotherapeutics. Cancer Cell 2007, 11, 431–435. [Google Scholar] [CrossRef]
- Bi, F.C.; Yang, X.H.; Cheng, X.Y.; Deng, W.B.; Guo, X.L.; Yang, H.; Wang, Y.; Li, J.; Yao, Y. Optimization of Cerebral Organoids: A More Qualified Model for Alzheimer’s Disease Research. Transl. Neurodegener. 2021, 10, 27. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Brain Organoids: A next Step for Humanized Alzheimer’s Disease Models? Mol. Psychiatry 2019, 24, 474–478. [Google Scholar] [CrossRef]
- Unnisa, A.; Greig, N.; Kamal, M. Nanotechnology-Based Gene Therapy as a Credible Tool in the Treatment of Alzheimer’s Disease. Neural Regen. Res. 2023, 18, 2127. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cao, Y.; Ye, J.; Yang, Z.; Chen, Q.; Liu, X.; Zhang, B.; Qiao, J.; Tang, Q.; Yang, H.; et al. Engineering Brain-Derived Neurotrophic Factor MRNA Delivery for the Treatment of Alzheimer’s Disease. Chem. Eng. J. 2023, 466, 143152. [Google Scholar] [CrossRef]
- Imran Sajid, M.; Sultan Sheikh, F.; Anis, F.; Nasim, N.; Sumbria, R.K.; Nauli, S.M.; Kumar Tiwari, R. SiRNA Drug Delivery across the Blood–Brain Barrier in Alzheimer’s Disease. Adv. Drug Deliv. Rev. 2023, 199, 114968. [Google Scholar] [CrossRef]
- Bhatnagar, D.; Ladhe, S.; Kumar, D. Discerning the Prospects of MiRNAs as a Multi-Target Therapeutic and Diagnostic for Alzheimer’s Disease. Mol. Neurobiol. 2023, 60, 5954–5974. [Google Scholar] [CrossRef]
- Mummery, C.J.; Börjesson-Hanson, A.; Blackburn, D.J.; Vijverberg, E.G.B.; De Deyn, P.P.; Ducharme, S.; Jonsson, M.; Schneider, A.; Rinne, J.O.; Ludolph, A.C.; et al. Tau-Targeting Antisense Oligonucleotide MAPTRx in Mild Alzheimer’s Disease: A Phase 1b, Randomized, Placebo-Controlled Trial. Nat. Med. 2023, 29, 1437. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greeny, A.; Nair, A.; Sadanandan, P.; Satarker, S.; Famurewa, A.C.; Nampoothiri, M. Epigenetic Alterations in Alzheimer’s Disease: Impact on Insulin Signaling and Advanced Drug Delivery Systems. Biology 2024, 13, 157. https://doi.org/10.3390/biology13030157
Greeny A, Nair A, Sadanandan P, Satarker S, Famurewa AC, Nampoothiri M. Epigenetic Alterations in Alzheimer’s Disease: Impact on Insulin Signaling and Advanced Drug Delivery Systems. Biology. 2024; 13(3):157. https://doi.org/10.3390/biology13030157
Chicago/Turabian StyleGreeny, Alosh, Ayushi Nair, Prashant Sadanandan, Sairaj Satarker, Ademola C. Famurewa, and Madhavan Nampoothiri. 2024. "Epigenetic Alterations in Alzheimer’s Disease: Impact on Insulin Signaling and Advanced Drug Delivery Systems" Biology 13, no. 3: 157. https://doi.org/10.3390/biology13030157
APA StyleGreeny, A., Nair, A., Sadanandan, P., Satarker, S., Famurewa, A. C., & Nampoothiri, M. (2024). Epigenetic Alterations in Alzheimer’s Disease: Impact on Insulin Signaling and Advanced Drug Delivery Systems. Biology, 13(3), 157. https://doi.org/10.3390/biology13030157