
The Involvement of YNR069C in Protein Synthesis in the Baker’s Yeast, Saccharomyces cerevisiae

 ,
,  , ,
, ,  and
and
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. Plasmids
2.3. β-Galactosidase Assay
2.4. qRT-PCR
2.5. Genetic Interaction Analysis
3. Results
3.1. Identification of YNR069C as a Potential Gene Involved in Protein Biosynthesis
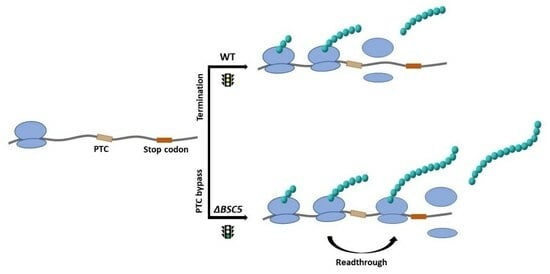
3.2. Deletion of YNR069C Is Linked to the Bypass of Premature Termination Codons
3.3. Deletion of YNR069C Reduces the Rate of Protein Biosynthesis
3.4. Genetic Interaction Analysis Further Connects the Activity of YNR069C to Translation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational readthrough potential of natural termination codons in eucaryotes—The impact of RNA sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Bertram, G.; Innes, S.; Minella, O.; Richardson, J.P.; Stansfield, I. Endless possibilities: Translation termination and stop codon recognition. Microbiology 2001, 147, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Hellen, C.U.T. Translation Termination and Ribosome Recycling in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2018, 10, a032656. Available online: http://cshperspectives.cshlp.org/content/10/10/a032656.abstract (accessed on 18 February 2024). [CrossRef] [PubMed]
- Nyikó, T.; Auber, A.; Szabadkai, L.; Benkovics, A.; Auth, M.; Mérai, Z.; Kerényi, Z.; Dinnyés, A.; Nagy, F.; Silhavy, D. Expression of the eRF1 translation termination factor is controlled by an autoregulatory circuit involving readthrough and nonsense-mediated decay in plants. Nucleic Acids Res. 2017, 45, 4174–4188. [Google Scholar] [CrossRef] [PubMed]
- Moskalenko, S.E.; Chabelskaya, S.V.; Inge-Vechtomov, S.G.; Philippe, M.; Zhouravleva, G.A. Viable nonsense mutants for the essential gene SUP45 of Saccharomyces cerevisiae. BMC Mol. Biol. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Eszter, C.; Ian, B.; Nerea, I. Modulation of Stop Codon Read-Through Efficiency and Its Effect on the Replication of Murine Leukemia Virus. J. Virol. 2014, 88, 10364–10376. [Google Scholar] [CrossRef][Green Version]
- Newburn, L.R.; Nicholson, B.L.; Yosefi, M.; Cimino, P.A.; White, K.A. Translational readthrough in Tobacco necrosis virus-D. Virology 2014, 450–451, 258–265. [Google Scholar] [CrossRef]
- Pezo, V.; Metzgar, D.; Hendrickson, T.L.; Waas, W.F.; Hazebrouck, S.; Döring, V.; Marliere, P.; Schimmel, P.; De Crécy-Lagard, V. Artificially ambiguous genetic code confers growth yield advantage. Proc. Natl. Acad. Sci. USA 2004, 101, 8593–8597. [Google Scholar] [CrossRef]
- Santos MA, S.; Cheesman, C.; Costa, V.; Moradas-Ferreira, P.; Tuite, M.F. Selective advantages created by codon ambiguity allowed for the evolution of an alternative genetic code in Candida spp. Mol. Microbiol. 1999, 31, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.C.; Miranda, I.; Silva, R.M.; Moura, G.R.; Thomas, B.; Akoulitchev, A.; Santos, M.A.S. A genetic code alteration generates a proteome of high diversity in the human pathogen Candida albicans. Genome Biol. 2007, 8, R206. [Google Scholar] [CrossRef] [PubMed]
- True, H.L.; Berlin, I.; Lindquist, S.L. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 2004, 431, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Haar, T.; Tuite, M. Regulated translation bypass of stop codons in yeast. Trends Microbiol. 2007, 15, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Samanfar, B.; Tan, L.H.; Shostak, K.; Chalabian, F.; Wu, Z.; Alamgir, M.; Sunba, N.; Burnside, D.; Omidi, K.; Hooshyar, M.; et al. A global investigation of gene deletion strains that affect premature stop codon bypass in yeast, Saccharomyces cerevisiae. Mol. BioSystems 2014, 10, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Novoselova, T.V.; Zahira, K.; Rose, R.S.; Sullivan, J.A. Bul Proteins, a Nonredundant, Antagonistic Family of Ubiquitin Ligase Regulatory Proteins. Eukaryot. Cell 2012, 11, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Zbieralski, K.; Wawrzycka, D. α-Arrestins and Their Functions: From Yeast to Human Health. Int. J. Mol. Sci. 2022, 23, 4988. [Google Scholar] [CrossRef]
- Tong, A.H.Y.; Evangelista, M.; Parsons, A.B.; Xu, H.; Bader, G.D.; Pagé, N.; Robinson, M.; Raghibizadeh, S.; Hogue, C.W.V.; Bussey, H.; et al. Systematic Genetic Analysis with Ordered Arrays of Yeast Deletion Mutants. Science 2001, 294, 2364–2368. [Google Scholar] [CrossRef]
- Gelperin, D.M.; White, M.A.; Wilkinson, M.L.; Kon, Y.; Kung, L.A.; Wise, K.J.; Lopez-Hoyo, N.; Jiang, L.; Piccirillo, S.; Yu, H.; et al. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005, 19, 2816–2826. [Google Scholar] [CrossRef]
- Dunham, M.; Gartenberg, M.; Brown, G.W. Methods in Yeast Genetics and Genomics, 2015 Edition: A CSHL Course Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Stansfield, I.; Akhmaloka; Tuite, M.F. A mutant allele of the SUP45 (SAL4) gene of Saccharomyces cerevisiae shows temperature-dependent allosuppressor and omnipotent suppressor phenotypes. Curr. Genet. 1995, 27, 417–426. [Google Scholar] [CrossRef]
- Alamgir, M.; Eroukova, V.; Jessulat, M.; Xu, J.; Golshani, A. Chemical-genetic profile analysis in yeast suggests that a previously uncharacterized open reading frame, YBR261C, affects protein synthesis. BMC Genom. 2008, 9, 583. [Google Scholar] [CrossRef]
- Mumberg, D.; Müller, R.; Funk, M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995, 156, 119–122. [Google Scholar] [CrossRef]
- Goldstein, A.L.; McCusker, J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 1999, 15, 1541–1553. [Google Scholar] [CrossRef]
- Fuxman Bass, J.I.; Reece-Hoyes, J.S.; Walhout, A.J.M. Colony Lift Colorimetric Assay for β-Galactosidase Activity. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot088963. Available online: http://cshprotocols.cshlp.org/content/2016/12/pdb.prot088963.abstract (accessed on 18 February 2024). [CrossRef][Green Version]
- Krogan, N.J.; Kim, M.; Tong, A.; Golshani, A.; Cagney, G.; Canadien, V.; Richards, D.P.; Beattie, B.K.; Emili, A.; Boone, C.; et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2003, 23, 4207–4218. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Yan Tong, A.H.; Boone, C. Synthetic Genetic Array Analysis in Saccharomyces cerevisiae. In Yeast Protocol; Xiao, W., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 171–191. [Google Scholar] [CrossRef]
- Tong, A.H.Y.; Boone, C. 16 High-Throughput Strain Construction Systematic Synthetic Lethal Screening in Saccharomycescerevisiae. In Yeast Gene Analysis; Stansfield, I., Stark, M.J.R., Eds.; Academic Press: Cambridge, MA, USA, 2007; Volume 36, pp. 369–707. [Google Scholar] [CrossRef]
- Memarian, N.; Jessulat, M.; Alirezaie, J.; Mir-Rashed, N.; Xu, J.; Zareie, M.; Smith, M.; Golshani, A. Colony size measurement of the yeast gene deletion strains for functional genomics. BMC Bioinform. 2007, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Wagih, O.; Usaj, M.; Baryshnikova, A.; VanderSluis, B.; Kuzmin, E.; Costanzo, M.; Myers, C.L.; Andrews, B.J.; Boone, C.M.; Parts, L. SGAtools: One-stop analysis and visualization of array-based genetic interaction screens. Nucleic Acids Res. 2013, 41, W591–W596. [Google Scholar] [CrossRef] [PubMed]
- Alamgir, M.; Erukova, V.; Jessulat, M.; Azizi, A.; Golshani, A. Chemical-genetic profile analysis of five inhibitory compounds in yeast. BMC Chem. Biol. 2010, 10, 6. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Samanfar, B.; Omidi, K.; Hooshyar, M.; Laliberte, B.; Alamgir, M.D.; Seal, A.J.; Ahmed-Muhsin, E.; Viteri, D.F.; Said, K.; Chalabian, F.; et al. Large-scale investigation of oxygen response mutants in Saccharomyces cerevisiae. Mol. BioSystems 2013, 9, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Sopko, R.; Huang, D.; Preston, N.; Chua, G.; Papp, B.; Kafadar, K.; Snyder, M.; Oliver, S.G.; Cyert, M.; Hughes, T.R.; et al. Mapping Pathways and Phenotypes by Systematic Gene Overexpression. Mol. Cell 2006, 21, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Namy, O.; Duchateau-Nguyen, G.; Hatin, I.; Hermann-Le Denmat, S.; Termier, M.; Rousset, J. Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res. 2003, 31, 2289–2296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gagarinova, A.; Stewart, G.; Samanfar, B.; Phanse, S.; White, C.A.; Aoki, H.; Deineko, V.; Beloglazova, N.; Yakunin, A.F.; Golshani, A.; et al. Systematic Genetic Screens Reveal the Dynamic Global Functional Organization of the Bacterial Translation Machinery. Cell Rep. 2016, 17, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.H.Y.; Lesage, G.; Bader, G.D.; Ding, H.; Xu, H.; Xin, X.; Young, J.; Berriz, G.F.; Brost, R.L.; Chang, M.; et al. Global Mapping of the Yeast Genetic Interaction Network. Science 2004, 303, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Baryshnikova, A.; Costanzo, M.; Dixon, S.; Vizeacoumar, F.J.; Myers, C.L.; Andrews, B.; Boone, C. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and schizosaccharomyces pombe. In Methods in Enzymology, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; Volume 470. [Google Scholar] [CrossRef]
- Jessulat, M.; Amin, S.; Hooshyar, M.; Malty, R.; Moutaoufik, M.T.; Zilocchi, M.; Istace, Z.; Phanse, S.; Aoki, H.; Omidi, K.; et al. The conserved Tpk1 regulates non-homologous end joining double-strand break repair by phosphorylation of Nej1, a homolog of the human XLF. Nucleic Acids Res. 2021, 49, 8145–8160. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, J.E.; Adames, N.R.; Rogers, M.F.; Kraikivski, P.; Ibele, A.; Nurzynski-Loth, K.; Kudlow, E.; Murali, T.M.; Tyson, J.J.; Peccoud, J. Genetic interactions derived from high-throughput phenotyping of 6589 yeast cell cycle mutants. Npj Syst. Biol. Appl. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Boone, C.; Bussey, H.; Andrews, B.J. Exploring genetic interactions and networks with yeast. Nat. Rev. Genet. 2007, 8, 437–449. [Google Scholar] [CrossRef]
- Wangen, J.R.; Green, R. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. eLife 2020, 9, e52611. [Google Scholar] [CrossRef]
- Valente, L.; Kinzy, T.G. Yeast as a sensor of factors affecting the accuracy of protein synthesis. Cell. Mol. Life Sci. CMLS 2003, 60, 2115–2130. [Google Scholar] [CrossRef]
- Röther, S.; Sträßer, K. The RNA polymerase II CTD kinase Ctk1 functions in translation elongation. Genes Dev. 2007, 21, 1409–1421. Available online: http://genesdev.cshlp.org/content/21/11/1409.abstract (accessed on 18 February 2024). [CrossRef] [PubMed]
- Keeling, K.M.; Salas-Marco, J.; Osherovich, L.Z.; Bedwell, D.M. Tpa1p Is Part of an mRNP Complex That Influences Translation Termination, mRNA Deadenylation, and mRNA Turnover in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 5237–5248. [Google Scholar] [CrossRef]
- Loenarz, C.; Sekirnik, R.; Thalhammer, A.; Ge, W.; Spivakovsky, E.; Mackeen, M.M.; McDonough, M.A.; Cockman, M.E.; Kessler, B.M.; Ratcliffe, P.J.; et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc. Natl. Acad. Sci. USA 2014, 111, 4019–4024. [Google Scholar] [CrossRef]
- Lee, K.; Sharma, R.; Shrestha, O.K.; Bingman, C.A.; Craig, E.A. Dual interaction of the Hsp70 J-protein cochaperone Zuotin with the 40S and 60S ribosomal subunits. Nat. Struct. Mol. Biol. 2016, 23, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, C.; Yuan, Y.; Zhu, J.; Li, N.; Chen, C.; Wu, S.; Yu, L.; Lei, J.; Gao, N. Structural basis for interaction of a cotranslational chaperone with the eukaryotic ribosome. Nat. Struct. Mol. Biol. 2014, 21, 1042–1046. [Google Scholar] [CrossRef]
- Peisker, K.; Braun, D.; Wölfle, T.; Hentschel, J.; Fünfschilling, U.; Fischer, G.; Sickmann, A.; Rospert, S. Ribosome-associated Complex Binds to Ribosomes in Close Proximity of Rpl31 at the Exit of the Polypeptide Tunnel in Yeast. Mol. Biol. Cell 2008, 19, 5279–5288. [Google Scholar] [CrossRef] [PubMed]
- Benko, A.L.; Vaduva, G.; Martin, N.C.; Hopper, A.K. Competition between a sterol biosynthetic enzyme and tRNA modification in addition to changes in the protein synthesis machinery causes altered nonsense suppression. Proc. Natl. Acad. Sci. USA 2000, 97, 61–66. [Google Scholar] [CrossRef]
- Anthony, R.A.; Liebman, S.W. Alterations in ribosomal protein RPS28 can diversely affect translational accuracy in Saccharomyces cerevisiae. Genetics 1995, 140, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Synetos, D.; Frantziou, C.P.; Alksne, L.E. Mutations in yeast ribosomal proteins S28 and S4 affect the accuracy of translation and alter the sensitivity of the ribosomes to paromomycin. Biochim. Et Biophys. Acta (BBA)-Gene Struct. Expr. 1996, 1309, 156–166. [Google Scholar] [CrossRef]
- Bez Batti Angulski, A.; Hosny, N.; Cohen, H.; Martin, A.A.; Hahn, D.; Bauer, J.; Metzger, J.M. Duchenne muscular dystrophy: Disease mechanism and therapeutic strategies. Front. Physiol. 2023, 14, 1183101. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Mendell, J.R. Aminoglycoside-induced mutation suppression (stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Ther. Adv. Neurol. Disord. 2010, 3, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Belakhov, V.; Werner, H.; Wegener, E.; Gärtner, J.; Nudelman, I.; Baasov, T.; Huppke, P. Readthrough of nonsense mutations in Rett syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. 2011, 89, 389–398. [Google Scholar] [CrossRef]
- Borgatti, M.; Altamura, E.; Salvatori, F.; D’Aversa, E.; Altamura, N. Screening Readthrough Compounds to Suppress Nonsense Mutations: Possible Application to β-Thalassemia. J. Clin. Med. 2020, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics Based on Stop Codon Readthrough. Annu. Rev. Genom. Hum. Genet. 2014, 15, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.A.; Muller, V.J.; Hopwood, J.J. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol. Med. 2006, 12, 367–373. [Google Scholar] [CrossRef]
- Zingman, L.V.; Park, S.; Olson, T.M.; Alekseev, A.E.; Terzic, A. Aminoglycoside-induced translational read-through in disease: Overcoming nonsense mutations by pharmacogenetic therapy. Clin. Pharmacol. Ther. 2007, 81, 99–103. [Google Scholar] [CrossRef]
- Keeling, K.M.; Bedwell, D.M. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip. Rev. RNA 2011, 2, 837–852. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takallou, S.; Hajikarimlou, M.; Al-gafari, M.; Wang, J.; Kazmirchuk, T.D.D.; Said, K.B.; Samanfar, B.; Golshani, A. The Involvement of YNR069C in Protein Synthesis in the Baker’s Yeast, Saccharomyces cerevisiae. Biology 2024, 13, 138. https://doi.org/10.3390/biology13030138
Takallou S, Hajikarimlou M, Al-gafari M, Wang J, Kazmirchuk TDD, Said KB, Samanfar B, Golshani A. The Involvement of YNR069C in Protein Synthesis in the Baker’s Yeast, Saccharomyces cerevisiae. Biology. 2024; 13(3):138. https://doi.org/10.3390/biology13030138
Chicago/Turabian StyleTakallou, Sarah, Maryam Hajikarimlou, Mustafa Al-gafari, Jiashu Wang, Thomas David Daniel Kazmirchuk, Kamaledin B. Said, Bahram Samanfar, and Ashkan Golshani. 2024. "The Involvement of YNR069C in Protein Synthesis in the Baker’s Yeast, Saccharomyces cerevisiae" Biology 13, no. 3: 138. https://doi.org/10.3390/biology13030138
APA StyleTakallou, S., Hajikarimlou, M., Al-gafari, M., Wang, J., Kazmirchuk, T. D. D., Said, K. B., Samanfar, B., & Golshani, A. (2024). The Involvement of YNR069C in Protein Synthesis in the Baker’s Yeast, Saccharomyces cerevisiae. Biology, 13(3), 138. https://doi.org/10.3390/biology13030138