
The Therapeutic Potential of Vitamins B1, B3 and B6 in Charcot–Marie–Tooth Disease with the Compromised Status of Vitamin-Dependent Processes


Abstract
Simple Summary
Abstract
1. Introduction
2. General Information on Charcot–Marie–Tooth Disease and Associated Mutations
3. CMT Disease upon Mutations of Vitamin-Dependent Enzymes
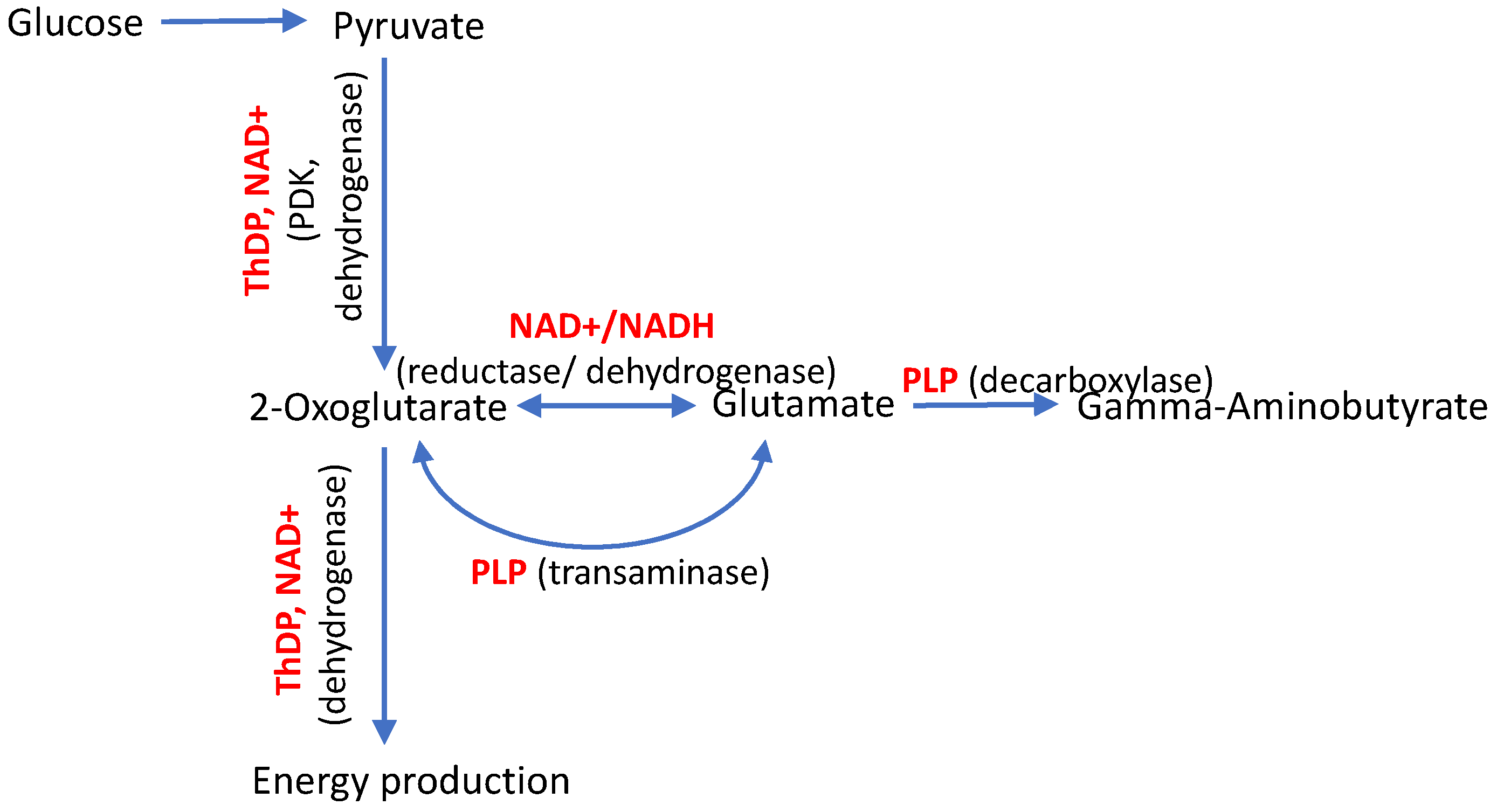
3.1. Vitamin-B6-Dependent Pyridoxal Kinase
3.2. Vitamin-B1-Dependent Enzymes

{kind=link}
{kind=link}
| Gene | Enzyme and Its Vitamin-Related Ligand | Gene Mutation | Protein Variant | Functional Significance of the Mutation | Primarily Affected Pathways | CMT Type | References |
|---|---|---|---|---|---|---|---|
| PDXK | Pyridoxal kinase, producer of the coenzyme form of vitamin B6 (PLP) | c.682G>A c.659G>A c.225T>A | Ala228Thr Arg220Gln Asn75Lys | Perturbed ATP binding, decreased enzyme activity, and low PLP levels are shown. Perturbed ATP binding, decreased enzyme activity, and low PLP levels are shown. Dimer destabilization and increased enzyme degradation are predicted. Very low levels of the enzyme activity and PLP are shown. | Transamination and decarboxylation of amino acids (decreased) | CMT6C (HMSN6C) CMT6C (HMSN6C) CMT6C (HMSN6C), early onset | [3,18] |
| PDK3 | Kinase of pyruvate dehydrogenase (isoform 3), inhibited by the coenzyme form of vitamin B1 (ThDP) | c.G473>A | Arg158His | A 5-fold increase in the enzyme activity and a 6-fold stronger binding to PDC are shown. In the C.elegans model, perturbed synaptic transmission and decreased ATP production are shown. | Mitochondrial oxidation of the glucose-generated pyruvate (decreased) | CMTX6 | [13,27,28] |
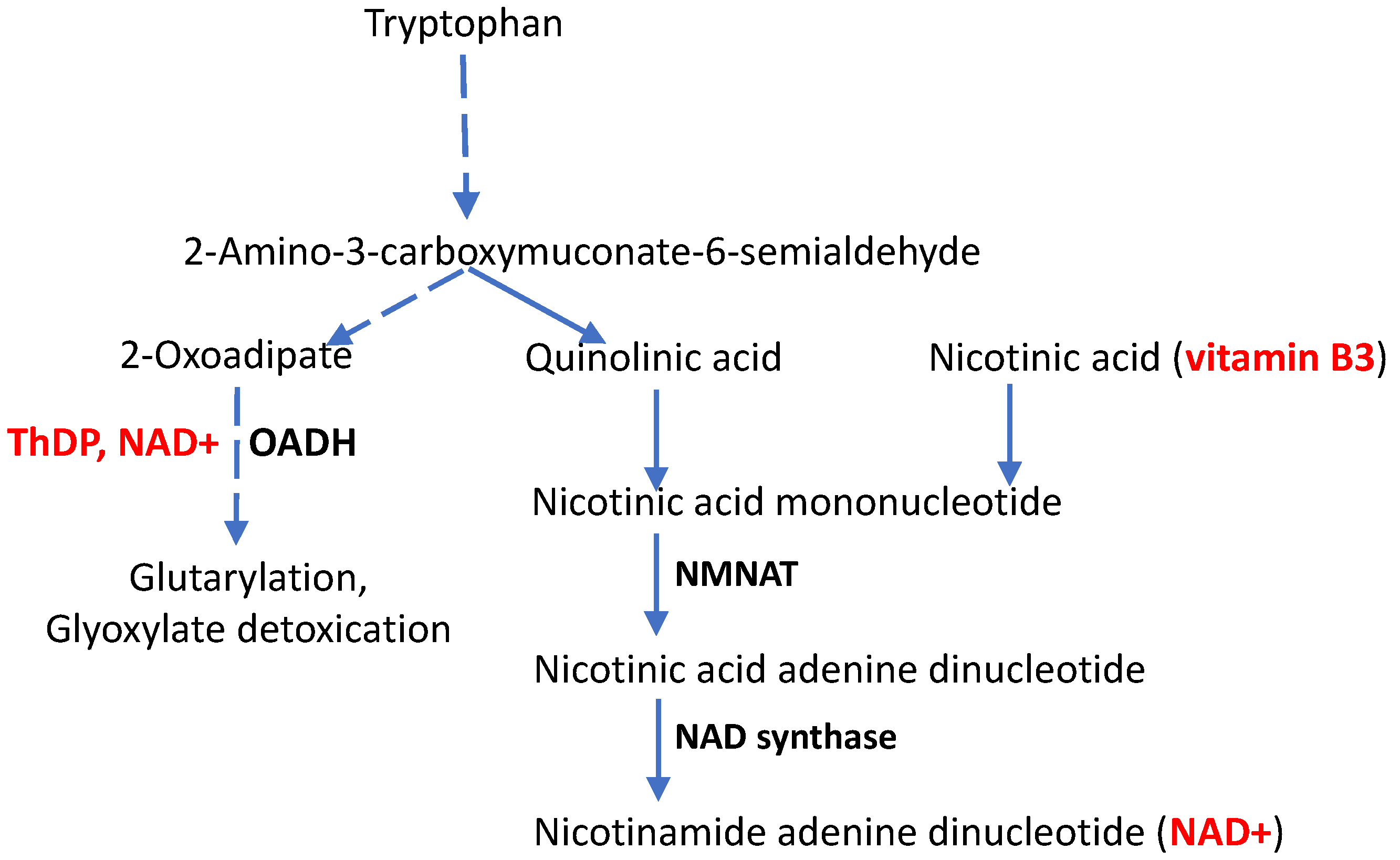
| DHTKD1 | 2-Oxoadipate dehydrogenase, requires the coenzyme form of vitamin B1 (ThDP) for its function | c.1455T>G | No protein from the mutated DNA | Preterm transcription termination at Tyr485 causes a 2-fold decrease in the enzyme mRNA. In HEK293 cells, rapid degradation of the truncated mRNA is shown. | Perturbed degradation of tryptophan and lysine | CMT2Q | [29] |
3.2.1. Isoenzyme 3 of Kinase of the ThDP-Dependent Pyruvate Dehydrogenase
3.2.2. Molecular Mechanisms of CMT Disease Caused by Mutations in the DHTKD1-Encoded ThDP-Dependent 2-Oxoadipate Dehydrogenase
3.3. The Shared Genetic Origin of the Vitamin-Related Forms of CMT and Other Neurological Disorders
4. CMT Disease, Aging and the Vitamin B3 Derivative NAD+
5. The Tissue-Specific Impact of Gene Mutations Perturbing Vitamin-Dependent Processes
6. Therapeutic Doses and Pharmacological Forms of Vitamins B1, B3, and B6
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gibson, G.E.; Feldman, H.H.; Zhang, S.; Flowers, S.A.; Luchsinger, J.A. Pharmacological thiamine levels as a therapeutic approach in alzheimer’s disease. Front. Med. 2022, 9, 1033272. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.O.; Kubota, M.; Terashima, H.; Ishiguro, A.; Kashii, H. Early administration of vitamins b1 and b6 and l-carnitine prevents a second attack of acute encephalopathy with biphasic seizures and late reduced diffusion: A case control study. Brain Dev. 2019, 41, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Chelban, V.; Wilson, M.P.; Warman Chardon, J.; Vandrovcova, J.; Zanetti, M.N.; Zamba-Papanicolaou, E.; Efthymiou, S.; Pope, S.; Conte, M.R.; Abis, G.; et al. Pdxk mutations cause polyneuropathy responsive to pyridoxal 5′-phosphate supplementation. Ann. Neurol. 2019, 86, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Rusch, C.T.; Wortmann, S.B.; Kovacs-Nagy, R.; Grehten, P.; Haberle, J.; Latal, B.; Stettner, G.M. Thiamine pyrophosphokinase deficiency due to mutations in the tpk1 gene: A rare, treatable neurodegenerative disorder. Neuropediatrics 2021, 52, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Qin, J.; Liu, D.; Wang, Y.; Shen, X.; Yang, N.; Zhou, H.; Cai, X.T.; Wang, Z.L.; Yu, D.; et al. Reduced thiamine binding is a novel mechanism for tpk deficiency disorder. Mol. Genet. Genom. 2019, 294, 409–416. [Google Scholar] [CrossRef]
- Bunik, V.; Aleshin, V.; Nogues, I.; Kahne, T.; Parroni, A.; Contestabile, R.; Salvo, M.L.; Graf, A.; Tramonti, A. Thiamine-dependent regulation of mammalian brain pyridoxal kinase in vitro and in vivo. J. Neurochem. 2022, 161, 20–39. [Google Scholar] [CrossRef]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo nad assay reveals the intracellular nad contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo nad(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Pan, X.; Fei, G.; Lu, J.; Jin, L.; Pan, S.; Chen, Z.; Wang, C.; Sang, S.; Liu, H.; Hu, W.; et al. Measurement of blood thiamine metabolites for alzheimer’s disease diagnosis. EBioMedicine 2016, 3, 155–162. [Google Scholar] [CrossRef]
- Pan, X.; Sang, S.; Fei, G.; Jin, L.; Liu, H.; Wang, Z.; Wang, H.; Zhong, C. Enhanced activities of blood thiamine diphosphatase and monophosphatase in alzheimer’s disease. PLoS ONE 2017, 12, e0167273. [Google Scholar] [CrossRef]
- Fattal-Valevski, A.; Kesler, A.; Sela, B.A.; Nitzan-Kaluski, D.; Rotstein, M.; Mesterman, R.; Toledano-Alhadef, H.; Stolovitch, C.; Hoffmann, C.; Globus, O.; et al. Outbreak of life-threatening thiamine deficiency in infants in israel caused by a defective soy-based formula. Pediatrics 2005, 115, e233–e238. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.J.; Guo, W.; Chen, L.; Wei, X.W.; He, Y.; Chen, Y.; Dang, S.Y.; Prior, R.; Li, X.; Kuang, Y.; et al. Cmt2q-causing mutation in the dhtkd1 gene lead to sensory defects, mitochondrial accumulation and altered metabolism in a knock-in mouse model. Acta Neuropathol. Commun. 2020, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.K.; Brewer, M.H.; Perez-Siles, G.; Ellis, M.; Ly, C.; Burgess, A.; Neumann, B.; Nicholson, G.A.; Vucic, S.; Kennerson, M.L. Charcot-marie-tooth disease causing mutation (p.R158h) in pyruvate dehydrogenase kinase 3 (pdk3) affects synaptic transmission, atp production and causes neurodegeneration in a cmtx6 c. Elegans model. Hum. Mol. Genet. 2021, 31, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Dohrn, M.F.; Glockle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hortnagel, K.; et al. Frequent genes in rare diseases: Panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J. Neurochem. 2017, 143, 507–522. [Google Scholar] [CrossRef]
- Vaur, P.; Brugg, B.; Mericskay, M.; Li, Z.; Schmidt, M.S.; Vivien, D.; Orset, C.; Jacotot, E.; Brenner, C.; Duplus, E. Nicotinamide riboside, a form of vitamin b(3), protects against excitotoxicity-induced axonal degeneration. FASEB J. 2017, 31, 5440–5452. [Google Scholar] [CrossRef]
- McGuinness, H.Y.; Gu, W.; Shi, Y.; Kobe, B.; Ve, T. Sarm1-dependent axon degeneration: Nucleotide signaling, neurodegenerative disorders, toxicity, and therapeutic opportunities. Neuroscientist 2023. [Google Scholar] [CrossRef]
- Balashova, N.V.; Zavileyskiy, L.G.; Artiukhov, A.V.; Shaposhnikov, L.A.; Sidorova, O.P.; Tishkov, V.I.; Tramonti, A.; Pometun, A.A.; Bunik, V.I. Efficient assay and marker significance of nad(+) in human blood. Front. Med. 2022, 9, 886485. [Google Scholar] [CrossRef]
- Keller, N.; Mendoza-Ferreira, N.; Maroofian, R.; Chelban, V.; Khalil, Y.; Mills, P.B.; Boostani, R.; Torbati, P.N.; Karimiani, E.G.; Thiele, H.; et al. Hereditary polyneuropathy with optic atrophy due to pdxk variant leading to impaired vitamin b6 metabolism. Neuromuscul. Disord. 2020, 30, 583–589. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Mkrtchyan, G.V.; Bunik, V.I. Mechanisms of non-coenzyme action of thiamine: Protein targets and medical significance. Biochemistry 2019, 84, 829–850. [Google Scholar] [CrossRef]
- Boyko, A.; Tsepkova, P.; Aleshin, V.; Artiukhov, A.; Mkrtchyan, G.; Ksenofontov, A.; Baratova, L.; Ryabov, S.; Graf, A.; Bunik, V. Severe spinal cord injury in rats induces chronic changes in the spinal cord and cerebral cortex metabolism, adjusted by thiamine that improves locomotor performance. Front. Mol. Neurosci. 2021, 14, 620593. [Google Scholar] [CrossRef]
- Mkrtchyan, G.V.; Ucal, M.; Mullebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 925–931. [Google Scholar] [CrossRef]
- Weidinger, A.; Milivojev, N.; Hosmann, A.; Duvigneau, J.C.; Szabo, C.; Toro, G.; Rauter, L.; Vaglio-Garro, A.; Mkrtchyan, G.V.; Trofimova, L.; et al. Oxoglutarate dehydrogenase complex controls glutamate-mediated neuronal death. Redox Biol. 2023, 62, 102669. [Google Scholar] [CrossRef] [PubMed]
- Tiamkao, S.; Boonsong, A.; Saepeung, K.; Kasemsap, N.; Apiwattanakul, M.; Suanprasert, N.; Hemachudha, T.; Pithak, P.; Juntee, K.; Waisaen, C.; et al. An outbreak of peripheral neuropathy in a prison. Case Rep. Neurol. 2019, 11, 53–60. [Google Scholar] [CrossRef]
- Bunik, V. Vitamin-Dependent Complexes of 2-Oxo Acid Dehydrogenases: Structure, Function, Regulation and Medical Implications; Nova Science Publishers: New York, NY, USA, 2017. [Google Scholar]
- Hanberry, B.S.; Berger, R.; Zastre, J.A. High-dose vitamin b1 reduces proliferation in cancer cell lines analogous to dichloroacetate. Cancer Chemother. Pharmacol. 2014, 73, 585–594. [Google Scholar] [CrossRef]
- Jonus, H.C.; Byrnes, C.C.; Kim, J.; Valle, M.L.; Bartlett, M.G.; Said, H.M.; Zastre, J.A. Thiamine mimetics sulbutiamine and benfotiamine as a nutraceutical approach to anticancer therapy. Biomed. Pharmacother. 2020, 121, 109648. [Google Scholar] [CrossRef] [PubMed]
- Kennerson, M.L.; Yiu, E.M.; Chuang, D.T.; Kidambi, A.; Tso, S.C.; Ly, C.; Chaudhry, R.; Drew, A.P.; Rance, G.; Delatycki, M.B.; et al. A new locus for x-linked dominant charcot-marie-tooth disease (cmtx6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (pdk3) gene. Hum. Mol. Genet. 2013, 22, 1404–1416. [Google Scholar] [CrossRef]
- Perez-Siles, G.; Cutrupi, A.; Ellis, M.; Screnci, R.; Mao, D.; Uesugi, M.; Yiu, E.M.; Ryan, M.M.; Choi, B.O.; Nicholson, G.; et al. Energy metabolism and mitochondrial defects in x-linked charcot-marie-tooth (cmtx6) ipsc-derived motor neurons with the p.R158h pdk3 mutation. Sci. Rep. 2020, 10, 9262. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.Y.; Gu, M.M.; Sun, L.H.; Guo, W.T.; Zhu, H.B.; Ma, J.F.; Yuan, W.T.; Kuang, Y.; Ji, B.J.; Wu, X.L.; et al. A nonsense mutation in dhtkd1 causes charcot-marie-tooth disease type 2 in a large chinese pedigree. Am. J. Hum. Genet. 2012, 91, 1088–1094. [Google Scholar] [CrossRef]
- Klyuyeva, A.; Tuganova, A.; Kedishvili, N.; Popov, K.M. Tissue-specific kinase expression and activity regulate flux through the pyruvate dehydrogenase complex. J. Biol. Chem. 2019, 294, 838–851. [Google Scholar] [CrossRef]
- Boyko, A.I.; Karlina, I.S.; Zavileyskiy, L.G.; Aleshin, V.A.; Artiukhov, A.V.; Kaehne, T.; Ksenofontov, A.L.; Ryabov, S.I.; Graf, A.V.; Tramonti, A.; et al. Delayed impact of 2-oxoadipate dehydrogenase inhibition on the rat brain metabolism is linked to protein glutarylation. Front. Med. 2022, 9, 896263. [Google Scholar] [CrossRef]
- Xie, L.; Xiao, Y.; Meng, F.; Li, Y.; Shi, Z.; Qian, K. Functions and mechanisms of lysine glutarylation in eukaryotes. Front. Cell Dev. Biol. 2021, 9, 667684. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.Y.; Zhu, H.; Shen, Y.; Wan, Y.H.; Tu, X.D.; Wu, W.T.; Tang, L.; Zhang, H.X.; Lu, S.Y.; Jin, X.L.; et al. Dhtkd1 deficiency causes charcot-marie-tooth disease in mice. Mol. Cell. Biol. 2018, 38, e00085-18. [Google Scholar] [CrossRef] [PubMed]
- Danhauser, K.; Sauer, S.W.; Haack, T.B.; Wieland, T.; Staufner, C.; Graf, E.; Zschocke, J.; Strom, T.M.; Traub, T.; Okun, J.G.; et al. Dhtkd1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria. Am. J. Hum. Genet. 2012, 91, 1082–1087. [Google Scholar] [CrossRef]
- Hagen, J.; te Brinke, H.; Wanders, R.J.; Knegt, A.C.; Oussoren, E.; Hoogeboom, A.J.; Ruijter, G.J.; Becker, D.; Schwab, K.O.; Franke, I.; et al. Genetic basis of alpha-aminoadipic and alpha-ketoadipic aciduria. J. Inherit. Metab. Dis. 2015, 38, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Stiles, A.R.; Venturoni, L.; Mucci, G.; Elbalalesy, N.; Woontner, M.; Goodman, S.; Abdenur, J.E. New cases of dhtkd1 mutations in patients with 2-ketoadipic aciduria. JIMD Rep. 2016, 25, 15–19. [Google Scholar] [PubMed]
- Sherrill, J.D.; Kc, K.; Wang, X.; Wen, T.; Chamberlin, A.; Stucke, E.M.; Collins, M.H.; Abonia, J.P.; Peng, Y.; Wu, Q.; et al. Whole-exome sequencing uncovers oxidoreductases dhtkd1 and ogdhl as linkers between mitochondrial dysfunction and eosinophilic esophagitis. JCI Insight 2018, 3, e99922. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Chen, Z.T.; Zhou, R.L.; Wang, Y.Z. A chinese pedigree with a novel mutation in gjb1 gene and a rare variation in dhtkd1 gene for diverse charcot-marie-tooth diseases. Mol. Med. Rep. 2019, 19, 4484–4490. [Google Scholar] [CrossRef]
- Fabrizi, G.M.; Hoyer, H.; Taioli, F.; Cavallaro, T.; Hilmarsen, H.T.; Squintani, G.M.; Zanette, G.; Braathen, G.J. Inherited motor-sensory neuropathy with upper limb predominance associated with the tropomyosin-receptor kinase fused gene. Neuromuscul. Disord. 2020, 30, 227–231. [Google Scholar] [CrossRef]
- Leandro, J.; Khamrui, S.; Wang, H.; Suebsuwong, C.; Nemeria, N.S.; Huynh, K.; Moustakim, M.; Secor, C.; Wang, M.; Dodatko, T.; et al. Inhibition and crystal structure of the human dhtkd1-thiamin diphosphate complex. ACS Chem. Biol. 2020, 15, 2041–2047. [Google Scholar] [CrossRef]
- Bezerra, G.A.; Foster, W.R.; Bailey, H.J.; Hicks, K.G.; Sauer, S.W.; Dimitrov, B.; McCorvie, T.J.; Okun, J.G.; Rutter, J.; Kolker, S.; et al. Crystal structure and interaction studies of human dhtkd1 provide insight into a mitochondrial megacomplex in lysine catabolism. IUCrJ 2020, 7, 693–706. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Kazantsev, A.V.; Lukashev, N.V.; Bellinzoni, M.; Bunik, V.I. Selective inhibition of 2-oxoglutarate and 2-oxoadipate dehydrogenases by the phosphonate analogs of their 2-oxo acid substrates. Front. Chem. 2020, 8, 596187. [Google Scholar] [CrossRef] [PubMed]
- Nemeria, N.S.; Nagy, B.; Sanchez, R.; Zhang, X.; Leandro, J.; Ambrus, A.; Houten, S.M.; Jordan, F. Functional versatility of the human 2-oxoadipate dehydrogenase in the l-lysine degradation pathway toward its non-cognate substrate 2-oxopimelic acid. Int. J. Mol. Sci. 2022, 23, 8213. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Degtyarev, D. Structure-function relationships in the 2-oxo acid dehydrogenase family: Substrate-specific signatures and functional predictions for the 2-oxoglutarate dehydrogenase-like proteins. Proteins 2008, 71, 874–890. [Google Scholar] [CrossRef] [PubMed]
- Ozohanics, O.; Zhang, X.; Nemeria, N.S.; Ambrus, A.; Jordan, F. Probing the e1o-e2o and e1a-e2o interactions in binary subcomplexes of the human 2-oxoglutarate dehydrogenase and 2-oxoadipate dehydrogenase complexes by chemical cross-linking mass spectrometry and molecular dynamics simulation. Int. J. Mol. Sci. 2023, 24, 4555. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Zhang, X.; Leandro, J.; Zhou, J.; Yang, L.; Houten, S.M.; Jordan, F. Toward an understanding of the structural and mechanistic aspects of protein-protein interactions in 2-oxoacid dehydrogenase complexes. Life 2021, 11, 407. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Gerfen, G.; Nareddy, P.R.; Yang, L.; Zhang, X.; Szostak, M.; Jordan, F. The mitochondrial 2-oxoadipate and 2-oxoglutarate dehydrogenase complexes share their e2 and e3 components for their function and both generate reactive oxygen species. Free Radic. Biol. Med. 2018, 115, 136–145. [Google Scholar] [CrossRef]
- Boyko, A.I.; Artiukhov, A.V.; Kaehne, T.; di Salvo, M.L.; Bonaccorsi di Patti, M.C.; Contestabile, R.; Tramonti, A.; Bunik, V.I. Isoforms of the dhtkd1-encoded 2-oxoadipate dehydrogenase, identified in animal tissues, are not observed upon the human dhtkd1 expression in bacterial or yeast systems. Biochemistry 2020, 85, 920–929. [Google Scholar] [CrossRef]
- Biagosch, C.; Ediga, R.D.; Hensler, S.V.; Faerberboeck, M.; Kuehn, R.; Wurst, W.; Meitinger, T.; Kolker, S.; Sauer, S.; Prokisch, H. Elevated glutaric acid levels in dhtkd1-/gcdh- double knockout mice challenge our current understanding of lysine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2220–2228. [Google Scholar] [CrossRef]
- Leandro, J.; Dodatko, T.; Aten, J.; Nemeria, N.S.; Zhang, X.; Jordan, F.; Hendrickson, R.C.; Sanchez, R.; Yu, C.; DeVita, R.J.; et al. Dhtkd1 and ogdh display substrate overlap in cultured cells and form a hybrid 2-oxo acid dehydrogenase complex in vivo. Hum. Mol. Genet. 2020, 29, 1168–1179. [Google Scholar] [CrossRef]
- Wu, Y.; Williams, E.G.; Dubuis, S.; Mottis, A.; Jovaisaite, V.; Houten, S.M.; Argmann, C.A.; Faridi, P.; Wolski, W.; Kutalik, Z.; et al. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 2014, 158, 1415–1430. [Google Scholar] [CrossRef]
- Wang, T.J.; Ngo, D.; Psychogios, N.; Dejam, A.; Larson, M.G.; Vasan, R.S.; Ghorbani, A.; O’Sullivan, J.; Cheng, S.; Rhee, E.P.; et al. 2-aminoadipic acid is a biomarker for diabetes risk. J. Clin. Investig. 2013, 123, 4309–4317. [Google Scholar] [CrossRef]
- Plubell, D.L.; Fenton, A.M.; Wilmarth, P.A.; Bergstrom, P.; Zhao, Y.; Minnier, J.; Heinecke, J.W.; Yang, X.; Pamir, N. Gm-csf driven myeloid cells in adipose tissue link weight gain and insulin resistance via formation of 2-aminoadipate. Sci. Rep. 2018, 8, 11485. [Google Scholar] [CrossRef]
- Lim, J.; Liu, Z.; Apontes, P.; Feng, D.; Pessin, J.E.; Sauve, A.A.; Angeletti, R.H.; Chi, Y. Dual mode action of mangiferin in mouse liver under high fat diet. PLoS ONE 2014, 9, e90137. [Google Scholar] [CrossRef]
- Tsepkova, P.M.; Artiukhov, A.V.; Boyko, A.I.; Aleshin, V.A.; Mkrtchyan, G.V.; Zvyagintseva, M.A.; Ryabov, S.I.; Ksenofontov, A.L.; Baratova, L.A.; Graf, A.V.; et al. Thiamine induces long-term changes in amino acid profiles and activities of 2-oxoglutarate and 2-oxoadipate dehydrogenases in rat brain. Biochemistry 2017, 82, 723–736. [Google Scholar] [CrossRef]
- Timmons, J.A.; Atherton, P.J.; Larsson, O.; Sood, S.; Blokhin, I.O.; Brogan, R.J.; Volmar, C.H.; Josse, A.R.; Slentz, C.; Wahlestedt, C.; et al. A coding and non-coding transcriptomic perspective on the genomics of human metabolic disease. Nucleic Acids Res. 2018, 46, 7772–7792. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Grabarska, A.; Gumbarewicz, E.; Aleshin, V.A.; Kahne, T.; Obata, T.; Kazantsev, A.V.; Lukashev, N.V.; Stepulak, A.; Fernie, A.R.; et al. Synthetic analogues of 2-oxo acids discriminate metabolic contribution of the 2-oxoglutarate and 2-oxoadipate dehydrogenases in mammalian cells and tissues. Sci. Rep. 2020, 10, 1886. [Google Scholar] [CrossRef]
- Xu, W.; Zhu, H.; Gu, M.; Luo, Q.; Ding, J.; Yao, Y.; Chen, F.; Wang, Z. Dhtkd1 is essential for mitochondrial biogenesis and function maintenance. FEBS Lett. 2013, 587, 3587–3592. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Panahi, H.K.S.; Kavyani, B.; Heng, B.; Tan, V.; Braidy, N.; Guillemin, G.J. The role of kynurenine pathway and nad(+) metabolism in myalgic encephalomyelitis/chronic fatigue syndrome. Aging Dis. 2022, 13, 698–711. [Google Scholar] [CrossRef]
- Birkisdottir, M.B.; van Galen, I.; Brandt, R.M.C.; Barnhoorn, S.; van Vliet, N.; van Dijk, C.; Nagarajah, B.; Imholz, S.; van Oostrom, C.T.; Reiling, E.; et al. The use of progeroid DNA repair-deficient mice for assessing anti-aging compounds, illustrating the benefits of nicotinamide riboside. Front. Aging 2022, 3, 1005322. [Google Scholar]
- Vreones, M.; Mustapic, M.; Moaddel, R.; Pucha, K.A.; Lovett, J.; Seals, D.R.; Kapogiannis, D.; Martens, C.R. Oral nicotinamide riboside raises nad+ and lowers biomarkers of neurodegenerative pathology in plasma extracellular vesicles enriched for neuronal origin. Aging Cell 2023, 22, e13754. [Google Scholar] [CrossRef]
- Osmanovic, A.; Gogol, I.; Martens, H.; Widjaja, M.; Muller, K.; Schreiber-Katz, O.; Feuerhake, F.; Langhans, C.D.; Schmidt, G.; Andersen, P.M.; et al. Heterozygous dhtkd1 variants in two european cohorts of amyotrophic lateral sclerosis patients. Genes 2021, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Storbeck, M.; Strathmann, E.A.; Delle Vedove, A.; Holker, I.; Altmueller, J.; Naghiyeva, L.; Schmitz-Steinkruger, L.; Vezyroglou, K.; Motameny, S.; et al. Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum. Mutat. 2018, 39, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; King, M.S.; Smith, A.C.; Olahova, M.; Bansagi, B.; Roos, A.; Eyassu, F.; Borchers, C.; Ramesh, V.; Lochmuller, H.; et al. Mitochondrial oxodicarboxylate carrier deficiency is associated with mitochondrial DNA depletion and spinal muscular atrophy-like disease. Genet. Med. 2018, 20, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Ævarsson, A.E.; Chuang, J.L.; Wynn, R.M.; Turley, S.; Chuang, D.T.; Hol, W.G. Crystal structure of human branched-chain alpha-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure 2000, 8, 277–291. [Google Scholar] [CrossRef]
- Chuang, J.L.; Wynn, R.M.; Moss, C.C.; Song, J.L.; Li, J.; Awad, N.; Mandel, H.; Chuang, D.T. Structural and biochemical basis for novel mutations in homozygous israeli maple syrup urine disease patients: A proposed mechanism for the thiamin-responsive phenotype. J. Biol. Chem. 2004, 279, 17792–17800. [Google Scholar] [CrossRef]
- Elstner, M.; Morris, C.M.; Heim, K.; Lichtner, P.; Bender, A.; Mehta, D.; Schulte, C.; Sharma, M.; Hudson, G.; Goldwurm, S.; et al. Single-cell expression profiling of dopaminergic neurons combined with association analysis identifies pyridoxal kinase as parkinson’s disease gene. Ann. Neurol. 2009, 66, 792–798. [Google Scholar] [CrossRef]
- Elstner, M.; Lichtner, P.; Schulte, C.; Gasser, T.; Meitinger, T.; Prokisch, H.; Turnbull, D.M. Reply. Ann. Neurol. 2010, 67, 412. [Google Scholar] [CrossRef]
- Vilarino-Guell, C.; Wider, C.; Aasly, J.O.; White, L.R.; Rajput, A.; Rajput, A.H.; Lynch, T.; Krygowska-Wajs, A.; Jasinska-Myga, B.; Opala, G.; et al. Association of pyridoxal kinase and parkinson disease. Ann. Neurol. 2010, 67, 409–411. [Google Scholar] [CrossRef]
- Guella, I.; Asselta, R.; Tesei, S.; Zini, M.; Pezzoli, G.; Duga, S. The pdxk rs2010795 variant is not associated with parkinson disease in italy. Ann. Neurol. 2010, 67, 411–412. [Google Scholar] [CrossRef]
- M’Angale, P.G.; Staveley, B.E. A loss of pdxk model of parkinson disease in drosophila can be suppressed by buffy. BMC Res. Notes 2017, 10, 205. [Google Scholar] [CrossRef]
- Xiong, L.L.; Qin, Y.X.; Xiao, Q.X.; Jin, Y.; Al-Hawwas, M.; Ma, Z.; Wang, Y.C.; Belegu, V.; Zhou, X.F.; Xue, L.L.; et al. Microrna339 targeting pdxk improves motor dysfunction and promotes neurite growth in the remote cortex subjected to spinal cord transection. Front. Cell Dev. Biol. 2020, 8, 577. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Jansen, P.R.; Savage, J.E.; Nandakumar, P.; Wang, X.; Hinds, D.A.; Gelernter, J.; Levey, D.F.; Polimanti, R.; Stein, M.B. Genome-wide meta-analysis of insomnia prioritizes genes associated with metabolic and psychiatric pathways. Nat. Genet. 2022, 54, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, V.A.; Artiukhov, A.V.; Kaehne, T.; Graf, A.V.; Bunik, V.I. Daytime dependence of the activity of the rat brain pyruvate dehydrogenase corresponds to the mitochondrial sirtuin 3 level and acetylation of brain proteins, all regulated by thiamine administration decreasing phosphorylation of pdha ser293. Int. J. Mol. Sci. 2021, 22, 8006. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Mkrtchyan, G.V.; Kaehne, T.; Graf, A.V.; Maslova, M.V.; Bunik, V.I. Diurnal regulation of the function of the rat brain glutamate dehydrogenase by acetylation and its dependence on thiamine administration. J. Neurochem. 2020, 153, 80–102. [Google Scholar] [CrossRef]
- Lee, D.; Kim, K.; Lee, Y.; Oh, K.; Jung, S.J. The relationship between thiamine intake and long sleep duration: Results from the korea national health and nutrition examination survey. J. Prev. Med. Public Health 2022, 55, 520–528. [Google Scholar] [CrossRef]
- Balzamo, E.; Vuillon-Cacciuttolo, G. [facilitation of a state of wakefulness by semi-chronic treatment with sulbutiamin (arcalion) in macaca mulatta]. Rev. Electroencephalogr. Neurophysiol. Clin. 1982, 12, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Kazamel, M.; Boes, C.J. Charcot marie tooth disease (cmt): Historical perspectives and evolution. J. Neurol. 2015, 262, 801–805. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. Nad+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Imai, S.I.; Guarente, L. It takes two to tango: Nad(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef]
- Johnson, S.; Imai, S.I. Nad (+) biosynthesis, aging, and disease. F1000Research 2018, 7, 132. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Nad(+) in aging: Molecular mechanisms and translational implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. Nad(+) intermediates: The biology and therapeutic potential of nmn and nr. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef]
- Kulikova, V.A.; Gromyko, D.V.; Nikiforov, A.A. The regulatory role of nad in human and animal cells. Biochemistry 2018, 83, 800–812. [Google Scholar] [CrossRef]
- Ying Cao, Y.W.; Yang, J. Nad+-dependent mechanism of pathological axon degeneration. Cell Insight 2022, 1, 100019. [Google Scholar]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear nad biosynthesis and sirt1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Moss, K.R.; Hoke, A. Targeting the programmed axon degeneration pathway as a potential therapeutic for charcot-marie-tooth disease. Brain Res. 2020, 1727, 146539. [Google Scholar] [CrossRef]
- Barile, A.; Nogues, I.; di Salvo, M.L.; Bunik, V.; Contestabile, R.; Tramonti, A. Molecular characterization of pyridoxine 5’-phosphate oxidase and its pathogenic forms associated with neonatal epileptic encephalopathy. Sci. Rep. 2020, 10, 13621. [Google Scholar] [CrossRef] [PubMed]
- Gachon, F.; Fonjallaz, P.; Damiola, F.; Gos, P.; Kodama, T.; Zakany, J.; Duboule, D.; Petit, B.; Tafti, M.; Schibler, U. The loss of circadian par bzip transcription factors results in epilepsy. Genes Dev. 2004, 18, 1397–1412. [Google Scholar] [CrossRef]
- Ghosh, R.; Mandal, A.; Roy, D.; Chatterjee, S.; Ghosh, M.K.; Dubey, S.; Lahiri, D.; Finsterer, J.; Ray, B.K. Seizure as a presenting manifestation of wernicke’s encephalopathy induced by hyperemesis gravidarum. J. Fam. Med. Prim. Care 2021, 10, 567–571. [Google Scholar]
- Mengi, T.; Beckmann, Y. Wernicke encephalopathy with epileptic seizures during pregnancy. Neurocase 2022, 28, 185–187. [Google Scholar] [CrossRef]
- Mimouni-Bloch, A.; Goldberg-Stern, H.; Strausberg, R.; Brezner, A.; Heyman, E.; Inbar, D.; Kivity, S.; Zvulunov, A.; Sztarkier, I.; Fogelman, R.; et al. Thiamine deficiency in infancy: Long-term follow-up. Pediatr. Neurol. 2014, 51, 311–316. [Google Scholar] [CrossRef]
- Szigeti, K.; Lupski, J.R. Charcot-marie-tooth disease. Eur. J. Hum. Genet. 2009, 17, 703–710. [Google Scholar] [CrossRef]
- Marce-Grau, A.; Marti-Sanchez, L.; Baide-Mairena, H.; Ortigoza-Escobar, J.D.; Perez-Duenas, B. Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies. J. Inherit. Metab. Dis. 2019, 42, 581–597. [Google Scholar] [CrossRef]
- Jensen, K.V.; Frid, M.; Stodberg, T.; Barbaro, M.; Wedell, A.; Christensen, M.; Bak, M.; Ek, J.; Madsen, C.G.; Darin, N.; et al. Diagnostic pitfalls in vitamin b6-dependent epilepsy caused by mutations in the plpbp gene. JIMD Rep. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T. Disorders affecting vitamin b(6) metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef]
- Alegre, G.F.S.; Pastore, G.M. Nad+ precursors nicotinamide mononucleotide (nmn) and nicotinamide riboside (nr): Potential dietary contribution to health. Curr. Nutr. Rep. 2023. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunik, V. The Therapeutic Potential of Vitamins B1, B3 and B6 in Charcot–Marie–Tooth Disease with the Compromised Status of Vitamin-Dependent Processes. Biology 2023, 12, 897. https://doi.org/10.3390/biology12070897
Bunik V. The Therapeutic Potential of Vitamins B1, B3 and B6 in Charcot–Marie–Tooth Disease with the Compromised Status of Vitamin-Dependent Processes. Biology. 2023; 12(7):897. https://doi.org/10.3390/biology12070897
Chicago/Turabian StyleBunik, Victoria. 2023. "The Therapeutic Potential of Vitamins B1, B3 and B6 in Charcot–Marie–Tooth Disease with the Compromised Status of Vitamin-Dependent Processes" Biology 12, no. 7: 897. https://doi.org/10.3390/biology12070897
APA StyleBunik, V. (2023). The Therapeutic Potential of Vitamins B1, B3 and B6 in Charcot–Marie–Tooth Disease with the Compromised Status of Vitamin-Dependent Processes. Biology, 12(7), 897. https://doi.org/10.3390/biology12070897