
Chromosomal Instability in Genome Evolution: From Cancer to Macroevolution


1
Andalusian Center for Molecular Biology and Regenerative Medicine—CABIMER, University of Pablo de Olavide—University of Seville—CSIC, Junta de Andalucía, 41092 Seville, Spain
2
Genetic Department, Faculty of Biology, University of Seville, 41080 Seville, Spain
*
Authors to whom correspondence should be addressed.
Biology 2023, 12(5), 671; https://doi.org/10.3390/biology12050671
Submission received: 29 March 2023
/
Revised: 21 April 2023
/
Accepted: 25 April 2023
/
Published: 28 April 2023
{kind=link}
{kind=link}
{kind=link}
{kind=link}
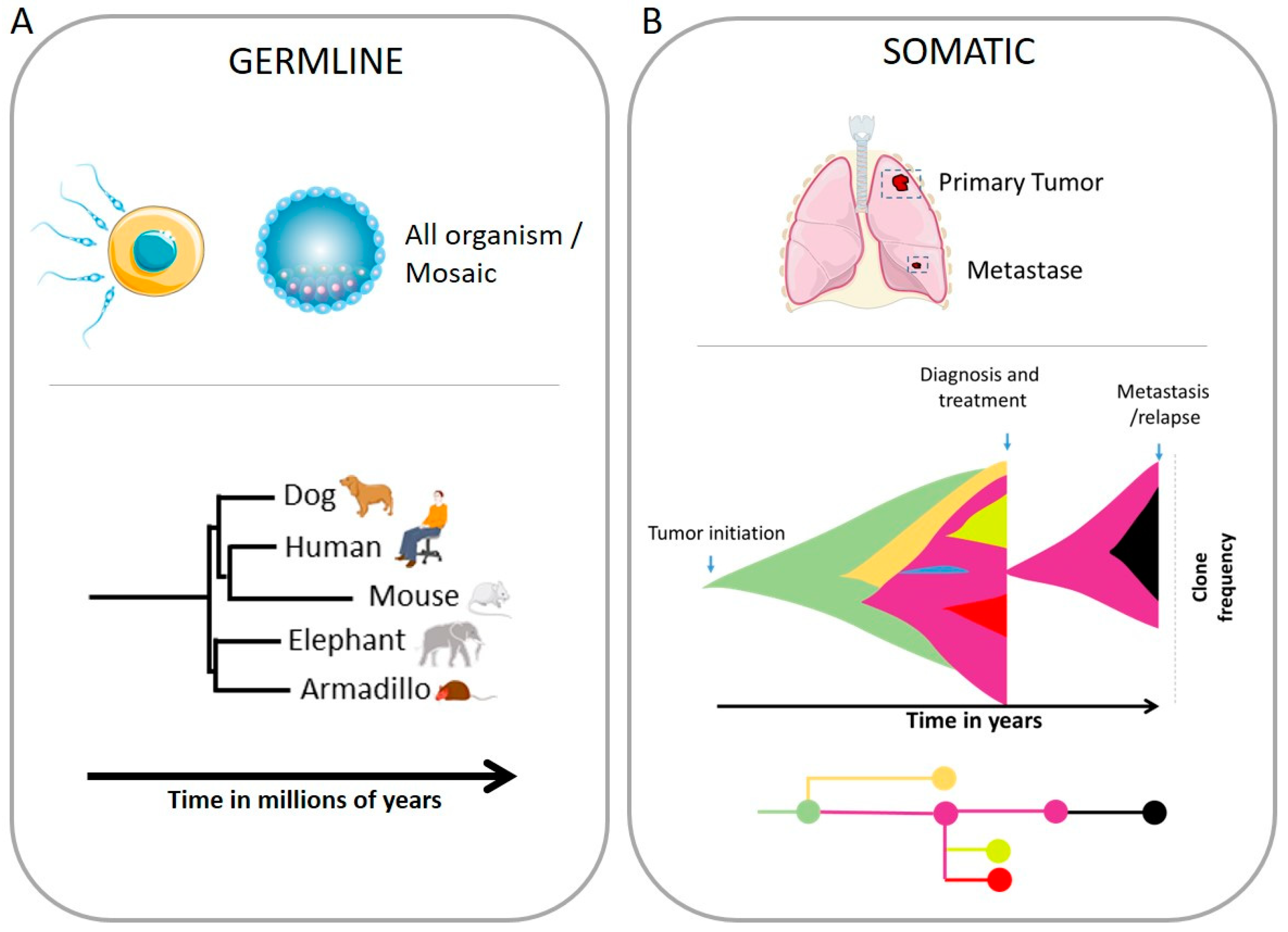
Simple Summary
The experimental/pathological observation of genome chaos (including massive and large-scale translocations, chromothripsis, and polyploidy cancer cells) highlighted the importance of genome reorganization in evolution. Recent advances in sequencing and bioinformatic analysis have highlighted such chromosomal diversity. The evolution of the genome has been studied in the field of macroevolution and speciation as well as in the context of cancer and tumor progression. Evolution is inherent to adaptation to the environment and preparing for future pressure to initiate survival. Human cells are plastic, and several mechanisms are involved in sporadic timing in view of generating genomic diversity under normal conditions, such as during gametogenesis or during pathology like cancer. Interestingly, patterns of chromosomal instability are strikingly similar in evolution and in cancer. Here we will discuss some events leading to several varieties of chromosomal patterns, from cancer to speciation, and discuss the disease associated with chromosomal instability.
Abstract
The integrity of the genome is crucial for the survival of all living organisms. However, genomes need to adapt to survive certain pressures, and for this purpose use several mechanisms to diversify. Chromosomal instability (CIN) is one of the main mechanisms leading to the creation of genomic heterogeneity by altering the number of chromosomes and changing their structures. In this review, we will discuss the different chromosomal patterns and changes observed in speciation, in evolutional biology as well as during tumor progression. By nature, the human genome shows an induction of diversity during gametogenesis but as well during tumorigenesis that can conclude in drastic changes such as the whole genome doubling to more discrete changes as the complex chromosomal rearrangement chromothripsis. More importantly, changes observed during speciation are strikingly similar to the genomic evolution observed during tumor progression and resistance to therapy. The different origins of CIN will be treated as the importance of double-strand breaks (DSBs) or the consequences of micronuclei. We will also explain the mechanisms behind the controlled DSBs, and recombination of homologous chromosomes observed during meiosis, to explain how errors lead to similar patterns observed during tumorigenesis. Then, we will also list several diseases associated with CIN, resulting in fertility issues, miscarriage, rare genetic diseases, and cancer. Understanding better chromosomal instability as a whole is primordial for the understanding of mechanisms leading to tumor progression.