


Humanin and Its Pathophysiological Roles in Aging: A Systematic Review
 , ,
, ,  , ,
, ,  and
and
Abstract
Simple Summary
Abstract
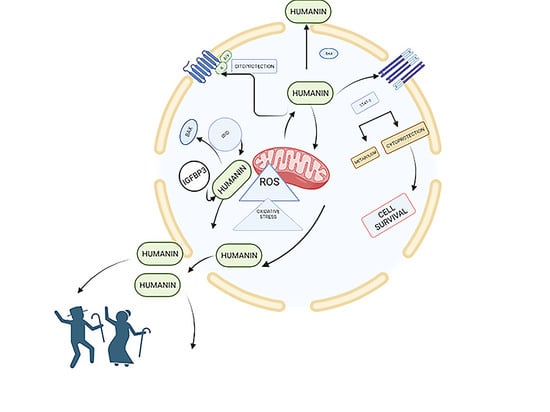
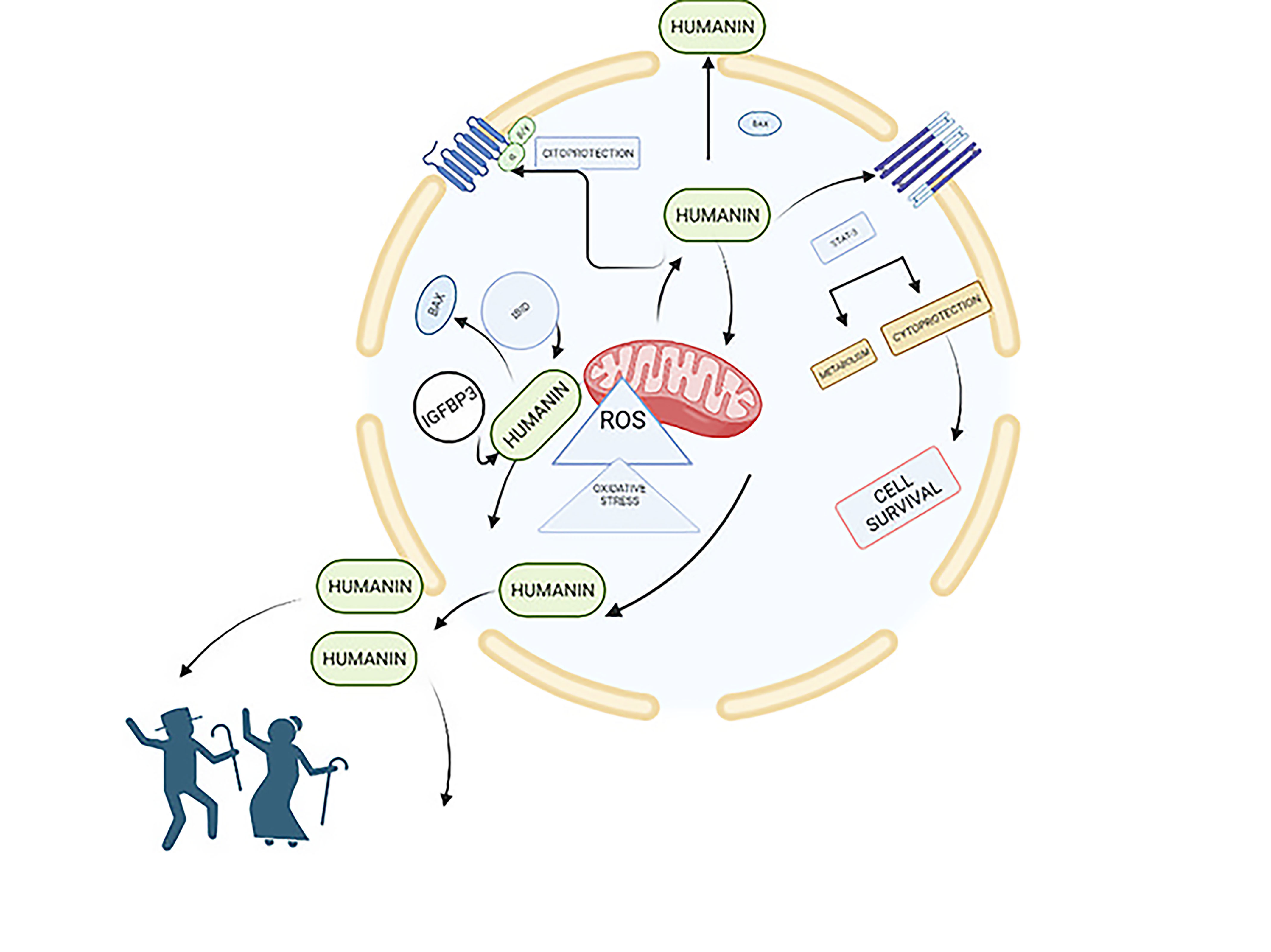
1. Introduction

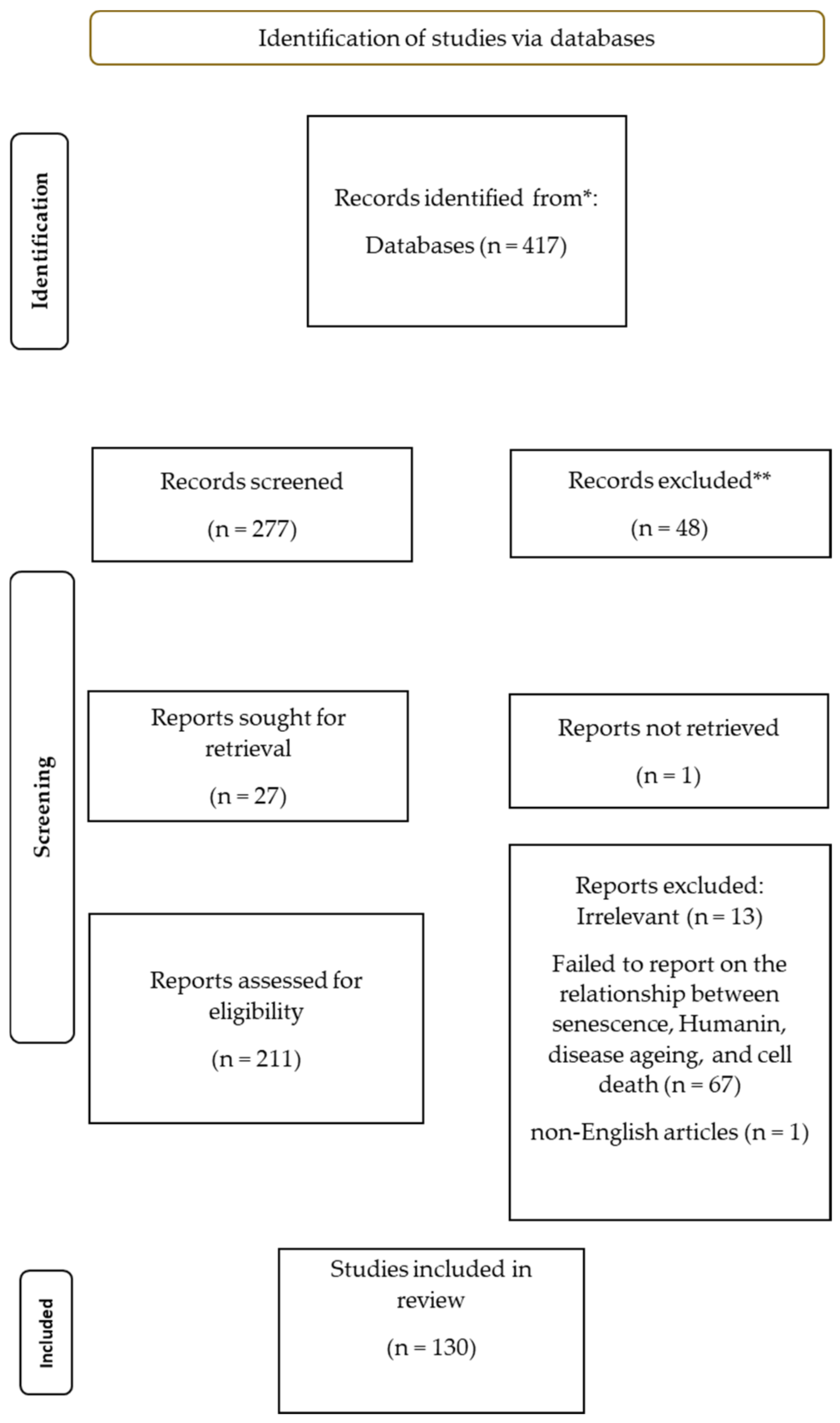
2. Materials and Methods
2.1. Search Strategy
2.2. Inclusion Criteria
2.3. Exclusion Criteria
2.4. Data Extraction
2.5. Quality Assessment
2.6. Analysis
3. Results and Discussion
3.1. Brain and Neurodegeneration
3.2. Heart and Cardiovascular Diseases

{kind=link}
{kind=link}
{kind=link}
| Article | Study Design | Population | Outcome Measures |
|---|---|---|---|
| Yen, K. et al. (2020) [40] | In vivo study | C. elegans, Mouse, Human | Circulating levels of humanin and their relation to diseases of aging and lifespan |
| Cai, H. et al. (2021) [72] | Review | N/A | Protective effect of humanin against oxidative stress |
| Conte, M. et al. (2019) [30] | In vivo study | Human | Aging; longevity. plasma levels of different mitokines |
| Gong, Z. et al. (2022) [49] | Review | Mouse, Porcine | Protective effect of humanin in myocardial ischemia-reperfusion. |
| Muzumdar, R.H. et al. (2010) [67] | In vivo study | Mouse | Intracardiac administration of a single dose of HNG at the time of ischemia or reperfusion reduces infarction size and improves cardiac function. Humanin attenuated protein levels of Bax in the heart following MI. |
| Thummasorn, S. et al. (2016) [68] | In vivo study | Mouse | HNG treatment reduced cardiac infarct size, improved the function of the left ventricle, and decreased cardiac arrhythmias during MI-R. HNG treatment improved cardiac mitochondrial function and decreased the mitochondrial ROS level in cardiac cells |
| Sharp, T.E., III et al. (2020) [71] | In vivo study | Porcine | A single dose of HNG, administered at the time of reperfusion, diminishes cardiac infarction size and inhibits cardiomyocyte apoptosis |
3.3. Immune System and Inflammation
3.4. Diabetes and Obesity
3.5. Potential Mechanisms Involved in the Protective Effects of Humanin
3.5.1. Autophagy
| Article | Study Design | Population | Outcome Measures |
|---|---|---|---|
| Miller, B. et al. (2022) [98] | Review | N/A | Aging; mitochondrial copy number; relative ratio of mtDNA to nuclear DNA, and autophagy |
| Li, P. et al. (2021) [107] | N/A | N/A | Autophagy in skeletal muscle |
| Sreekumar, P.G. et al. (2016) [114] | In vitro study | hRPE cells | Expression of humanin and its effect on oxidative stress-induced cell death; mitochondrial bioenergetics, and senescence. |
| Gong, Z. et al. (2018) [115] | In vitro study | N/A | Chaperone-mediated autophagy |
| Kim, S.J. et al. (2022) [116] | In vivo and in vitro study | HEK293 cells; Mouse; C. elegans; Human | Role of humanin in the activation and regulation of autophagy |
| Kim, S.J. et al. (2018) [124] | In vitro study | Primary senescent human fibroblasts | Number of mitochondria; levels of mitochondrial respiration; mtDNA methylation, and mitochondria-encoded peptides |
3.5.2. Cytoprotective Activity
| Article | Study Design | Population | Outcome Measures |
|---|---|---|---|
| Alsanousi, N. et al. (2016) [125] | In vitro study | N/A | Inhibitory effect against amyloid-β fibrillation of humanin |
| Zaman, F. et al. (2019) [126] | In vitro study | N/A | Humanin regulator of Hedgehog signaling and prevents glucocorticoid-induced bone growth impairment |
| Qin, Q. et al. (2018) [129] | In vivo study | Mouse | Effect of exogenous humanin to prevent and reverse cardiac fibrosis and apoptosis in the aging heart |
| Liu, C. et al. (2019) [106] | In vivo study | Human | Expression levels of humanin and MOTS-C in skeletal muscle and serum levels in CKD |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jakovljevic, M.M.; Netz, Y.; Buttigieg, S.C.; Adany, R.; Laaser, U.; Varjacic, M. Population aging and migration–history and UN forecasts in the EU-28 and its east and south near neighborhood–one century perspective 1950–2050. Glob. Health 2018, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Ryan, B.; Sonntag, W.; Kavvada, A.; Friedl, L. Earth observation in service of the 2030 Agenda for Sustainable Development. Geo-Spat. Inf. Sci. 2017, 20, 77–96. [Google Scholar] [CrossRef]
- Frias, L. Highlights from ICOPA 2022. Lancet Microbe 2022, 3, e813. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I. Which is the most significant cause of aging? Antioxidants 2015, 4, 793–810. [Google Scholar] [CrossRef]
- Sthijns, M.M.; van Blitterswijk, C.A.; LaPointe, V.L. Redox regulation in regenerative medicine and tissue engineering: The paradox of oxygen. J. Tissue Eng. Regen. Med. 2018, 12, 2013–2020. [Google Scholar] [CrossRef]
- Coradduzza, D.; Congiargiu, A.; Chen, Z.; Zinellu, A.; Carru, C.; Medici, S. Ferroptosis and Senescence: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 3658. [Google Scholar] [CrossRef]
- Coradduzza, D.; Arru, C.; Culeddu, N.; Congiargiu, A.; Azara, E.G.; Scanu, A.M.; Zinellu, A.; Muroni, M.R.; Rallo, V.; Medici, S. Quantitative Metabolomics to Explore the Role of Plasma Polyamines in Colorectal Cancer. Int. J. Mol. Sci. 2022, 24, 101. [Google Scholar] [CrossRef]
- Coradduzza, D.; Ghironi, A.; Azara, E.; Culeddu, N.; Cruciani, S.; Zinellu, A.; Maioli, M.; De Miglio, M.R.; Medici, S.; Fozza, C. Role of Polyamines as Biomarkers in Lymphoma Patients: A Pilot Study. Diagnostics 2022, 12, 2151. [Google Scholar] [CrossRef]
- Coradduzza, D.; Solinas, T.; Azara, E.; Culeddu, N.; Cruciani, S.; Zinellu, A.; Medici, S.; Maioli, M.; Madonia, M.; Carru, C. Plasma polyamine biomarker panels: Agmatine in support of prostate cancer diagnosis. Biomolecules 2022, 12, 514. [Google Scholar] [CrossRef]
- Coradduzza, D.; Azara, E.; Medici, S.; Arru, C.; Solinas, T.; Madonia, M.; Zinellu, A.; Carru, C. A preliminary study procedure for detection of polyamines in plasma samples as a potential diagnostic tool in prostate cancer. J. Chromatogr. B 2021, 1162, 122468. [Google Scholar] [CrossRef]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.; Madeo, F.; Kroemer, G. Metabolic control of longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular communication: Shaping health and disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018, 28, 516–524.e517. [Google Scholar] [CrossRef]
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 2016, 8, 796. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.-J.; Mehta, H.; Hevener, A.L.; de Cabo, R. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ito, Y.; Niikura, T.; Shao, Z.; Hata, M.; Oyama, F.; Nishimoto, I. Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem. Biophys. Res. Commun. 2001, 283, 460–468. [Google Scholar] [CrossRef]
- Nishimoto, I.; Matsuoka, M.; Niikura, T. Unravelling the role of Humanin. Trends Mol. Med. 2004, 10, 102–105. [Google Scholar] [CrossRef]
- Matsuoka, M.; Hashimoto, Y.; Aiso, S.; Nishimoto, I. Humanin and colivelin: Neuronal-death-suppressing peptides for Alzheimer’s disease and amyotrophic lateral sclerosis. CNS Drug Rev. 2006, 12, 113–122. [Google Scholar] [CrossRef]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef]
- Yasuda, H.; Nakatani, S.; Stugaard, M.; Tsujita-Kuroda, Y.; Bando, K.; Kobayashi, J.; Yamagishi, M.; Kitakaze, M.; Kitamura, S.; Miyatake, K. Failure to prevent progressive dilation of ascending aorta by aortic valve replacement in patients with bicuspid aortic valve: Comparison with tricuspid aortic valve. Circulation 2003, 108, II-291–II-294. [Google Scholar] [CrossRef] [PubMed]
- Bodzioch, M.; Lapicka-Bodzioch, K.; Zapala, B.; Kamysz, W.; Kiec-Wilk, B.; Dembinska-Kiec, A. Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics 2009, 94, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.-S.; Duan, D.-X.; Ma, R.-H.; Shen, J.-Y.; Li, H.-L.; Ma, Z.-W.; Luo, Y.; Wang, L.; Qi, X.-H.; Wang, Q. Humanin attenuates Alzheimer-like cognitive deficits and pathological changes induced by amyloid β-peptide in rats. Neurosci. Bull. 2014, 30, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, H.; Wu, J.; Yin, L.; Yan, L.-J.; Zhang, C. Humanin attenuates NMDA-induced excitotoxicity by inhibiting ROS-dependent JNK/p38 MAPK pathway. Int. J. Mol. Sci. 2018, 19, 2982. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Luciano, F.; Zhu, X.; Guo, B.; Satterthwait, A.C.; Reed, J.C. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J. Biol. Chem. 2005, 280, 15815–15824. [Google Scholar] [CrossRef] [PubMed]
- Njomen, E.; Evans, H.G.; Gedara, S.H.; Heyl, D.L. Humanin peptide binds to insulin-like growth factor-binding protein 3 (IGFBP3) and regulates its interaction with importin-β. Protein Pept. Lett. 2015, 22, 869–876. [Google Scholar] [CrossRef]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.-W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef]
- Gong, Z.; Tasset, I. Humanin enhances the cellular response to stress by activation of chaperone-mediated autophagy. Oncotarget 2018, 9, 10832. [Google Scholar] [CrossRef]
- Muzumdar, R.H.; Huffman, D.M.; Atzmon, G.; Buettner, C.; Cobb, L.J.; Fishman, S.; Budagov, T.; Cui, L.; Einstein, F.H.; Poduval, A. Humanin: A novel central regulator of peripheral insulin action. PLoS ONE 2009, 4, e6334. [Google Scholar] [CrossRef]
- Conte, M.; Ostan, R.; Fabbri, C.; Santoro, A.; Guidarelli, G.; Vitale, G.; Mari, D.; Sevini, F.; Capri, M.; Sandri, M. Human aging and longevity are characterized by high levels of mitokines. J. Gerontol. Ser. A 2019, 74, 600–607. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Mitochondrial-derived peptides exacerbate senescence. Rejuvenation Res. 2018, 21, 369–373. [Google Scholar] [CrossRef]
- Klein, L.E.; Cui, L.; Gong, Z.; Su, K.; Muzumdar, R. A humanin analog decreases oxidative stress and preserves mitochondrial integrity in cardiac myoblasts. Biochem. Biophys. Res. Commun. 2013, 440, 197–203. [Google Scholar] [CrossRef]
- Minasyan, L.; Sreekumar, P.G.; Hinton, D.R.; Kannan, R. Protective mechanisms of the mitochondrial-derived peptide humanin in oxidative and endoplasmic reticulum stress in RPE cells. Oxid. Med. Cell. Longev. 2017, 2017, 1675230. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kim, S.-J.; Miller, B.; Kumagai, H.; Silverstein, A.R.; Flores, M.; Yen, K. Mitochondrial-derived peptides in aging and age-related diseases. Geroscience 2021, 43, 1113–1121. [Google Scholar] [CrossRef]
- Yen, K.; Wan, J.; Mehta, H.H.; Miller, B.; Christensen, A.; Levine, M.E.; Salomon, M.P.; Brandhorst, S.; Xiao, J.; Kim, S.-J. Humanin prevents age-related cognitive decline in mice and is associated with improved cognitive age in humans. Sci. Rep. 2018, 8, 14212. [Google Scholar] [CrossRef]
- Lorenzini, A.; Salmon, A.B.; Lerner, C.; Torres, C.; Ikeno, Y.; Motch, S.; McCarter, R.; Sell, C. Mice producing reduced levels of insulin-like growth factor type 1 display an increase in maximum, but not mean, life span. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, 410–419. [Google Scholar] [CrossRef]
- Yen, K.; Mehta, H.H.; Kim, S.-J.; Lue, Y.; Hoang, J.; Guerrero, N.; Port, J.; Bi, Q.; Navarrete, G.; Brandhorst, S. The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging 2020, 12, 11185. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Uzsoy, R.; Martin-Vega, L.A. Efficient algorithms for scheduling semiconductor burn-in operations. Oper. Res. 1992, 40, 764–775. [Google Scholar] [CrossRef]
- Caso, V.M.; Manzo, V.; Pecchillo Cimmino, T.; Conti, V.; Caso, P.; Esposito, G.; Russo, V.; Filippelli, A.; Ammendola, R.; Cattaneo, F. Regulation of inflammation and oxidative stress by formyl peptide receptors in cardiovascular disease progression. Life 2021, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, D.G.; Kim, S.G.; Massat, A.E.; Bachar, A.R.; Oh, Y.K.; Herrmann, J.; Rodriguez-Porcel, M.; Cohen, P.; Lerman, L.O.; Lerman, A. Humanin, a cytoprotective peptide, is expressed in carotid artherosclerotic plaques in humans. PLoS ONE 2012, 7, e31065. [Google Scholar] [CrossRef]
- Yacila, G.; Sari, Y. Potential therapeutic drugs and methods for the treatment of amyotrophic lateral sclerosis. Curr. Med. Chem. 2014, 21, 3583–3593. [Google Scholar] [CrossRef]
- Yen, K.; Lee, C.; Mehta, H.; Cohen, P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J. Mol. Endocrinol. 2013, 50, R11. [Google Scholar] [CrossRef]
- Boutari, C.; Pappas, P.D.; Theodoridis, T.D.; Vavilis, D. Humanin and diabetes mellitus: A review of in vitro and in vivo studies. World J. Diabetes 2022, 13, 213. [Google Scholar] [CrossRef]
- Wang, X.; Wu, Z.; He, Y.; Zhang, H.; Tian, L.; Zheng, C.; Shang, T.; Zhu, Q.; Li, D.; He, Y. Humanin prevents high glucose-induced monocyte adhesion to endothelial cells by targeting KLF2. Mol. Immunol. 2018, 101, 245–250. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Nikiforov, N.G.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. The role of mitochondria-derived peptides in cardiovascular diseases and their potential as therapeutic targets. Int. J. Mol. Sci. 2021, 22, 8770. [Google Scholar] [CrossRef]
- Gong, Z.; Goetzman, E.; Muzumdar, R.H. Cardio-protective role of Humanin in myocardial ischemia-reperfusion. Biochim. Biophys. Acta BBA Gen. Subj. 2022, 1866, 130066. [Google Scholar] [CrossRef]
- Zárate, S.C.; Traetta, M.E.; Codagnone, M.G.; Seilicovich, A.; Reinés, A.G. Humanin, a mitochondrial-derived peptide released by astrocytes, prevents synapse loss in hippocampal neurons. Front. Aging Neurosci. 2019, 11, 123. [Google Scholar] [CrossRef]
- Gong, Z.; Tas, E.; Muzumdar, R. Humanin and age-related diseases: A new link? Front. Endocrinol. 2014, 5, 210. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Aβ. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef]
- Zhang, W.; Du, Y.; Bai, M.; Xi, Y.; Li, Z.; Miao, J. Retracted: S14G-humanin inhibits Aβ1–42 fibril formation, disaggregates preformed fibrils, and protects against Aβ-induced cytotoxicity in vitro. J. Pept. Sci. 2013, 19, 159–165. [Google Scholar] [CrossRef]
- Romeo, M.; Stravalaci, M.; Beeg, M.; Rossi, A.; Fiordaliso, F.; Corbelli, A.; Salmona, M.; Gobbi, M.; Cagnotto, A.; Diomede, L. Humanin specifically interacts with amyloid-β oligomers and counteracts their in vivo toxicity. J. Alzheimers Dis. 2017, 57, 857–871. [Google Scholar] [CrossRef]
- Park, T.-Y.; Kim, S.-H.; Shin, Y.-C.; Lee, N.-H.; Lee, R.-K.C.; Shim, J.-H.; Glimcher, L.H.; Mook-Jung, I.; Cheong, E.; Kim, W.-K. Amelioration of neurodegenerative diseases by cell death-induced cytoplasmic delivery of humanin. J. Control. Release 2013, 166, 307–315. [Google Scholar] [CrossRef]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Cai, H.; Liu, Y.; Men, H.; Zheng, Y. Protective mechanism of humanin against oxidative stress in aging-related cardiovascular diseases. Front. Endocrinol. 2021, 12, 680. [Google Scholar] [CrossRef]
- Li, Y.; Lin, R.; Peng, X.; Wang, X.; Liu, X.; Li, L.; Bai, R.; Wen, S.; Ruan, Y.; Chang, X. The Role of Mitochondrial Quality Control in Anthracycline-Induced Cardiotoxicity: From Bench to Bedside. Oxid. Med. Cell. Longev. 2022, 2022, 3659278. [Google Scholar] [CrossRef]
- Deng, J.; Jiang, Y.; Chen, Z.B.; Rhee, J.-W.; Deng, Y.; Wang, Z.V. Mitochondrial Dysfunction in Cardiac Arrhythmias. Cells 2023, 12, 679. [Google Scholar] [CrossRef]
- Ji, A.; Meyer, J.M.; Cai, L.; Akinmusire, A.; de Beer, M.C.; Webb, N.R.; Van der Westhuyzen, D.R. Scavenger receptor SR-BI in macrophage lipid metabolism. Atherosclerosis 2011, 217, 106–112. [Google Scholar] [CrossRef]
- Ding, Y.; Feng, Y.; Zhu, W.; Zou, Y.; Xie, Y.; Wang, F.; Liu, C.F.; Zhang, Y.; Liu, H. [Gly14]-Humanin Prevents Lipid Deposition and Endothelial Cell Apoptosis in a Lectin-like Oxidized Low-density Lipoprotein Receptor-1-Dependent Manner. Lipids 2019, 54, 697–705. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, Z.-h.; Li, X.-y.; Xu, X.; Hu, L.-f.; Zhang, Y.-l.; Liu, C.-f. Protection effect of [Gly14]-Humanin from apoptosis induced by high glucose in human umbilical vein endothelial cells. Diabetes Res. Clin. Pract. 2014, 106, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-Y.; Chang, H.-J.; Choi, S.-I.; Kim, K.-I.; Cho, Y.-S.; Youn, T.-J.; Chung, W.-Y.; Chae, I.-H.; Choi, D.-J.; Kim, H.-S. Long-term exercise training attenuates age-related diastolic dysfunction: Association of myocardial collagen cross-linking. J. Korean Med. Sci. 2009, 24, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Wan, J.; Miyazaki, B.; Fang, Y.; Guevara-Aguirre, J.; Yen, K.; Longo, V.; Bartke, A.; Cohen, P. IGF-I regulates the age-dependent signaling peptide humanin. Aging Cell 2014, 13, 958–961. [Google Scholar] [CrossRef]
- Thummasorn, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. High-dose Humanin analogue applied during ischemia exerts cardioprotection against ischemia/reperfusion injury by reducing mitochondrial dysfunction. Cardiovasc. Ther. 2017, 35, e12289. [Google Scholar] [CrossRef]
- Muzumdar, R.H.; Huffman, D.M.; Calvert, J.W.; Jha, S.; Weinberg, Y.; Cui, L.; Nemkal, A.; Atzmon, G.; Klein, L.; Gundewar, S. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1940–1948. [Google Scholar] [CrossRef]
- Thummasorn, S.; Apaijai, N.; Kerdphoo, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; Chattipakorn, N. Humanin exerts cardioprotection against cardiac ischemia/reperfusion injury through attenuation of mitochondrial dysfunction. Cardiovasc. Ther. 2016, 34, 404–414. [Google Scholar] [CrossRef]
- Wu, D.; Kampmann, E.; Qian, G. Novel insights into the role of mitochondria-derived peptides in myocardial infarction. Front. Physiol. 2021, 12, 750177. [Google Scholar] [CrossRef]
- Frontiers Editorial Office. Retraction: Novel insights into the role of mitochondria-derived peptides in myocardial infarction. Front. Physiol. 2022, 13, 1006441. [Google Scholar] [CrossRef]
- Sharp, T.E., III; Gong, Z.; Scarborough, A.; Goetzman, E.S.; Ali, M.J.; Spaletra, P.; Lefer, D.J.; Muzumdar, R.H.; Goodchild, T.T. Efficacy of a novel mitochondrial-derived peptide in a porcine model of myocardial ischemia/reperfusion injury. Basic Transl. Sci. 2020, 5, 699–714. [Google Scholar] [CrossRef]
- Coradduzza, D.; Cruciani, S.; Arru, C.; Garroni, G.; Pashchenko, A.; Jedea, M.; Zappavigna, S.; Caraglia, M.; Amler, E.; Carru, C.; et al. Role of miRNA-145, 148, and 185 and Stem Cells in Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 1626. [Google Scholar] [CrossRef]
- Bajracharya, R.; Ballard, J.W.O. Low protein to carbohydrate ratio diet delays onset of Parkinsonism like phenotype in Drosophila melanogaster parkin null mutants. Mech. Ageing Dev. 2016, 160, 19–27. [Google Scholar] [CrossRef]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef]
- Fujita, Y.; Taniguchi, Y.; Shinkai, S.; Tanaka, M.; Ito, M. Secreted growth differentiation factor 15 as a potential biomarker for mitochondrial dysfunctions in aging and age-related disorders. Geriatr. Gerontol. Int. 2016, 16, 17–29. [Google Scholar] [CrossRef]
- Hashimoto, H.; Niikura, T.; Tajima, H.; Yasukawa, M.; Kouyama, K.; Doyu, M.; Sobue, G.; Koide, T.; Tsuji, D.; Lang, J. Erratum: A rescue factor abolishing nueronal cell death by a wide spectrum of familial Alzheimer’s diseas genes and Aβ (Proceedings of the National Academy of Science of the United States of America (May 22, 2001) 98: 11 (6336-6341)). Proc. Natl. Acad. Sci. USA 2001, 98, 12854. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How increased Hormesis in Aging and Neurodegeneration—A Prodigy Awaiting Dissection mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Klaus, S.; Ost, M. Mitochondrial uncoupling and longevity—A role for mitokines? Exp. Gerontol. 2020, 130, 110796. [Google Scholar] [CrossRef]
- Esterhuizen, K.; Van der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef]
- Kanfer, G.; Peterka, M.; Arzhanik, V.K.; Drobyshev, A.L.; Ataullakhanov, F.I.; Volkov, V.A.; Kornmann, B. CENP-F couples cargo to growing and shortening microtubule ends. Mol. Biol. Cell 2017, 28, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Bachar, A.R.; Scheffer, L.; Schroeder, A.S.; Nakamura, H.K.; Cobb, L.J.; Oh, Y.K.; Lerman, L.O.; Pagano, R.E.; Cohen, P.; Lerman, A. Humanin is expressed in human vascular walls and has a cytoprotective effect against oxidized LDL-induced oxidative stress. Cardiovasc. Res. 2010, 88, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.-T.; Zhao, L.; Li, J.-H. Neuroprotective peptide humanin inhibits inflammatory response in astrocytes induced by lipopolysaccharide. Neurochem. Res. 2013, 38, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, M.F.; Jaita, G.; Magri, M.L.; Zárate, S.; Moreno Ayala, M.; Ferraris, J.; Eijo, G.; Pisera, D.; Candolfi, M.; Seilicovich, A. Antiapoptotic factor humanin is expressed in normal and tumoral pituitary cells and protects them from TNF-α-induced apoptosis. PLoS ONE 2014, 9, e111548. [Google Scholar] [CrossRef]
- Varma Shrivastav, S.; Bhardwaj, A.; Pathak, K.A.; Shrivastav, A. Insulin-like growth factor binding protein-3 (IGFBP-3): Unraveling the role in mediating IGF-independent effects within the cell. Front. Cell Dev. Biol. 2020, 8, 286. [Google Scholar] [CrossRef]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef]
- Conte, M.; Martucci, M.; Chiariello, A.; Franceschi, C.; Salvioli, S. Mitochondria, immunosenescence and inflammaging: A role for mitokines? In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2020; pp. 607–617. [Google Scholar]
- De Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory cascade in Alzheimer’s disease pathogenesis: A review of experimental findings. Cells 2021, 10, 2581. [Google Scholar] [CrossRef]
- Conte, M.; Sabbatinelli, J.; Chiariello, A.; Martucci, M.; Santoro, A.; Monti, D.; Arcaro, M.; Galimberti, D.; Scarpini, E.; Bonfigli, A.R. Disease-specific plasma levels of mitokines FGF21, GDF15, and Humanin in type II diabetes and Alzheimer’s disease in comparison with healthy aging. Geroscience 2021, 43, 985–1001. [Google Scholar] [CrossRef]
- Li, W.; Zhang, D.; Yuan, W.; Wang, C.; Huang, Q.; Luo, J. Humanin ameliorates free fatty acid-induced endothelial inflammation by suppressing the NLRP3 inflammasome. ACS Omega 2020, 5, 22039–22045. [Google Scholar] [CrossRef]
- McLaughlin, K.; Lytvyn, Y.; Luca, M.C.; Liuni, A.; Gori, T.; Parker, J.D. Repeated daily dosing with sildenafil provides sustained protection from endothelial dysfunction caused by ischemia and reperfusion: A human in vivo study. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H888–H894. [Google Scholar] [CrossRef]
- Salemi, M.; Ridolfo, F.; Salluzzo, M.G.; Cannarrella, R.; Giambirtone, M.; Caniglia, S.; Tirolo, C.; Ferri, R.; Romano, C. Humanin gene expression in fibroblast of Down syndrome subjects. Int. J. Med. Sci. 2020, 17, 320. [Google Scholar] [CrossRef]
- Gensous, N.; Bacalini, M.G.; Franceschi, C.; Garagnani, P. Down syndrome, accelerated aging and immunosenescence. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2020; pp. 635–645. [Google Scholar]
- Nashine, S.; Cohen, P.; Wan, J.; Kenney, M.C. Effect of Humanin G (HNG) on inflammation in age-related macular degeneration (AMD). Aging 2022, 14, 4247. [Google Scholar] [CrossRef]
- Merry, T.L.; Chan, A.; Woodhead, J.S.; Reynolds, J.C.; Kumagai, H.; Kim, S.-J.; Lee, C. Mitochondrial-derived peptides in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E659–E666. [Google Scholar] [CrossRef]
- Mehta, H.H.; Xiao, J.; Ramirez, R.; Miller, B.; Kim, S.-J.; Cohen, P.; Yen, K. Metabolomic profile of diet-induced obesity mice in response to humanin and small humanin-like peptide 2 treatment. Metabolomics 2019, 15, 88. [Google Scholar] [CrossRef]
- Cruciani, S.; Garroni, G.; Pala, R.; Coradduzza, D.; Cossu, M.L.; Ginesu, G.C.; Capobianco, G.; Dessole, S.; Ventura, C.; Maioli, M. Metformin and vitamin D modulate adipose-derived stem cell differentiation towards the beige phenotype. Adipocyte 2022, 11, 356–365. [Google Scholar] [CrossRef]
- Miller, B.; Kim, S.J.; Kumagai, H.; Yen, K.; Cohen, P. Mitochondria-derived peptides in aging and healthspan. J. Clin. Investig. 2022, 132, e158449. [Google Scholar] [CrossRef]
- Ng, L.F.; Ng, L.T.; van Breugel, M.; Halliwell, B.; Gruber, J. Mitochondrial DNA damage does not determine C. elegans lifespan. Front. Genet. 2019, 10, 311. [Google Scholar] [CrossRef]
- Hazafa, A.; Batool, A.; Ahmad, S.; Amjad, M.; Chaudhry, S.N.; Asad, J.; Ghuman, H.F.; Khan, H.M.; Naeem, M.; Ghani, U. Humanin: A mitochondrial-derived peptide in the treatment of apoptosis-related diseases. Life Sci. 2021, 264, 118679. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 105–137. [Google Scholar] [CrossRef]
- Wan, W.; Zhang, L.; Lin, Y.; Rao, X.; Wang, X.; Hua, F.; Ying, J. Mitochondria-derived peptide MOTS-c: Effects and mechanisms related to stress, metabolism and aging. J. Transl. Med. 2023, 21, 36. [Google Scholar] [CrossRef]
- Chen, T.-H.; Koh, K.-Y.; Lin, K.M.-C.; Chou, C.-K. Mitochondrial dysfunction as an underlying cause of skeletal muscle disorders. Int. J. Mol. Sci. 2022, 23, 12926. [Google Scholar] [CrossRef] [PubMed]
- Woodhead, J.S.; D’Souza, R.F.; Hedges, C.P.; Wan, J.; Berridge, M.V.; Cameron-Smith, D.; Cohen, P.; Hickey, A.J.; Mitchell, C.J.; Merry, T.L. High-intensity interval exercise increases humanin, a mitochondrial encoded peptide, in the plasma and muscle of men. J. Appl. Physiol. 2020, 128, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Alser, M.; Ramanjaneya, M.; Anwardeen, N.R.; Donati, F.; Botrè, F.; Jerobin, J.; Bettahi, I.; Mohamed, N.A.; Abou-Samra, A.B.; Elrayess, M.A. The Effect of Chronic Endurance Exercise on Serum Levels of MOTS-c and Humanin in Professional Athletes. Rev. Cardiovasc. Med. 2022, 23, 181. [Google Scholar] [CrossRef]
- Liu, C.; Gidlund, E.-K.; Witasp, A.; Qureshi, A.R.; Söderberg, M.; Thorell, A.; Nader, G.A.; Barany, P.; Stenvinkel, P.; von Walden, F. Reduced skeletal muscle expression of mitochondrial-derived peptides humanin and MOTS-C and Nrf2 in chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2019, 317, F1122–F1131. [Google Scholar] [CrossRef]
- Li, P.; Ma, Y.; Yu, C.; Wu, S.; Wang, K.; Yi, H.; Liang, W. Autophagy and aging: Roles in skeletal muscle, eye, brain and hepatic tissue. Front. Cell Dev. Biol. 2021, 9, 2925. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Angius, A.; Pira, G.; Cossu-Rocca, P.; Sotgiu, G.; Saderi, L.; Muroni, M.R.; Virdis, P.; Piras, D.; Vincenzo, R.; Carru, C. Deciphering clinical significance of BCL11A isoforms and protein expression roles in triple-negative breast cancer subtype. J. Cancer Res. Clin. Oncol. 2022. epub ahead of print. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Y.; Shi, Y.; Zhang, Z.; Huang, C.; He, W.; Wang, C.; Shen, H.M. Autophagy in health and disease: From molecular mechanisms to therapeutic target. MedComm 2022, 3, e150. [Google Scholar] [CrossRef]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial adaptation in cancer drug resistance: Prevalence, mechanisms, and management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1238–1253. [Google Scholar] [CrossRef]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 2018, 217, 635–647. [Google Scholar] [CrossRef]
- Kim, S.-J.; Devgan, A.; Miller, B.; Lee, S.M.; Kumagai, H.; Wilson, K.A.; Wassef, G.; Wong, R.; Mehta, H.H.; Cohen, P. Humanin-induced autophagy plays important roles in skeletal muscle function and lifespan extension. Biochim. Biophys. Acta BBA Gen. Subj. 2022, 1866, 130017. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Kannan, R. Mechanisms of protection of retinal pigment epithelial cells from oxidant injury by humanin and other mitochondrial-derived peptides: Implications for age-related macular degeneration. Redox Biol. 2020, 37, 101663. [Google Scholar] [CrossRef]
- Ghosh, R.; Vinod, V.; Symons, J.D.; Boudina, S. Protein and mitochondria quality control mechanisms and cardiac aging. Cells 2020, 9, 933. [Google Scholar] [CrossRef]
- Bahar, E.; Han, S.-Y.; Kim, J.-Y.; Yoon, H. Chemotherapy resistance: Role of mitochondrial and autophagic components. Cancers 2022, 14, 1462. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Han, J.-S.; Jung, Y.; Lee, S.-M.; Park, S.-H.; Park, M.; Shin, M.-G.; Kim, N.; Kang, M.S.; Kim, S. A new AMPK isoform mediates glucose-restriction induced longevity non-cell autonomously by promoting membrane fluidity. Nat. Commun. 2023, 14, 288. [Google Scholar] [CrossRef]
- Burtscher, J.; Soltany, A.; Visavadiya, N.P.; Burtscher, M.; Millet, G.P.; Khoramipour, K.; Khamoui, A.V. Mitochondrial stress and mitokines in aging. Aging Cell 2023, 22, e13770. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.-H. AMPK, mitochondrial function, and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Triolo, M.; Oliveira, A.N.; Kumari, R.; Hood, D.A. The influence of age, sex, and exercise on autophagy, mitophagy, and lysosome biogenesis in skeletal muscle. Skelet. Muscle 2022, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Mehta, H.H.; Wan, J.; Kuehnemann, C.; Chen, J.; Hu, J.-F.; Hoffman, A.R.; Cohen, P. Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging 2018, 10, 1239. [Google Scholar] [CrossRef] [PubMed]
- Alsanousi, N.; Sugiki, T.; Furuita, K.; So, M.; Lee, Y.-H.; Fujiwara, T.; Kojima, C. Solution NMR structure and inhibitory effect against amyloid-β fibrillation of Humanin containing a d-isomerized serine residue. Biochem. Biophys. Res. Commun. 2016, 477, 647–653. [Google Scholar] [CrossRef]
- Zaman, F.; Zhao, Y.; Celvin, B.; Mehta, H.H.; Wan, J.; Chrysis, D.; Ohlsson, C.; Fadeel, B.; Cohen, P.; Sävendahl, L. Humanin is a novel regulator of Hedgehog signaling and prevents glucocorticoid-induced bone growth impairment. FASEB J. 2019, 33, 4962. [Google Scholar] [CrossRef]
- Aghajanian, P.; Mohan, S. The art of building bone: Emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018, 6, 19. [Google Scholar] [CrossRef]
- Chen, H.; Tan, X.-N.; Hu, S.; Liu, R.-Q.; Peng, L.-H.; Li, Y.-M.; Wu, P. Molecular mechanisms of chondrocyte proliferation and differentiation. Front. Cell Dev. Biol. 2021, 9, 664168. [Google Scholar] [CrossRef]
- Qin, Q.; Mehta, H.; Yen, K.; Navarrete, G.; Brandhorst, S.; Wan, J.; Delrio, S.; Zhang, X.; Lerman, L.O.; Cohen, P. Chronic treatment with the mitochondrial peptide humanin prevents age-related myocardial fibrosis in mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1127–H1136. [Google Scholar] [CrossRef]

| Article | Study Design | Population | Outcome Measures |
|---|---|---|---|
| Zárate, S.C. et al. (2019) [50] | In vivo study | Rat | Neuroprotective effect of humanin and relationship with ovarian hormones |
| Yen, K. et al. (2018) [38] | In vitro and in vivo study | SH-SY5Y cells, Mouse | Neuroprotective effect of humanin |
| Yen, K. et al. (2020) [40] | In vivo study | C. elegans, Mouse, Human | Circulating levels of humanin and their relation to diseases of aging and lifespan |
| Gong, Z. et al. (2022) [49] | Review | Mouse, Porcine | Protective effect of humanin in myocardial ischemia-reperfusion. |
| Kim, S.J. et al. (2021) [37] | Review | N/A | Humanin in age-related disease |
| Gong, Z. et al. (2014) [51] | Review | N/A | Role of humanin in age-related disease |
| Caso, V.M. et al. (2021) [42] | In vitro study | N/A | Neuroprotective effects of humanin and its homologs (HNG) from Aβ |
| Zacharias, D.G. et al. (2012) [43] | In vivo study | Mouse | Humanin reduced plaque accumulation in Alzheimer’s disease and has a cytoprotective action in stroke |
| Park, T. et al. (2013) [55] | In vivo study | Middle-aged APPswe/PS1dE9 mice | Treatment with HNG significantly improves spatial learning and memory deficits, reduces Aβ plaque accumulation and insoluble Aβ concentration; decreases neuro-inflammatory responses |
| Article | Study Design | Population | Outcome Measures |
|---|---|---|---|
| Esterhuizen, K. et al. (2017) [80] | In vitro study | N/A | Role of HN against oxidative stress, apoptosis, and inflammatory response |
| Bachar, A.R. et al. (2010) [82] | In vitro study | N/A | HN attenuates inflammation and macrophage infiltration, reduces in vitro production of IL-6, IL-1β, and TNF-α |
| Nashine, S. et al. (2022) [94] | In vivo study | AMD patients AMD RPE transmitochondrial cybrid cells | Treatment with HNG can reduce plasma levels of inflammation-associated marker protein. In AMD RPE cybrid cells, treatment with HNG reduced CD62E/E-Selectin, CD62P/P-Selectin, ICAM-1, TNF-α, MIP-1α, IFN–γ, IL-1β, IL-13. and IL-17A |
| Conte, M. et al. (2021) [89] | In vivo study | Patients with T2D and AD | Plasma levels of fibroblast growth factor 21 (FGF21), growth differentiation factor 15 (GDF15), humanin, and mitochondrial stress-related mitokines |
| Conte, M. et al. (2019) [30] | In vivo study | Human | Aging; longevity; plasma levels of different mitokines |
| Merry, T.L. et al. (2020) [95] | In vivo study | Human | Plasma humanin levels in long-lived subjects |
| Article | Study Design | Population | Outcome Measures |
|---|---|---|---|
| Boutari, C. et al. (2022) [46] | Review | N/A | Role of humanin in age-related diseases |
| Mehta, H.H. et al. (2019) [96] | In vivo study | DIO Mouse | Humanin effect on metabolism |
| Merry, T.L. et al. (2020) [95] | Review | N/A | Role of humanin in metabolism; relationship with (GH)/IGF-1 |
| Muzumdar, R.H. et al. (2009) [29] | In vivo study | Rat | Central effects of HN on insulin action |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coradduzza, D.; Congiargiu, A.; Chen, Z.; Cruciani, S.; Zinellu, A.; Carru, C.; Medici, S. Humanin and Its Pathophysiological Roles in Aging: A Systematic Review. Biology 2023, 12, 558. https://doi.org/10.3390/biology12040558
Coradduzza D, Congiargiu A, Chen Z, Cruciani S, Zinellu A, Carru C, Medici S. Humanin and Its Pathophysiological Roles in Aging: A Systematic Review. Biology. 2023; 12(4):558. https://doi.org/10.3390/biology12040558
Chicago/Turabian StyleCoradduzza, Donatella, Antonella Congiargiu, Zhichao Chen, Sara Cruciani, Angelo Zinellu, Ciriaco Carru, and Serenella Medici. 2023. "Humanin and Its Pathophysiological Roles in Aging: A Systematic Review" Biology 12, no. 4: 558. https://doi.org/10.3390/biology12040558
APA StyleCoradduzza, D., Congiargiu, A., Chen, Z., Cruciani, S., Zinellu, A., Carru, C., & Medici, S. (2023). Humanin and Its Pathophysiological Roles in Aging: A Systematic Review. Biology, 12(4), 558. https://doi.org/10.3390/biology12040558