

Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
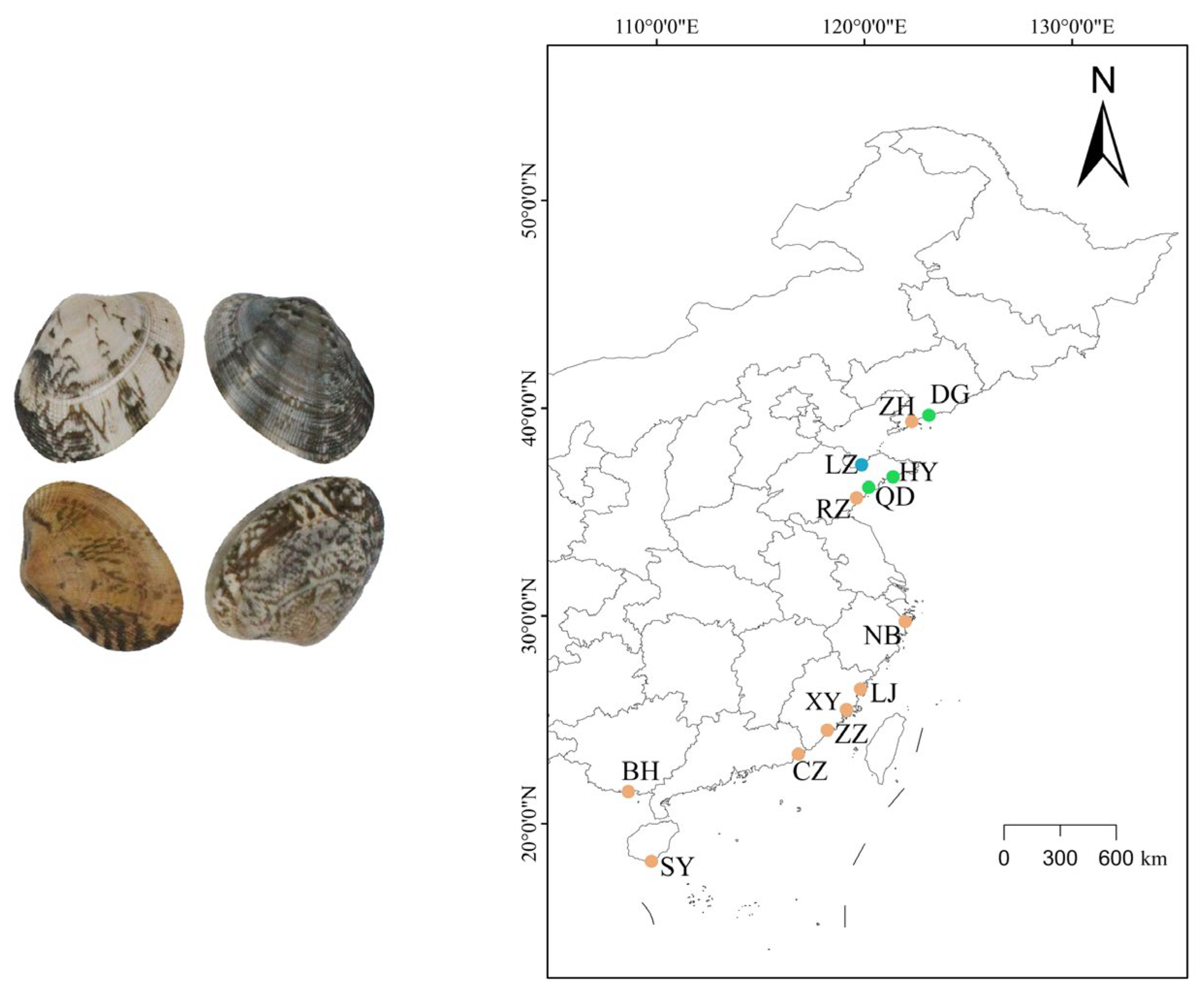
2.1. Sample Collection and DNA Extraction
2.2. Primer Screening and PCR Amplification
2.3. Data Processing
3. Results
3.1. Genetic Diversity within Populations
3.2. Genetic Differentiation among Populations
3.3. Estimation of Effective Population Size (Ne)
4. Discussion
4.1. Genetic Diversity of Manila Clams in Different Populations from North to South
4.2. The Genetic Differentiation among Clam Populations
4.3. Estimation of Effective Population Sizes in Clam Populations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Montaudouin, X.; Lucia, M.; Binias, C.; Lassudrie, M.; Baudrimont, M.; Legeay, A.; Raymond, N.; Jude-Lemeilleur, F.; Lambert, C.; Le Goïc, N. Why is Asari (=Manila) clam Ruditapes philippinarum fitness poor in Arcachon Bay: A meta-analysis to answer? Estuar. Coast. Shelf Sci. 2016, 179, 226–235. [Google Scholar] [CrossRef]
- Tan, Y.; Fang, L.; Qiu, M.; Huo, Z.; Yan, X. Population genetics of the Manila clam (Ruditapes philippinarum) in East Asia. Sci. Rep. 2020, 10, 21890. [Google Scholar] [CrossRef]
- DOF. China Fisheries Statistic Yearbook; China Agriculture Press: Beijing, China, 2021. [Google Scholar]
- Liu, X.; Zhenmin, B.; Jingjie, H.; Shi, W.; Aibin, Z.; Hui, L.; Jianguang, F.; Rucai, W. AFLP analysis revealed differences in genetic diversity of four natural populations of Manila clam (Ruditapes philippinarum) in China. Acta Oceanol. Sin. 2007, 1, 150–158. [Google Scholar]
- Kristensen, T.N.; Hoffmann, A.A.; Pertoldi, C.; Stronen, A.V. What can livestock breeders learn from conservation genetics and vice versa? Front. Genet. 2015, 6, 38. [Google Scholar] [CrossRef]
- Conover, D.; Clarke, L.; Munch, S.; Wagner, G. Spatial and temporal scales of adaptive divergence in marine fishes and the implications for conservation. J. Fish Biol. 2006, 69, 21–47. [Google Scholar] [CrossRef]
- Okumuş, İ.; Çiftci, Y. Fish population genetics and molecular markers: II-molecular markers and their applications in fisheries and aquaculture. Turk. J. Fish. Aquat. Sci. 2003, 3, 51–79. [Google Scholar]
- Askari, G.; Shabani, A.; Kolangi Miandare, H. Application of molecular markers in fisheries and aquaculture. Sci. J. Anim. Sci. 2013, 2, 82–88. [Google Scholar]
- Nie, H.; Niu, H.; Zhao, L.; Yang, F.; Yan, X.; Zhang, G. Genetic diversity and structure of Manila clam (Ruditapes philippinarum) populations from Liaodong peninsula revealed by SSR markers. Biochem. Syst. Ecol. 2015, 59, 116–125. [Google Scholar] [CrossRef]
- An, H.; Lee, J.; Park, J. Population genetics of the Pacific abalone (Haliotis discus hannai) in Korea inferred from microsatellite marker analysis. Genet. Mol. Res. 2012, 11, 3904–3922. [Google Scholar] [CrossRef]
- Bai, Z.; Zheng, H.; Lin, J.; Wang, G.; Li, J. Comparative analysis of the transcriptome in tissues secreting purple and white nacre in the pearl mussel Hyriopsis cumingii. PLoS ONE 2013, 8, e53617. [Google Scholar] [CrossRef]
- Mandal, S.; Jena, J.; Singh, R.K.; Mohindra, V.; Lakra, W.; Deshmukhe, G.; Pathak, A.; Lal, K.K. De novo development and characterization of polymorphic microsatellite markers in a schilbid catfish, Silonia silondia (Hamilton, 1822) and their validation for population genetic studies. Mol. Biol. Rep. 2016, 43, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cui, F.; Hu, P.; Yi, G.; Ge, Y.; Liu, W.; Yan, H.; Wang, L.; Liu, H.; Song, J. Using of microsatellite DNA profiling to identify hatchery-reared seed and assess potential genetic risks associated with large-scale release of swimming crab Portunus trituberculatus in Panjin, China. Fish. Res. 2018, 207, 187–196. [Google Scholar] [CrossRef]
- Wang, L.; Yu, H.; Li, Q. Development of microsatellite markers and analysis of genetic diversity of Barbatia virescens in the southern coasts of China. Genes Genom. 2019, 41, 407–416. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Chen, P.; Liu, P.; He, Y. Transcriptome analysis of eyestalk and hemocytes in the ridgetail white prawn Exopalaemon carinicauda: Assembly, annotation and marker discovery. Mol. Biol. Rep. 2015, 42, 135–147. [Google Scholar] [CrossRef]
- DeWoody, J.; Nason, J.D.; Hipkins, V.D. Mitigating scoring errors in microsatellite data from wild populations. Mol. Ecol. Notes 2006, 6, 951–957. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Hayden, M.J.; Nguyen, T.M.; Waterman, A.; Chalmers, K.J. Multiplex-ready PCR: A new method for multiplexed SSR and SNP genotyping. BMC Genom. 2008, 9, 80. [Google Scholar] [CrossRef]
- Guichoux, E.; Lagache, L.; Wagner, S.; Chaumeil, P.; Léger, P.; Lepais, O.; Lepoittevin, C.; Malausa, T.; Revardel, E.; Salin, F. Current trends in microsatellite genotyping. Mol. Ecol. Resour. 2011, 11, 591–611. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.L.J.H.; Zh; Guo, W.; Yan, X. Analysis of genetic variability in selected lines and a wild population of Ruditapes philippinarum using microsatellite markers. J. Fish. Sci. China 2016, 23, 538–546. [Google Scholar]
- Yu, Z.; Yan, X.; Yang, F.; Wang, J.; Zhang, Y.; Yang, F.; Zhang, G. Genetic diversity of different generations of the Dalian population of Manila clam Ruditapes philippinarum through selective breeding. Acta Ecol. Sin. 2011, 31, 4199–4206. [Google Scholar]
- Dieringer, D.; Schlötterer, C. Microsatellite analyser (MSA): A platform independent analysis tool for large microsatellite data sets. Mol. Ecol. Notes 2003, 3, 167–169. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3). 2001. Available online: https://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 19 October 2021.).
- Sokal, R.R.; Rohlf, F.J.; Rohlf, J.F. Biometry; Macmillan: New York, NY, USA, 1995. [Google Scholar]
- Raymond, M.; Rousset, F. An exact test for population differentiation. Evolution 1995, 49, 1280–1283. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (Version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Waples, R.S.; Do, C. LDNE: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 2008, 8, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef]
- An, H.S.; Park, K.J.; Cho, K.C.; Han, H.S.; Myeong, J.-I. Genetic structure of Korean populations of the clam Ruditapes philippinarum inferred from microsatellite marker analysis. Biochem. Syst. Ecol. 2012, 44, 186–195. [Google Scholar] [CrossRef]
- Vargas, K.; Asakura, Y.; Ikeda, M.; Taniguchi, N.; Obata, Y.; Hamasaki, K.; Tsuchiya, K.; Kitada, S. Allozyme variation of littleneck clam Ruditapes philippinarum and genetic mixture analysis of foreign clams in Ariake Sea and Shiranui Sea off Kyushu Island, Japan. Fish. Sci. 2008, 74, 533–543. [Google Scholar] [CrossRef]
- Sekine, Y.; Yamakawa, H.; Takazawa, S.; Lin, Y.; Toba, M. Geographic variation of the COX1 gene of the short-neck clam Ruditapes philippinarum in coastal regions of Japan and China. Venus 2006, 65, 229–240. [Google Scholar]
- Melo, M.A.D.; da Silva, A.R.B.; Varela, E.S.; Sampaio, I.; Tagliaro, C.H. Development and characterization of ten microsatellite markers for population studies of the native Brazilian oyster Crassostrea gasar. Conserv. Genet. Resour. 2012, 4, 583–586. [Google Scholar] [CrossRef]
- Xiao, J.; Cordes, J.F.; Wang, H.; Guo, X.; Reece, K.S. Population genetics of Crassostrea ariakensis in Asia inferred from microsatellite markers. Mar. Biol. 2010, 157, 1767–1781. [Google Scholar] [CrossRef]
- Hoshino, A.A.; Bravo, J.P.; Nobile, P.M.; Morelli, K.A. Microsatellites as Tools for Genetic Diversity Analysis. 2012. Available online: https://www.researchgate.net/profile/Andrea-Hoshino/publication/221925456_Microsatellites_as_Tools_for_Genetic_Diversity_Analysis/links/0d1c84f492b24ee605000000/Microsatellites-as-Tools-for-Genetic-Diversity-Analysis.pdf (accessed on 26 October 2021).
- Hedgecock, D.; Li, G.; Hubert, S.; Bucklin, K.; Ribes, V. Widespread null alleles and poor cross-species amplification of microsatellite DNA loci cloned from the Pacific oyster, Crassostrea gigas. J. Shellfish Res. 2004, 23, 379–386. [Google Scholar]
- Hedgecock, D.; Launey, S.; Pudovkin, A.; Naciri, Y.; Lapegue, S.; Bonhomme, F. Small effective number of parents (Nb) inferred for a naturally spawned cohort of juvenile European flat oysters Ostrea edulis. Mar. Biol. 2007, 150, 1173–1182. [Google Scholar] [CrossRef]
- VanTassel, N.M.; Morris, T.J.; Wilson, C.G.; Zanatta, D.T. Genetic diversity maintained in comparison of captive-propagated and wild populations of Lampsilis fasciola and Ptychobranchus fasciolaris (Bivalvia: Unionidae). Can. J. Fish. Aquat. 2021, 78, 1312–1320. [Google Scholar] [CrossRef]
- Mathias, P.T.; Hoffman, J.R.; Wilson, C.C.; Zanatta, D.T. Signature of postglacial colonization on contemporary genetic structure and diversity of Quadrula quadrula (Bivalvia: Unionidae). Hydrobiologia 2018, 810, 207–225. [Google Scholar] [CrossRef]
- Chacón, G.M.; Arias-Pérez, A.; Freire, R.; Martínez, L.; Ojea, J.; Insua, A. Genetic characterization of wild, broodstock and seed samples of Polititapes rhomboides (Bivalvia: Veneridae): Implications for hatchery seed production. Aquac. Rep. 2021, 20, 100658. [Google Scholar] [CrossRef]
- Plough, L.V.; Hedgecock, D. Quantitative trait locus analysis of stage-specific inbreeding depression in the Pacific oyster Crassostrea gigas. Genetics 2011, 189, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Plough, L.; Shin, G.; Hedgecock, D. Genetic inviability is a major driver of type III survivorship in experimental families of a highly fecund marine bivalve. Mol. Ecol. 2016, 25, 895–910. [Google Scholar] [CrossRef]
- Lallias, D.; Taris, N.; Boudry, P.; Bonhomme, F.; Lapegue, S. Variance in the reproductive success of flat oyster Ostrea edulis L. assessed by parentage analyses in natural and experimental conditions. Genet. Res. 2010, 92, 175–187. [Google Scholar] [CrossRef]
- Mackintosh, A.; Laetsch, D.R.; Hayward, A.; Charlesworth, B.; Waterfall, M.; Vila, R.; Lohse, K. The determinants of genetic diversity in butterflies. Nat. Commun. 2019, 10, 3466. [Google Scholar] [CrossRef]
- Cordero, D.; Delgado, M.; Liu, B.; Ruesink, J.; Saavedra, C. Population genetics of the Manila clam (Ruditapes philippinarum) introduced in North America and Europe. Sci. Rep. 2017, 7, 39745. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.C.; Huber, C.D. The inflated significance of neutral genetic diversity in conservation genetics. Proc. Natl. Acad. Sci. USA 2021, 118, e2015096118. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, A.D.; Wasson, K.; Waters, T.; Adams, J.; Donatuto, J.; Fleener, G.; Froehlich, H.; Govender, R.; Kornbluth, A.; Lorda, J. Conservation aquaculture as a tool for imperiled marine species: Evaluation of opportunities and risks for Olympia oysters, Ostrea lurida. PLoS ONE 2021, 16, e0252810. [Google Scholar] [CrossRef]
- Wang, J.; Santiago, E.; Caballero, A. Prediction and estimation of effective population size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef]
- Waples, R.S.; Hindar, K.; Karlsson, S.; Hard, J.J. Evaluating the Ryman–Laikre effect for marine stock enhancement and aquaculture. Curr. Zool. 2016, 62, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.R.; Marine, M.; French, R.; Waples, R.S.; Blouin, M. Effective size of a wild salmonid population is greatly reduced by hatchery supplementation. Heredity 2012, 109, 254–260. [Google Scholar] [CrossRef]
- Morvezen, R.; Boudry, P.; Laroche, J.; Charrier, G. Stock enhancement or sea ranching? Insights from monitoring the genetic diversity, relatedness and effective population size in a seeded great scallop population (Pecten maximus). Heredity 2016, 117, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Baskett, M.L.; Burgess, S.C.; Waples, R.S. Assessing strategies to minimize unintended fitness consequences of aquaculture on wild populations. Evol. Appl. 2013, 6, 1090–1108. [Google Scholar] [CrossRef]
- Hold, N.; Murray, L.G.; Kaiser, M.J.; Hinz, H.; Beaumont, A.R.; Taylor, M.I. Potential effects of stock enhancement with hatchery-reared seed on genetic diversity and effective population size. Can. J. Fish. Aquat. Sci. 2013, 70, 330–338. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.; Briscoe, D. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Franklin, I.; Frankham, R. How large must populations be to retain evolutionary potential? Anim. Conserv. Forum 1998, 1, 69–70. [Google Scholar] [CrossRef]
- Waples, R.S.; Do, C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef]
- Lallias, D.; Boudry, P.; Lapegue, S.; King, J.W.; Beaumont, A.R. Strategies for the retention of high genetic variability in European flat oyster (Ostrea edulis) restoration programmes. Conserv. Genet. 2010, 11, 1899–1910. [Google Scholar] [CrossRef]
- Hedgecock, D.; Pudovkin, A.I. Sweepstakes reproductive success in highly fecund marine fish and shellfish: A review and commentary. Bull. Mar. Sci. 2011, 87, 971–1002. [Google Scholar] [CrossRef]
- England, P.R.; Cornuet, J.-M.; Berthier, P.; Tallmon, D.A.; Luikart, G. Estimating effective population size from linkage disequilibrium: Severe bias in small samples. Conserv. Genet. 2006, 7, 303–308. [Google Scholar] [CrossRef]
- Waples, R.S. A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv. Genet. 2006, 7, 167–184. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Name | Location | Collection Date | Sample Size |
|---|---|---|---|---|
| CZ | Chaozhou | Chaozhou, Guangdong Province | July 2020 | 32 |
| LZ | Laizhou | Laizhou, Shandong Province | August 2020 | 32 |
| LJ | Lianjiang | Lianjiang, Fujian Province | July 2020 | 32 |
| NB | Ningbo | Ningbo, Zhejiang Province | July 2020 | 32 |
| SY | Sanya | Sanya, Hainan Province | June 2020 | 32 |
| RZ | Rizhao | Rizhao, Shandong Province | August 2020 | 32 |
| ZZ | Zhangzhou | Zhangzhou, Fujian Province | July 2020 | 32 |
| BH | Beihai | Beihai, Guangxi Province | June 2020 | 32 |
| QD | Qingdao | Qingdao, Shandong Province | August 2020 | 29 |
| HY | Haiyang | Haiyang, Shandong Province | August 2020 | 28 |
| DG | Donggang | Donggang, Liaoning Province | August 2020 | 24 |
| ZH | Zhuanghe | Zhuanghe, Liaoning Province | August 2020 | 29 |
| XY | Selected population | Putian, Fujian Province | August 2020 | 40 |
| Sequence | Locus | Accession | Primer (5′-3′) | Repeat Unit | Tm (°C) | Fluorescent Labelling | Size Ranges/ bp |
|---|---|---|---|---|---|---|---|
| 1 | Rpt23 | KC811247 | F: AGCGTGTTGCTGCTCTTC | (AGC)6 | 48 | FAM | 81–117 |
| R: ATTACTCCCACTGTTCGT | |||||||
| 2 | Rp-07 | AM874000 | F: TATGGCTGGTTTGGACTG | (AT)7 | 51 | TAM | 119–151 |
| R: TCCCGTTACACTTACTTTCA | |||||||
| 3 | Asari16 | AB257421 | F: GCTCGAGTCTGATTGGCTACTGAA | (CT)12 | 55 | ROX | 151–174 |
| R: GGTATCTAGTCAGCTCTTGCAGTA | |||||||
| 4 | Rp-03 | AM873616 | F: CCGCTGTGAGGAGACCAA | (TTG)6 | 58 | FAM | 170–213 |
| R: CCGCCTATGTGACAAAATGA | |||||||
| 5 | Rpt36 | KC811251 | F: TTGAGGCATCAATAACTTTC | (TTG)8 | 50 | TAM | 230–268 |
| R: ACTTCTGCATCTCGGCTA | |||||||
| 6 | Rpt100 | KC811260 | F: TCATTTCCAAGGCAGGTA | (ATG)5 | 50 | ROX | 237–274 |
| R: GAGGTGTTGAAGGAGCAG | |||||||
| 7 | Rpt106 | KC811263 | F: ACCTCAGTTCAAATGTCT | (AGT)6 | 48 | HEX | 373–409 |
| R: AATACTAACGCTGTGGAT | |||||||
| 8 | Rpt105 | KC811262 | F: GGTATGGTGGTAAATGGA | (GTT)5 | 46 | FAM | 375–411 |
| R: TCATAGGTAGGGTGGTTT | |||||||
| 9 | Rpt67 | KC811255 | F: GGGTTCTTCTGTAGTTGG | (GAA)5 | 46 | TAM | 379–415 |
| R: TGAGAAATCAGACCCAAT | |||||||
| 10 | Rpt32 | KC811249 | F: TCACTTTCTGCTCCTACA | (CAT)5 | 47 | ROX | 415–451 |
| R: AAAGGGAATCTCGTGGTG | |||||||
| 11 | Rpt83 | KC811257 | F: GGTCGCCTAATTTCGTAG | (TGT)7 | 46 | HEX | 429–472 |
| R: TAATAATTTTCCTGGAGCTCTGGCG |
| Population | CZ | LZ | LJ | NB | SY | RZ | ZZ | BH | QD | HY | DG | ZH | XY |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CZ | 0 | 0.3291 | 0.0446 | 0.0927 | 0.1573 | 0.1847 | 0.1353 | 0.0810 | 0.2450 | 0.2361 | 0.2355 | 0.1407 | 0.0973 |
| LZ | 0.1084 ** | 0 | 0.3005 | 0.3113 | 0.3484 | 0.3194 | 0.3144 | 0.2863 | 0.4629 | 0.4265 | 0.4702 | 0.4178 | 0.3777 |
| LJ | 0.0048 | 0.1020 ** | 0 | 0.0492 | 0.1161 | 0.1349 | 0.1117 | 0.0556 | 0.1863 | 0.2015 | 0.2198 | 0.1525 | 0.0956 |
| NB | 0.0270 ** | 0.1021 ** | 0.0046 | 0 | 0.1050 | 0.1422 | 0.1141 | 0.0639 | 0.1517 | 0.1835 | 0.2201 | 0.1461 | 0.0899 |
| SY | 0.0474 ** | 0.1089 ** | 0.0285 ** | 0.0227 ** | 0 | 0.0835 | 0.1874 | 0.1731 | 0.1091 | 0.1284 | 0.1954 | 0.1685 | 0.1320 |
| RZ | 0.0648 ** | 0.1065 ** | 0.0435 ** | 0.0434 ** | 0.0260 ** | 0 | 0.1324 | 0.1868 | 0.1898 | 0.1660 | 0.2514 | 0.1676 | 0.1701 |
| ZZ | 0.0561 ** | 0.1181 ** | 0.0423 ** | 0.0375 ** | 0.0593 ** | 0.0484 ** | 0 | 0.0979 | 0.2433 | 0.2337 | 0.2641 | 0.1674 | 0.1423 |
| BH | 0.0425 ** | 0.1084 ** | 0.0252 ** | 0.0145 ** | 0.0348 ** | 0.0654 ** | 0.0413 ** | 0 | 0.1775 | 0.1881 | 0.2136 | 0.1410 | 0.1035 |
| QD | 0.1349 ** | 0.1983 ** | 0.1157 ** | 0.0857 ** | 0.0809 ** | 0.1048 ** | 0.1224 ** | 0.0794 ** | 0 | 0.1417 | 0.2242 | 0.2310 | 0.1688 |
| HY | 0.0982 ** | 0.1453 ** | 0.0864 ** | 0.0725 ** | 0.0488 ** | 0.0773 ** | 0.0904 ** | 0.0478 ** | 0.0679 ** | 0 | 0.2373 | 0.1507 | 0.1284 |
| DG | 0.1058 ** | 0.1653 ** | 0.0947 ** | 0.0827 ** | 0.0716 ** | 0.0977 ** | 0.1060 ** | 0.0716 ** | 0.0742 ** | 0.0559 ** | 0 | 0.2263 | 0.1750 |
| ZH | 0.0610 ** | 0.1444 ** | 0.0612 ** | 0.0510 ** | 0.0519 ** | 0.0599 ** | 0.0679 ** | 0.0580 ** | 0.0933 ** | 0.0508 ** | 0.0801 ** | 0 | 0.1510 |
| XY | 0.0415 ** | 0.1096 ** | 0.0405 ** | 0.0290 ** | 0.0410 ** | 0.0561 ** | 0.0557 ** | 0.0332 ** | 0.0602 ** | 0.0427 ** | 0.0510 ** | 0.0490 ** | 0 |
| Source of Variation | d.f. | Sum of Squares | MS | Est. Var. | Percentage Variation |
|---|---|---|---|---|---|
| Among Populations | 12 | 260.237 | 21.686 | 0.273 | 6.97% |
| Among Individuals | 393 | 1828.026 | 4.651 | 1.007 | 25.70% |
| Within Individuals | 406 | 1071.000 | 2.638 | 2.638 | 67.33% |
| Total | 811 | 3159.262 | 3.918 | 100.00% |
| LD | Cn | ||||||
|---|---|---|---|---|---|---|---|
| Pop | n | Ne (95% CI) | Pop | n | Ne (95% CI) | ||
| CZ | 28.5 | 0.0483 | 0.0391 | 31.7 (20.3–58.8) | CZ | 30.1 | 7.2 (3.4–12.5) |
| LZ | 31.0 | 0.0384 | 0.0355 | 114.9 (50.1–Infinite) | LZ | 31.5 | Infinite (Infinite–Infinite) |
| LJ | 29.5 | 0.0373 | 0.0377 | −876.7 (92.3–Infinite) | LJ | 30.8 | Infinite (Infinite–Infinite) |
| NB | 28.8 | 0.0375 | 0.0387 | −258.6 (129.9–Infinite) | NB | 30.4 | 32.8 (0–164.9) |
| SY | 28.6 | 0.0434 | 0.0390 | 68.2 (33.3–510.2) | SY | 29.8 | 20.7 (1.5–64.4) |
| RZ | 29.0 | 0.0418 | 0.0384 | 88.3 (36.7–Infinite) | RZ | 30.5 | 18.9 (0–94.8) |
| ZZ | 29.2 | 0.0374 | 0.0380 | −513.7 (74.7–Infinite) | ZZ | 30.3 | Infinite (Infinite–Infinite) |
| BH | 24.7 | 0.0419 | 0.0458 | −79.9 (290.4–Infinite) | BH | 27.9 | 54.3 (0.1–272.7) |
| QD | 17.7 | 0.0526 | 0.0672 | 22.7 (-39.9–Infinite) | QD | 19.0 | 11.9 (2–30.6) |
| HY | 18.2 | 0.0526 | 0.0649 | −26.5 (-47.9–Infinite) | HY | 22.4 | 11.1 (2.7–25.4) |
| DG | 14.9 | 0.0776 | 0.0827 | −62.1 (58.3–Infinite) | DG | 19.5 | 6.2 (4.2–8.5) |
| ZH | 22.9 | 0.0497 | 0.0498 | −4158.7 (47.7–Infinite) | ZH | 26.5 | 17.0 (2.8–43.7) |
| XY | 30.3 | 0.0374 | 0.0365 | 375.4 (73.5–Infinite) | XY | 34.8 | Infinite (Infinite–Infinite) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, S.; Zhang, T.; Tu, K.; Li, L.; Liu, Z.; Wu, B.; Zhou, L.; Sun, X. Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers. Biology 2023, 12, 557. https://doi.org/10.3390/biology12040557
Zheng S, Zhang T, Tu K, Li L, Liu Z, Wu B, Zhou L, Sun X. Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers. Biology. 2023; 12(4):557. https://doi.org/10.3390/biology12040557
Chicago/Turabian StyleZheng, Sichen, Tianshi Zhang, Kang Tu, Li Li, Zhihong Liu, Biao Wu, Liqing Zhou, and Xiujun Sun. 2023. "Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers" Biology 12, no. 4: 557. https://doi.org/10.3390/biology12040557
APA StyleZheng, S., Zhang, T., Tu, K., Li, L., Liu, Z., Wu, B., Zhou, L., & Sun, X. (2023). Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers. Biology, 12(4), 557. https://doi.org/10.3390/biology12040557