
Advances in Mass Spectrometry-Based Single Cell Analysis



Abstract
Simple Summary
Abstract
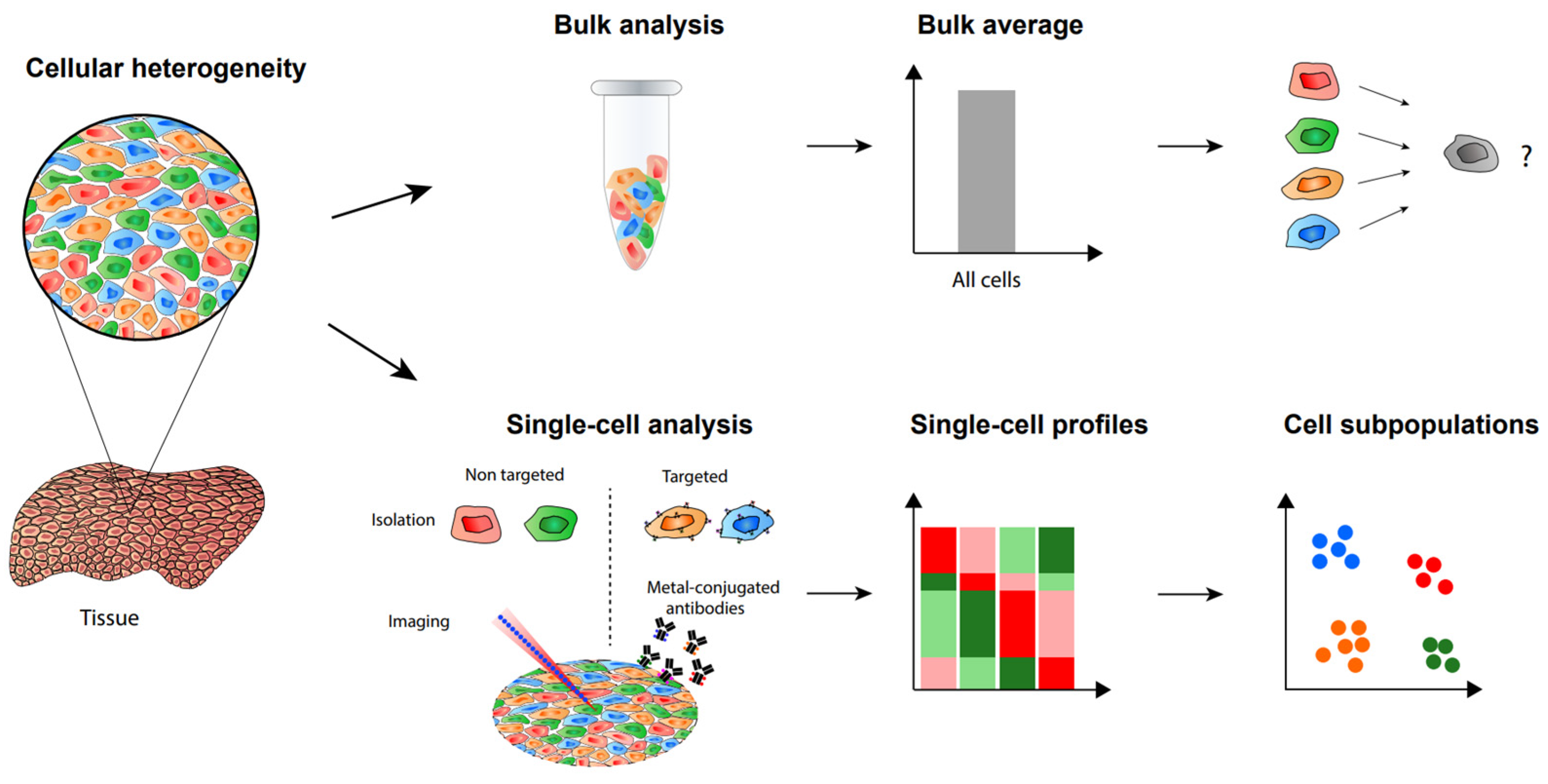
1. Introduction
2. Label-Free Single-Cell Proteomic Analysis
3. TMT-Assisted Single-Cell Proteomics
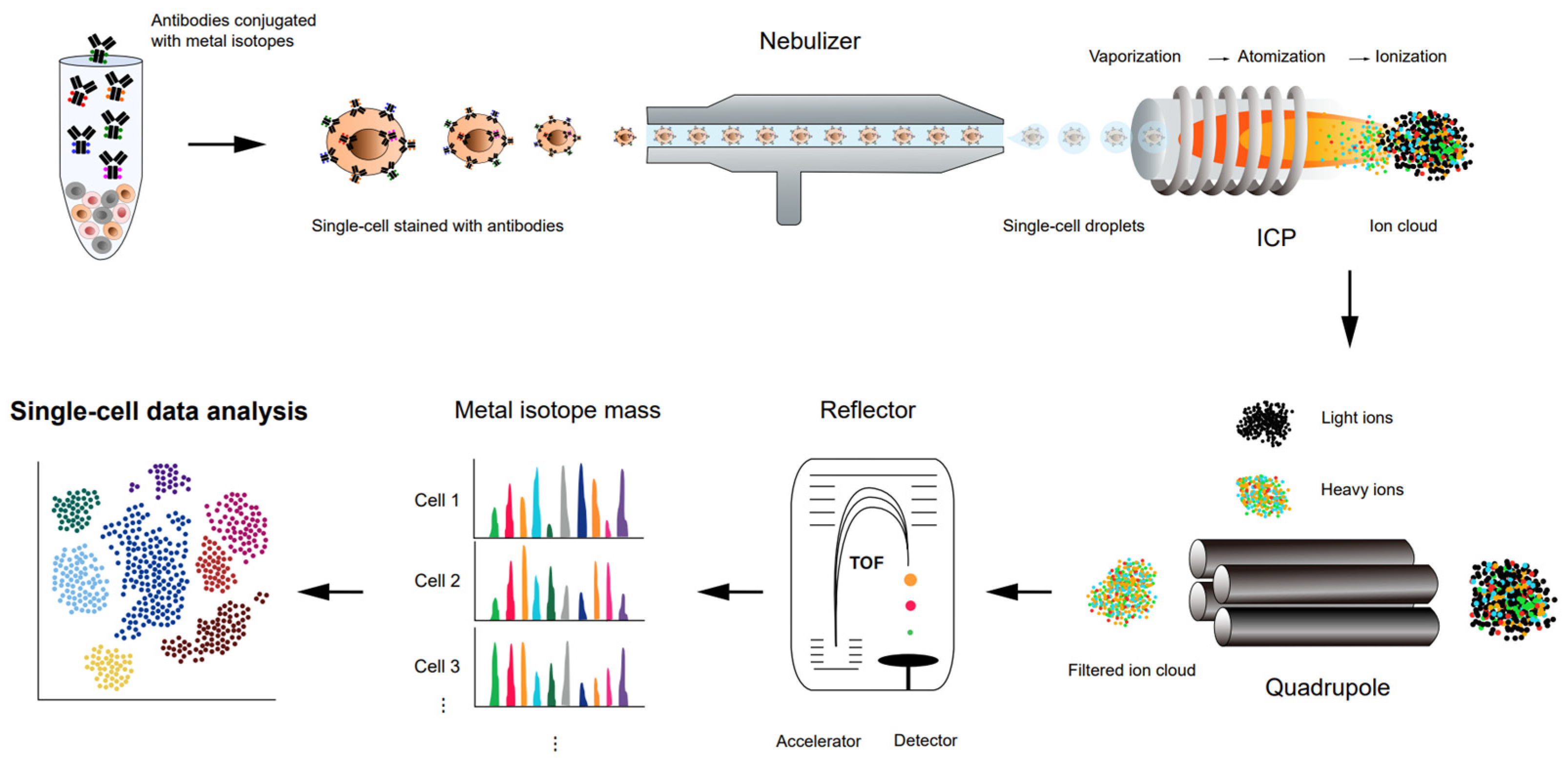
4. CyTOF for Single-Cell Proteomics
5. SIMS for Single-Cell Mass Spectrometry Imaging
6. ESI-Based MS for Single-Cell Metabolomic Analysis
7. DESI for Single-Cell Metabolomic Mass Spectrometry Imaging
8. Nano-DESI for Single-Cell Metabolomic Mass Spectrometry Imaging
9. LAESI for Single-Cell Metabolomic Mass Spectrometry Imaging
10. MALDI for Single-Cell Mass Spectrometry Imaging
11. Application of Single-Cell Proteomic Analysis
12. Application of Single-Cell Metabolomic Analysis
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elsasser, W.M. Outline of a theory of cellular heterogeneity. Proc. Natl. Acad. Sci. USA 1984, 81, 5126–5129. [Google Scholar] [CrossRef] [PubMed]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Slavov, N. Unpicking the proteome in single cells. Science 2020, 367, 512–513. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Ding, X. The Intriguing Landscape of Single-Cell Protein Analysis. Adv. Sci. 2022, 9, 2105932. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.-I. Immunocytochemistry: Theory and Practice; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Burry, R.W. Controls for Immunocytochemistry. J. Histochem. Cytochem. 2011, 59, 6–12. [Google Scholar] [CrossRef]
- Ramos-Vara, J.A. Technical Aspects of Immunohistochemistry. Vet. Pathol. 2005, 42, 405–426. [Google Scholar] [CrossRef]
- Herzenberg, L.A.; De Rosa, S.C.; Herzenberg, L.A. Monoclonal antibodies and the FACS: Complementary tools for immunobiology and medicine. Immunol. Today 2000, 21, 383–390. [Google Scholar] [CrossRef]
- Herzenberg, L.A.; Parks, D.; Sahaf, B.; Perez, O.; Roederer, M.; Herzenberg, L.A. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin. Chem. 2002, 48, 1819–1827. [Google Scholar] [CrossRef]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Jindal, A.; Gupta, P.; Jayadeva; Sengupta, D. Discovery of rare cells from voluminous single cell expression data. Nat. Commun. 2018, 9, 4719. [Google Scholar] [CrossRef]
- Wegmann, R.; Neri, M.; Schuierer, S.; Bilican, B.; Hartkopf, H.; Nigsch, F.; Mapa, F.; Waldt, A.; Cuttat, R.; Salick, M.R.; et al. CellSIUS provides sensitive and specific detection of rare cell populations from complex single-cell RNA-seq data. Genome Biol. 2019, 20, 142. [Google Scholar] [CrossRef]
- Pratapa, A.; Jalihal, A.P.; Law, J.N.; Bharadwaj, A.; Murali, T.M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 2020, 17, 147–154. [Google Scholar] [CrossRef]
- Jackson, C.A.; Castro, D.M.; Saldi, G.-A.; Bonneau, R.; Gresham, D. Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. eLife 2020, 9, e51254. [Google Scholar] [CrossRef] [PubMed]
- Iacono, G.; Massoni-Badosa, R.; Heyn, H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019, 20, 110. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, H. Accounting for cell type hierarchy in evaluating single cell RNA-seq clustering. Genome Biol. 2020, 21, 123. [Google Scholar] [CrossRef] [PubMed]
- Tajik, M.; Baharfar, M.; Donald, W.A. Single-cell mass spectrometry. Trends Biotechnol. 2022, 40, 1374–1392. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, Z.; Liu, Y.; Gao, Y.; Gu, J. Recent advances in single-cell analysis by mass spectrometry. Analyst 2019, 144, 824–845. [Google Scholar] [CrossRef]
- Brunner, A.D.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18, e10798. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Motamedchaboki, K.; Misal, S.A.; Liang, Y.; Guise, A.J.; Truong, T.; Huguet, R.; Plowey, E.D.; Zhu, Y.; Lopez-Ferrer, D.; et al. Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 2020, 12, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Clair, G.C.; Williams, S.M.; Feng, S.; Tsai, C.F.; Moore, R.J.; Chrisler, W.B.; Smith, R.D.; Kelly, R.T.; Paša-Tolić, L.; et al. Three-dimensional feature matching improves coverage for single-cell proteomics based on ion mobility filtering. Cell Syst. 2022, 13, 426–434.e424. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Liang, Y.; Motamedchaboki, K.; Huguet, R.; Truong, T.; Zhao, R.; Shen, Y.; Lopez-Ferrer, D.; Zhu, Y.; Kelly, R.T. Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem. 2020, 92, 2665–2671. [Google Scholar] [CrossRef] [PubMed]
- Kalxdorf, M.; Müller, T.; Stegle, O.; Krijgsveld, J. IceR improves proteome coverage and data completeness in global and single-cell proteomics. Nat. Commun. 2021, 12, 4787. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Cox, J.; Mann, M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 2011, 10, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Jang, W.E.; Park, J.H.; Park, G.; Bang, G.; Na, C.H.; Kim, J.Y.; Kim, K.Y.; Kim, K.P.; Shin, C.Y.; An, J.Y.; et al. Cntnap2-dependent molecular networks in autism spectrum disorder revealed through an integrative multi-omics analysis. Mol. Psychiatry 2023, 28, 810–821. [Google Scholar] [CrossRef]
- Mohammad, H.B.; Park, J.H.; Lee, J.H.; Vu, M.H.; Lee, J.-Y.; Jeong, W.-H.; Kim, M.-S. Comprehensive identification of VX-adducted plasma proteins using high-resolution mass spectrometry. Bull. Korean Chem. Soc. 2022, 43, 1217–1222. [Google Scholar] [CrossRef]
- Lee, J.H.; Jang, W.E.; Park, J.H.; Mohammad, H.B.; Lee, J.-Y.; Jeong, W.-H.; Kim, M.-S. Identification of organophosphate modifications by high-resolution mass spectrometry. Bull. Korean Chem. Soc. 2022, 43, 444–449. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.-D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Röst, H.L.; Malmström, L.; Aebersold, R. Reproducible quantitative proteotype data matrices for systems biology. Mol. Biol. Cell 2015, 26, 3926–3931. [Google Scholar] [CrossRef]
- Gebreyesus, S.T.; Siyal, A.A.; Kitata, R.B.; Chen, E.S.; Enkhbayar, B.; Angata, T.; Lin, K.I.; Chen, Y.J.; Tu, H.L. Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat. Commun. 2022, 13, 37. [Google Scholar] [CrossRef]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Ryu, S.J.; Kim, B.J.; Cho, H.J.; Park, C.H.; Choi, H.J.C.; Jang, E.J.; Yang, E.J.; Hwang, J.A.; Woo, S.H.; et al. Disruption of nucleocytoplasmic trafficking as a cellular senescence driver. Exp. Mol. Med. 2021, 53, 1092–1108. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, D.Y.; Nam, D.; Han, Y.; Kim, D.K.; Kim, G.; Kim, D.; Bae, J.; Back, S.; Mun, D.G.; Madar, I.H.; et al. Proteogenomic landscape of human pancreatic ductal adenocarcinoma in an Asian population reveals tumor cell-enriched and immune-rich subtypes. Nat. Cancer 2022, 4, 290–307. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, Z.; Bomgarden, R.D.; Pike, I.; Kuhn, K.; Rogers, J.C.; Roberts, T.M.; Gygi, S.P.; Paulo, J.A. TMTpro-18plex: The expanded and complete set of TMTpro reagents for sample multiplexing. J. Proteome Res. 2021, 20, 2964–2972. [Google Scholar] [CrossRef]
- Isasa, M.; Rose, C.M.; Elsasser, S.; Navarrete-Perea, J.; Paulo, J.A.; Finley, D.J.; Gygi, S.P. Multiplexed, proteome-wide protein expression profiling: Yeast deubiquitylating enzyme knockout strains. J. Proteome Res. 2015, 14, 5306–5317. [Google Scholar] [CrossRef]
- O’Connell, J.D.; Paulo, J.A.; O’Brien, J.J.; Gygi, S.P. Proteome-Wide Evaluation of Two Common Protein Quantification Methods. J. Proteome Res. 2018, 17, 1934–1942. [Google Scholar] [CrossRef]
- Brenes, A.; Hukelmann, J.; Bensaddek, D.; Lamond, A.I. Multibatch TMT Reveals False Positives, Batch Effects and Missing Values. Mol. Cell. Proteom. 2019, 18, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Hamood, F.; Bayer, F.P.; Wilhelm, M.; Kuster, B.; The, M. SIMSI-Transfer: Software-assisted reduction of missing values in phosphoproteomic and proteomic isobaric labeling data using tandem mass spectrum clustering. Mol. Cell. Proteom. 2022, 21, 100238. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.T. Single-cell Proteomics: Progress and Prospects. Mol. Cell. Proteom. 2020, 19, 1739–1748. [Google Scholar] [CrossRef]
- Wang, H.; Qian, W.-J.; Mottaz, H.M.; Clauss, T.R.W.; Anderson, D.J.; Moore, R.J.; Camp, D.G.; Khan, A.H.; Sforza, D.M.; Pallavicini, M.; et al. Development and Evaluation of a Micro- and Nanoscale Proteomic Sample Preparation Method. J. Proteome Res. 2005, 4, 2397–2403. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.L.; Piehowski, P.D.; Orton, D.J.; Moore, R.J.; Qian, W.-J.; Casey, C.P.; Sun, X.; Dey, S.K.; Burnum-Johnson, K.E.; Smith, R.D. SNaPP: Simplified Nanoproteomics Platform for Reproducible Global Proteomic Analysis of Nanogram Protein Quantities. Endocrinology 2016, 157, 1307–1314. [Google Scholar] [CrossRef]
- Chen, Q.; Yan, G.; Gao, M.; Zhang, X. Ultrasensitive Proteome Profiling for 100 Living Cells by Direct Cell Injection, Online Digestion and Nano-LC-MS/MS Analysis. Anal. Chem. 2015, 87, 6674–6680. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef]
- Slavov, N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2021, 60, 1–9. [Google Scholar] [CrossRef]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22, 50. [Google Scholar] [CrossRef]
- Petelski, A.A.; Emmott, E.; Leduc, A.; Huffman, R.G.; Specht, H.; Perlman, D.H.; Slavov, N. Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc. 2021, 16, 5398–5425. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.; Harmange, G.; Perlman, D.H.; Emmott, E.; Niziolek, Z.; Budnik, B.; Slavov, N. Automated sample preparation for high-throughput single-cell proteomics. bioRxiv 2018, 399774. [Google Scholar] [CrossRef]
- Zhu, Y.; Piehowski, P.D.; Zhao, R.; Chen, J.; Shen, Y.; Moore, R.J.; Shukla, A.K.; Petyuk, V.A.; Campbell-Thompson, M.; Mathews, C.E.; et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018, 9, 882. [Google Scholar] [CrossRef] [PubMed]
- Dou, M.; Clair, G.; Tsai, C.F.; Xu, K.; Chrisler, W.B.; Sontag, R.L.; Zhao, R.; Moore, R.J.; Liu, T.; Pasa-Tolic, L.; et al. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labeling in a Nanodroplet Sample Preparation Platform. Anal. Chem. 2019, 91, 13119–13127. [Google Scholar] [CrossRef]
- Williams, S.M.; Liyu, A.V.; Tsai, C.-F.; Moore, R.J.; Orton, D.J.; Chrisler, W.B.; Gaffrey, M.J.; Liu, T.; Smith, R.D.; Kelly, R.T.; et al. Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography–Mass Spectrometry for High-Throughput Single-Cell Proteomics. Anal. Chem. 2020, 92, 10588–10596. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Waanders, L.F.; Chwalek, K.; Monetti, M.; Kumar, C.; Lammert, E.; Mann, M. Quantitative proteomic analysis of single pancreatic islets. Proc. Natl. Acad. Sci. USA 2009, 106, 18902–18907. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Ostasiewicz, P.; Mann, M. High Recovery FASP Applied to the Proteomic Analysis of Microdissected Formalin Fixed Paraffin Embedded Cancer Tissues Retrieves Known Colon Cancer Markers. J. Proteome Res. 2011, 10, 3040–3049. [Google Scholar] [CrossRef]
- Woo, J.; Williams, S.M.; Markillie, L.M.; Feng, S.; Tsai, C.F.; Aguilera-Vazquez, V.; Sontag, R.L.; Moore, R.J.; Hu, D.; Mehta, H.S.; et al. High-throughput and high-efficiency sample preparation for single-cell proteomics using a nested nanowell chip. Nat. Commun. 2021, 12, 6246. [Google Scholar] [CrossRef]
- Gross, A.; Schoendube, J.; Zimmermann, S.; Steeb, M.; Zengerle, R.; Koltay, P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015, 16, 16897–16919. [Google Scholar] [CrossRef]
- Kassem, S.; van der Pan, K.; de Jager, A.L.; Naber, B.A.E.; de Laat, I.F.; Louis, A.; van Dongen, J.J.M.; Teodosio, C.; Díez, P. Proteomics for Low Cell Numbers: How to Optimize the Sample Preparation Workflow for Mass Spectrometry Analysis. J. Proteome Res. 2021, 20, 4217–4230. [Google Scholar] [CrossRef] [PubMed]
- Swensen, A.C.; Veličković, D.; Williams, S.M.; Moore, R.J.; Day, L.Z.; Niessen, S.; Hennessy, S.; Posso, C.; Monetti, M.; Qian, W.-J.; et al. Proteomic Profiling of Intra-Islet Features Reveals Substructure-Specific Protein Signatures. Mol. Cell. Proteom. 2022, 21, 100426. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dou, M.; Piehowski, P.D.; Liang, Y.; Wang, F.; Chu, R.K.; Chrisler, W.B.; Smith, J.N.; Schwarz, K.C.; Shen, Y.; et al. Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol. Cell. Proteom. 2018, 17, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tian, T.; Shi, Y.; Liu, W.; Zou, Y.; Khajvand, T.; Wang, S.; Zhu, Z.; Yang, C. Enrichment and single-cell analysis of circulating tumor cells. Chem. Sci. 2017, 8, 1736–1751. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwängler, B.; Üresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; Dick, J.E.; Porse, B.T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 3341. [Google Scholar] [CrossRef]
- Zhang, L.; Vertes, A. Single-Cell Mass Spectrometry Approaches to Explore Cellular Heterogeneity. Angew. Chem. Int. Ed. 2018, 57, 4466–4477. [Google Scholar] [CrossRef]
- Russell, C.L.; Heslegrave, A.; Mitra, V.; Zetterberg, H.; Pocock, J.M.; Ward, M.A.; Pike, I. Combined tissue and fluid proteomics with Tandem Mass Tags to identify low-abundance protein biomarkers of disease in peripheral body fluid: An Alzheimer’s Disease case study. Rapid Commun. Mass Spectrom. 2017, 31, 153–159. [Google Scholar] [CrossRef]
- McKinnon, K.M. Flow Cytometry: An Overview. Curr. Protoc. Immunol. 2018, 120, 5.1.1–5.1.11. [Google Scholar] [CrossRef]
- Adan, A.; Alizada, G.; Kiraz, Y.; Baran, Y.; Nalbant, A. Flow cytometry: Basic principles and applications. Crit. Rev. Biotechnol. 2017, 37, 163–176. [Google Scholar] [CrossRef]
- Hoffman, R.A. Flow Cytometry: Instrumentation, Applications, Future Trends and Limitations; Springer: Berlin/Heidelberg, Germany, 2008; Volume 6, pp. 307–342. [Google Scholar]
- Roederer, M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry 2001, 45, 194–205. [Google Scholar] [CrossRef]
- Garry, M. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef]
- Nowicka, M.; Krieg, C.; Weber, L.M.; Hartmann, F.J.; Guglietta, S.; Becher, B.; Levesque, M.P.; Robinson, M.D. CyTOF workflow: Differential discovery in high-throughput high-dimensional cytometry datasets. F1000Research 2017, 6, 748. [Google Scholar] [CrossRef]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir el, A.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Samusik, N.; Good, Z.; Spitzer, M.H.; Davis, K.L.; Nolan, G.P. Automated mapping of phenotype space with single-cell data. Nat. Methods 2016, 13, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Luo, D.; Zhong, X.; Choi, J.H.; Ma, Y.; Wang, S.; Mahrt, E.; Guo, W.; Stawiski, E.W.; Modrusan, Z.; et al. SCINA: A Semi-Supervised Subtyping Algorithm of Single Cells and Bulk Samples. Genes 2019, 10, 531. [Google Scholar] [CrossRef] [PubMed]
- Li, S.K.H.; Majonis, D.; Bagwell, C.B.; Hunsberger, B.C.; Baranov, V.; Ornatsky, O. A robust human immunophenotyping workflow using CyTOF technology coupled with Maxpar Pathsetter, an automated data analysis software. J. Immunol. 2019, 202, 131.2. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Chang, Q.; Ornatsky, O.I.; Siddiqui, I.; Loboda, A.; Baranov, V.I.; Hedley, D.W. Imaging mass cytometry. Cytom. Part A 2017, 91, 160–169. [Google Scholar] [CrossRef]
- Baharlou, H.; Canete, N.P.; Cunningham, A.L.; Harman, A.N.; Patrick, E. Mass Cytometry Imaging for the Study of Human Diseases—Applications and Data Analysis Strategies. Front. Immunol. 2019, 10, 2657. [Google Scholar] [CrossRef]
- Olsen, L.R.; Leipold, M.D.; Pedersen, C.B.; Maecker, H.T. The anatomy of single cell mass cytometry data. Cytom. Part A 2019, 95, 156–172. [Google Scholar] [CrossRef]
- Frei, A.P.; Bava, F.-A.; Zunder, E.R.; Hsieh, E.W.; Chen, S.-Y.; Nolan, G.P.; Gherardini, P.F. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat. Methods 2016, 13, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Sahaf, B.; Rahman, A.; Maecker, H.T.; Bendall, S.C. High-Parameter Immune Profiling with CyTOF. In Biomarkers for Immunotherapy of Cancer. Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2055, pp. 351–368. [Google Scholar]
- Bjornson, Z.B.; Nolan, G.P.; Fantl, W.J. Single-cell mass cytometry for analysis of immune system functional states. Curr. Opin. Immunol. 2013, 25, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Strauss-Albee, D.M.; Leipold, M.; Kubo, J.; Nemat-Gorgani, N.; Dogan, O.C.; Dekker, C.L.; Mackey, S.; Maecker, H.; Swan, G.E.; et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl. Med. 2013, 5, 208ra145. [Google Scholar] [CrossRef]
- Mason, G.M.; Lowe, K.; Melchiotti, R.; Ellis, R.; De Rinaldis, E.; Peakman, M.; Heck, S.; Lombardi, G.; Tree, T.I.M. Phenotypic Complexity of the Human Regulatory T Cell Compartment Revealed by Mass Cytometry. J. Immunol. 2015, 195, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.W.; Strauss-Albee, D.M.; Blish, C.A. Application of Mass Cytometry (CyTOF) for Functional and Phenotypic Analysis of Natural Killer Cells. Methods Mol. Biol. 2016, 1441, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Bouzekri, A.; Esch, A.; Ornatsky, O. Multidimensional profiling of drug-treated cells by Imaging Mass Cytometry. FEBS Open Bio 2019, 9, 1652–1669. [Google Scholar] [CrossRef]
- Lingblom, C.M.D.; Kowli, S.; Swaminathan, N.; Maecker, H.T.; Lambert, S.L. Baseline immune profile by CyTOF can predict response to an investigational adjuvanted vaccine in elderly adults. J. Transl. Med. 2018, 16, 153. [Google Scholar] [CrossRef]
- Sen, N.; Mukherjee, G.; Arvin, A.M. Single cell mass cytometry reveals remodeling of human T cell phenotypes by varicella zoster virus. Methods 2015, 90, 85–94. [Google Scholar] [CrossRef]
- Cavrois, M.; Banerjee, T.; Mukherjee, G.; Raman, N.; Hussien, R.; Rodriguez, B.A.; Vasquez, J.; Spitzer, M.H.; Lazarus, N.H.; Jones, J.J.; et al. Mass Cytometric Analysis of HIV Entry, Replication, and Remodeling in Tissue CD4+ T Cells. Cell Rep. 2017, 20, 984–998. [Google Scholar] [CrossRef]
- Nair, N.; Mei, H.E.; Chen, S.-Y.; Hale, M.; Nolan, G.P.; Maecker, H.T.; Genovese, M.; Fathman, C.G.; Whiting, C.C. Mass cytometry as a platform for the discovery of cellular biomarkers to guide effective rheumatic disease therapy. Arthritis Res. Ther. 2015, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Morilla, S.; Villarroel-Espindola, F.; Wong, P.F.; Toki, M.I.; Aung, T.N.; Pelekanou, V.; Bourke-Martin, B.; Schalper, K.A.; Kluger, H.M.; Rimm, D.L. Biomarker Discovery in Patients with Immunotherapy-Treated Melanoma with Imaging Mass Cytometry. Clin. Cancer Res. 2021, 27, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, R.E.; Rahman, A.; Pak, T.R.; Maringer, K.; Mena, I.; Bernal-Rubio, D.; Potla, U.; Maestre, A.M.; Fredericks, A.C.; Amir, E.-A.D.; et al. High-dimensional CyTOF analysis of dengue virus–infected human DCs reveals distinct viral signatures. JCI Insight 2017, 2, e92424. [Google Scholar] [CrossRef]
- van Unen, V.; Li, N.; Molendijk, I.; Temurhan, M.; Höllt, T.; van der Meulen-de, A.E.; Verspaget, H.W.; Mearin, M.L.; Mulder, C.J.; van Bergen, J.; et al. Mass Cytometry of the Human Mucosal Immune System Identifies Tissue- and Disease-Associated Immune Subsets. Immunity 2016, 44, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Geanon, D.; Lee, B.; Kelly, G.; Handler, D.; Upadhyaya, B.; Leech, J.; Herbinet, M.; Valle, D.D.; Gnjatic, S.; Kim-Schulze, S.; et al. A Streamlined CyTOF Workflow To Facilitate Standardized Multi-Site Immune Profiling of COVID-19 Patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Wang, W.; Su, B.; Pang, L.; Qiao, L.; Feng, Y.; Ouyang, Y.; Guo, X.; Shi, H.; Wei, F.; Su, X.; et al. High-dimensional immune profiling by mass cytometry revealed immunosuppression and dysfunction of immunity in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 650–652. [Google Scholar] [CrossRef]
- Stewart, E.; Wang, X.; Chupp, G.L.; Montgomery, R.R. Profiling cellular heterogeneity in asthma with single cell multiparameter CyTOF. J. Leukoc. Biol. 2020, 108, 1555–1564. [Google Scholar] [CrossRef]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir el, A.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef]
- Gadalla, R.; Noamani, B.; MacLeod, B.L.; Dickson, R.J.; Guo, M.; Xu, W.; Lukhele, S.; Elsaesser, H.J.; Razak, A.R.A.; Hirano, N.; et al. Validation of CyTOF Against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Front. Oncol. 2019, 9, 415. [Google Scholar] [CrossRef]
- Wiedeman, A.E.; Muir, V.S.; Rosasco, M.G.; Deberg, H.A.; Presnell, S.; Haas, B.; Dufort, M.J.; Speake, C.; Greenbaum, C.J.; Serti, E.; et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J. Clin. Investig. 2019, 130, 480–490. [Google Scholar] [CrossRef]
- Galli, E.; Hartmann, F.J.; Schreiner, B.; Ingelfinger, F.; Arvaniti, E.; Diebold, M.; Mrdjen, D.; Van Der Meer, F.; Krieg, C.; Nimer, F.A.; et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat. Med. 2019, 25, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, C.; Van Der Poel, M.; Fernández-Zapata, C.; Schlickeiser, S.; Leman, J.K.H.; Hsiao, C.-C.; Mizee, M.R.; Adelia; Vincenten, M.C.J.; Kunkel, D.; et al. Single-cell mass cytometry reveals complex myeloid cell composition in active lesions of progressive multiple sclerosis. Acta Neuropathol. Commun. 2020, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.P.; Eggert, T.; Araujo, D.J.; Vijayanand, P.; Ottensmeier, C.H.; Hedrick, C.C. CyTOF mass cytometry reveals phenotypically distinct human blood neutrophil populations differentially correlated with melanoma stage. J. Immunother. Cancer 2020, 8, e000473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lou, Y.; Yang, J.; Wang, J.; Feng, J.; Zhao, Y.; Wang, L.; Huang, X.; Fu, Q.; Ye, M.; et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 2019, 68, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Neuperger, P.; Balog, J.Á.; Tiszlavicz, L.; Furák, J.; Gémes, N.; Kotogány, E.; Szalontai, K.; Puskás, L.G.; Szebeni, G.J. Analysis of the Single-Cell Heterogeneity of Adenocarcinoma Cell Lines and the Investigation of Intratumor Heterogeneity Reveals the Expression of Transmembrane Protein 45A (TMEM45A) in Lung Adenocarcinoma Cancer Patients. Cancers 2021, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- Benninghoven, A.; Rudenauer, F.; Werner, H.W. Secondary Ion Mass Spectrometry: Basic Concepts, Instrumental Aspects, Applications and Trends; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Benninghoven, A. Surface analysis by secondary ion mass spectrometry (SIMS). Surf. Sci. 1994, 299, 246–260. [Google Scholar] [CrossRef]
- Dowsett, M.; Barlow, R.; Allen, P. Secondary ion mass spectrometry analysis of ultrathin impurity layers in semiconductors and their use in quantification, instrumental assessment, and fundamental measurements. J. Vac. Sci. Technol. B Microelectron. Nanometer. Struct. Process. Meas. Phenom. 1994, 12, 186–198. [Google Scholar] [CrossRef]
- Benninghoven, A. Chemical analysis of inorganic and organic surfaces and thin films by static time-of-flight secondary ion mass spectrometry (TOF-SIMS). Angew. Chem. Int. Ed. Engl. 1994, 33, 1023–1043. [Google Scholar] [CrossRef]
- Ninomiya, S.; Ichiki, K.; Yamada, H.; Nakata, Y.; Seki, T.; Aoki, T.; Matsuo, J. Molecular depth profiling of multilayer structures of organic semiconductor materials by secondary ion mass spectrometry with large argon cluster ion beams. Rapid Commun. Mass Spectrom. 2009, 23, 3264–3268. [Google Scholar] [CrossRef]
- Davies, N.; Weibel, D.; Blenkinsopp, P.; Lockyer, N.; Hill, R.; Vickerman, J. Development and experimental application of a gold liquid metal ion source. Appl. Surf. Sci. 2003, 203, 223–227. [Google Scholar] [CrossRef]
- Kollmer, F. Cluster primary ion bombardment of organic materials. Appl. Surf. Sci. 2004, 231, 153–158. [Google Scholar] [CrossRef]
- Yamada, I.; Matsuo, J.; Toyoda, N.; Kirkpatrick, A. Materials processing by gas cluster ion beams. Mater. Sci. Eng. R Rep. 2001, 34, 231–295. [Google Scholar] [CrossRef]
- Rabbani, S.; Barber, A.M.; Fletcher, J.S.; Lockyer, N.P.; Vickerman, J.C. TOF-SIMS with argon gas cluster ion beams: A comparison with C60+. Anal. Chem. 2011, 83, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Winograd, N. Gas cluster ion beams for secondary ion mass spectrometry. Annu. Rev. Anal. Chem. 2018, 11, 29–48. [Google Scholar] [CrossRef]
- Passarelli, M.K.; Winograd, N. Lipid imaging with time-of-flight secondary ion mass spectrometry (ToF-SIMS). Biochim. Biophys. Acta 2011, 1811, 976–990. [Google Scholar] [CrossRef]
- Kraft, M.L.; Klitzing, H.A. Imaging lipids with secondary ion mass spectrometry. Biochim. Biophys. Acta 2014, 1841, 1108–1119. [Google Scholar] [CrossRef]
- Agüi-Gonzalez, P.; Jähne, S.; Phan, N.T. SIMS imaging in neurobiology and cell biology. J. Anal. At. Spectrom. 2019, 34, 1355–1368. [Google Scholar] [CrossRef]
- Passarelli, M.K.; Pirkl, A.; Moellers, R.; Grinfeld, D.; Kollmer, F.; Havelund, R.; Newman, C.F.; Marshall, P.S.; Arlinghaus, H.; Alexander, M.R. The 3D OrbiSIMS—label-free metabolic imaging with subcellular lateral resolution and high mass-resolving power. Nat. Methods 2017, 14, 1175–1183. [Google Scholar] [CrossRef]
- Zhang, J.; Brown, J.; Scurr, D.J.; Bullen, A.; MacLellan-Gibson, K.; Williams, P.; Alexander, M.R.; Hardie, K.R.; Gilmore, I.S.; Rakowska, P.D. Cryo-OrbiSIMS for 3D molecular imaging of a bacterial biofilm in its native state. Anal. Chem. 2020, 92, 9008–9015. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Aoyagi, S.; Fujii, M.; Matsuo, J.; Fletcher, J.S.; Lockyer, N.P.; Vickerman, J.C.; Passarelli, M.K.; Havelund, R.; Seah, M.P. Peptide fragmentation and surface structural analysis by means of ToF-SIMS using large cluster ion sources. Anal. Chem. 2016, 88, 3592–3597. [Google Scholar] [CrossRef]
- Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.; Liu, S.D.; Zhao, S. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442. [Google Scholar] [CrossRef]
- Keren, L.; Bosse, M.; Thompson, S.; Risom, T.; Vijayaragavan, K.; McCaffrey, E.; Marquez, D.; Angoshtari, R.; Greenwald, N.F.; Fienberg, H. MIBI-TOF: A multiplexed imaging platform relates cellular phenotypes and tissue structure. Sci. Adv. 2019, 5, eaax5851. [Google Scholar] [CrossRef]
- Keren, L.; Bosse, M.; Marquez, D.; Angoshtari, R.; Jain, S.; Varma, S.; Yang, S.-R.; Kurian, A.; Van Valen, D.; West, R. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 2018, 174, 1373–1387.e1319. [Google Scholar] [CrossRef]
- Liu, C.C.; McCaffrey, E.F.; Greenwald, N.F.; Soon, E.; Risom, T.; Vijayaragavan, K.; Oliveria, J.-P.; Mrdjen, D.; Bosse, M.; Tebaykin, D. Multiplexed Ion Beam Imaging: Insights into Pathobiology. Annu. Rev. Pathol. 2022, 17, 403–423. [Google Scholar] [CrossRef]
- Moon, D.W.; Park, Y.H.; Lee, S.Y.; Lim, H.; Kwak, S.; Kim, M.S.; Kim, H.; Kim, E.; Jung, Y.; Hoe, H.-S. Multiplex protein imaging with secondary ion mass spectrometry using metal oxide nanoparticle-conjugated antibodies. ACS Appl. Mater. Interfaces 2020, 12, 18056–18064. [Google Scholar] [CrossRef]
- Kotowska, A.M.; Trindade, G.F.; Mendes, P.M.; Williams, P.M.; Aylott, J.W.; Shard, A.G.; Alexander, M.R.; Scurr, D.J. Protein identification by 3D OrbiSIMS to facilitate in situ imaging and depth profiling. Nat. Commun. 2020, 11, 5832. [Google Scholar] [CrossRef]
- Chandra, S.; Morrison, G.H. Sample preparation of animal tissues and cell cultures for secondary ion mass spectrometry (SIMS) microscopy. Biol. Cell 1992, 74, 31–42. [Google Scholar] [CrossRef]
- Schaepe, K.; Kokesch-Himmelreich, J.; Rohnke, M.; Wagner, A.-S.; Schaaf, T.; Wenisch, S.; Janek, J. Assessment of different sample preparation routes for mass spectrometric monitoring and imaging of lipids in bone cells via ToF-SIMS. Biointerphases 2015, 10, 019016. [Google Scholar] [CrossRef] [PubMed]
- Winograd, N.; Bloom, A. Sample preparation for 3D SIMS chemical imaging of cells. In Mass Spectrometry Imaging of Small Molecules; Springer: Berlin/Heidelberg, Germany, 2015; pp. 9–19. [Google Scholar]
- Lee, S.Y.; Lim, H.; Moon, D.W.; Kim, J.Y. Improved ion imaging of slowly dried neurons and skin cells by graphene cover in time-of-flight secondary ion mass spectrometry. Biointerphases 2019, 14, 051001. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Lee, S.Y.; Moon, D.W.; Kim, J.Y. Preparation of cellular samples using graphene cover and air-plasma treatment for time-of-flight secondary ion mass spectrometry imaging. RSC Adv. 2019, 9, 28432–28438. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Jeong, W.; Lim, H.; Cho, S.; Lee, H.; Jang, Y.; Cho, J.; Bae, S.; Lin, Y.T.; Tsai, L.H.; et al. APOE4-carrying human astrocytes oversupply cholesterol to promote neuronal lipid raft expansion and Aβ generation. Stem Cell Rep. 2021, 16, 2128–2137. [Google Scholar] [CrossRef]
- Intisar, A.; Kim, W.-H.; Shin, H.Y.; Kim, M.Y.; Kim, Y.S.; Lim, H.; Kang, H.G.; Mo, Y.J.; Aly, M.A.S.; Lee, Y.-I.; et al. An electroceutical approach enhances myelination via upregulation of lipid biosynthesis in the dorsal root ganglion. Biofabrication 2022, 14, 015017. [Google Scholar] [CrossRef]
- Intisar, A.; Shin, H.Y.; Kim, W.-H.; Kang, H.G.; Kim, M.Y.; Kim, Y.S.; Cho, Y.; Mo, Y.J.; Lim, H.; Lee, S.; et al. Implantable Electroceutical Approach Improves Myelination by Restoring Membrane Integrity in a Mouse Model of Peripheral Demyelinating Neuropathy. Adv. Sci. 2022, 9, 2201358. [Google Scholar] [CrossRef]
- Lim, H.; Lee, S.Y.; Park, Y.; Jin, H.; Seo, D.; Jang, Y.H.; Moon, D.W. Mass spectrometry imaging of untreated wet cell membranes in solution using single-layer graphene. Nat. Methods 2021, 18, 316–320. [Google Scholar] [CrossRef]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Alexandrov, M.L.; Gall, L.N.; Krasnov, N.V.; Nikolaev, V.I.; Pavlenko, V.A.; Shkurov, V.A. Extraction of ions from solutions under atmospheric pressure as a method for mass spectrometric analysis of bioorganic compounds. Rapid Commun. Mass Spectrom. 2008, 22, 267–270. [Google Scholar] [CrossRef]
- Konermann, L.; Ahadi, E.; Rodriguez, A.D.; Vahidi, S. Unraveling the Mechanism of Electrospray Ionization. Anal. Chem. 2013, 85, 2–9. [Google Scholar] [CrossRef]
- Ho, C.S.; Lam, C.W.; Chan, M.H.; Cheung, R.C.; Law, L.K.; Lit, L.C.; Ng, K.F.; Suen, M.W.; Tai, H.L. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clin. Biochem. Rev. 2003, 24, 3–12. [Google Scholar]
- Banerjee, S.; Mazumdar, S. Electrospray Ionization Mass Spectrometry: A Technique to Access the Information beyond the Molecular Weight of the Analyte. Int. J. Anal. Chem. 2012, 2012, 282574. [Google Scholar] [CrossRef]
- Zhu, G.; Shao, Y.; Liu, Y.; Pei, T.; Li, L.; Zhang, D.; Guo, G.; Wang, X. Single-cell metabolite analysis by electrospray ionization mass spectrometry. TrAC Trends Anal. Chem. 2021, 143, 116351. [Google Scholar] [CrossRef]
- Masujima, T. Live Single-cell Mass Spectrometry. Anal. Sci. 2009, 25, 953–960. [Google Scholar] [CrossRef]
- Gong, X.; Zhao, Y.; Cai, S.; Fu, S.; Yang, C.; Zhang, S.; Zhang, X. Single Cell Analysis with Probe ESI-Mass Spectrometry: Detection of Metabolites at Cellular and Subcellular Levels. Anal. Chem. 2014, 86, 3809–3816. [Google Scholar] [CrossRef]
- Onjiko, R.M.; Plotnick, D.O.; Moody, S.A.; Nemes, P. Metabolic comparison of dorsal versus ventral cells directly in the live 8-cell frog embryo by microprobe single-cell CE-ESI-MS. Anal. Methods 2017, 9, 4964–4970. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Eberlin, L.S.; Hallett, J.E.; Cooks, R.G. Single oocyte and single embryo lipid analysis by desorption electrospray ionization mass spectrometry. J. Mass Spectrom. 2012, 47, 29–33. [Google Scholar] [CrossRef]
- González-Serrano, A.F.; Pirro, V.; Ferreira, C.R.; Oliveri, P.; Eberlin, L.S.; Heinzmann, J.; Lucas-Hahn, A.; Niemann, H.; Cooks, R.G. Desorption Electrospray Ionization Mass Spectrometry Reveals Lipid Metabolism of Individual Oocytes and Embryos. PLoS ONE 2013, 8, e74981. [Google Scholar] [CrossRef]
- Pirro, V.; Oliveri, P.; Ferreira, C.R.; González-Serrano, A.F.; Machaty, Z.; Cooks, R.G. Lipid characterization of individual porcine oocytes by dual mode DESI-MS and data fusion. Anal. Chim. Acta 2014, 848, 51–60. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Pirro, V.; Jarmusch, A.K.; Alfaro, C.M.; Cooks, R.G. Ambient Lipidomic Analysis of Single Mammalian Oocytes and Preimplantation Embryos Using Desorption Electrospray Ionization (DESI) Mass Spectrometry. In Single Cell Metabolism: Methods and Protocols; Shrestha, B., Ed.; Springer: New York, NY, USA, 2020; pp. 159–179. [Google Scholar]
- Bergman, H.-M.; Lanekoff, I. Profiling and quantifying endogenous molecules in single cells using nano-DESI MS. Analyst 2017, 142, 3639–3647. [Google Scholar] [CrossRef]
- Stopka, S.A.; Khattar, R.; Agtuca, B.J.; Anderton, C.R.; Paša-Tolić, L.; Stacey, G.; Vertes, A. Metabolic Noise and Distinct Subpopulations Observed by Single Cell LAESI Mass Spectrometry of Plant Cells in situ. Front. Plant Sci. 2018, 9, 1646. [Google Scholar] [CrossRef]
- Taylor, M.J.; Liyu, A.; Vertes, A.; Anderton, C.R. Ambient Single-Cell Analysis and Native Tissue Imaging Using Laser-Ablation Electrospray Ionization Mass Spectrometry with Increased Spatial Resolution. J. Am. Soc. Mass Spectrom. 2021, 32, 2490–2494. [Google Scholar] [CrossRef]
- Takáts, Z.; Wiseman, J.M.; Gologan, B.; Cooks, R.G. Mass Spectrometry Sampling Under Ambient Conditions with Desorption Electrospray Ionization. Science 2004, 306, 471–473. [Google Scholar] [CrossRef]
- Chernetsova, E.S.; Morlock, G.E. Ambient desorption ionization mass spectrometry (DART, DESI) and its bioanalytical applications. Bioanal. Rev. 2011, 3, 1–9. [Google Scholar] [CrossRef]
- Parrot, D.; Papazian, S.; Foil, D.; Tasdemir, D. Imaging the Unimaginable: Desorption Electrospray Ionization – Imaging Mass Spectrometry (DESI-IMS) in Natural Product Research. Planta Med. 2018, 84, 584–593. [Google Scholar] [CrossRef]
- Morelato, M.; Beavis, A.; Kirkbride, P.; Roux, C. Forensic applications of desorption electrospray ionisation mass spectrometry (DESI-MS). Forensic Sci. Int. 2013, 226, 10–21. [Google Scholar] [CrossRef]
- Wójtowicz, A.; Wietecha-Posłuszny, R. DESI-MS analysis of human fluids and tissues for forensic applications. Appl. Phys. A 2019, 125, 312. [Google Scholar] [CrossRef]
- Hemalatha, R.G.; Pradeep, T. Understanding the Molecular Signatures in Leaves and Flowers by Desorption Electrospray Ionization Mass Spectrometry (DESI MS) Imaging. J. Agric. Food Chem. 2013, 61, 7477–7487. [Google Scholar] [CrossRef]
- Tata, A.; Perez, C.J.; Hamid, T.S.; Bayfield, M.A.; Ifa, D.R. Analysis of Metabolic Changes in Plant Pathosystems by Imprint Imaging DESI-MS. J. Am. Soc. Mass Spectrom. 2015, 26, 641–648. [Google Scholar] [CrossRef]
- Song, Y.; Talaty, N.; Datsenko, K.; Wanner, B.L.; Cooks, R.G. In vivo recognition of Bacillus subtilis by desorption electrospray ionization mass spectrometry (DESI-MS). Analyst 2009, 134, 838–841. [Google Scholar] [CrossRef]
- Pirro, V.; Guffey, S.C.; Sepúlveda, M.S.; Mahapatra, C.T.; Ferreira, C.R.; Jarmusch, A.K.; Cooks, R.G. Lipid dynamics in zebrafish embryonic development observed by DESI-MS imaging and nanoelectrospray-MS. Mol. Biosyst. 2016, 12, 2069–2079. [Google Scholar] [CrossRef]
- Bodzon-Kulakowska, A.; Cichon, T.; Golec, A.; Drabik, A.; Ner, J.; Suder, P. DESI–MS as a tool for direct lipid analysis in cultured cells. Cytotechnology 2015, 67, 1085–1091. [Google Scholar] [CrossRef]
- Wiseman, J.M.; Ifa, D.R.; Song, Q.; Cooks, R.G. Tissue Imaging at Atmospheric Pressure Using Desorption Electrospray Ionization (DESI) Mass Spectrometry. Angew. Chem. Int. Ed. 2006, 45, 7188–7192. [Google Scholar] [CrossRef]
- Manicke, N.E.; Dill, A.L.; Ifa, D.R.; Cooks, R.G. High-resolution tissue imaging on an orbitrap mass spectrometer by desorption electrospray ionization mass spectrometry. J. Mass Spectrom. 2010, 45, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Abbassi-Ghadi, N.; Jones, E.A.; Veselkov, K.A.; Huang, J.; Kumar, S.; Strittmatter, N.; Golf, O.; Kudo, H.; Goldin, R.D.; Hanna, G.B.; et al. Repeatability and reproducibility of desorption electrospray ionization-mass spectrometry (DESI-MS) for the imaging analysis of human cancer tissue: A gateway for clinical applications. Anal. Methods 2015, 7, 71–80. [Google Scholar] [CrossRef]
- Hong, Y.; Birse, N.; Quinn, B.; Montgomery, H.; Wu, D.; Rosas da Silva, G.; van Ruth, S.M.; Elliott, C.T. Identification of milk from different animal and plant sources by desorption electrospray ionisation high-resolution mass spectrometry (DESI-MS). NPJ Sci. Food 2022, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Claude, E.; Jones, E.A.; Pringle, S.D. DESI Mass Spectrometry Imaging (MSI). Methods Mol. Biol. 2017, 1618, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Towers, M.W.; Karancsi, T.; Jones, E.A.; Pringle, S.D.; Claude, E. Optimised Desorption Electrospray Ionisation Mass Spectrometry Imaging (DESI-MSI) for the Analysis of Proteins/Peptides Directly from Tissue Sections on a Travelling Wave Ion Mobility Q-ToF. J. Am. Soc. Mass Spectrom. 2018, 29, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J.; Laskin, J.; Laskin, A. Nanospray desorption electrospray ionization: An ambient method for liquid-extraction surface sampling in mass spectrometry. Analyst 2010, 135, 2233–2236. [Google Scholar] [CrossRef]
- Bergman, H.-M.; Lundin, E.; Andersson, M.; Lanekoff, I. Quantitative mass spectrometry imaging of small-molecule neurotransmitters in rat brain tissue sections using nanospray desorption electrospray ionization. Analyst 2016, 141, 3686–3695. [Google Scholar] [CrossRef]
- Lanekoff, I.; Thomas, M.; Laskin, J. Shotgun approach for quantitative imaging of phospholipids using nanospray desorption electrospray ionization mass spectrometry. Anal. Chem. 2014, 86, 1872–1880. [Google Scholar] [CrossRef]
- Lanekoff, I.; Thomas, M.; Carson, J.P.; Smith, J.N.; Timchalk, C.; Laskin, J. Imaging nicotine in rat brain tissue by use of nanospray desorption electrospray ionization mass spectrometry. Anal. Chem. 2013, 85, 882–889. [Google Scholar] [CrossRef]
- Nguyen, S.N.; Liyu, A.V.; Chu, R.K.; Anderton, C.R.; Laskin, J. Constant-Distance Mode Nanospray Desorption Electrospray Ionization Mass Spectrometry Imaging of Biological Samples with Complex Topography. Anal. Chem. 2017, 89, 1131–1137. [Google Scholar] [CrossRef]
- Nguyen, S.N.; Sontag, R.L.; Carson, J.P.; Corley, R.A.; Ansong, C.; Laskin, J. Towards High-Resolution Tissue Imaging Using Nanospray Desorption Electrospray Ionization Mass Spectrometry Coupled to Shear Force Microscopy. J. Am. Soc. Mass Spectrom. 2018, 29, 316–322. [Google Scholar] [CrossRef]
- Yin, R.; Burnum-Johnson, K.E.; Sun, X.; Dey, S.K.; Laskin, J. High spatial resolution imaging of biological tissues using nanospray desorption electrospray ionization mass spectrometry. Nat. Protoc. 2019, 14, 3445–3470. [Google Scholar] [CrossRef] [PubMed]
- Nemes, P.; Vertes, A. Laser Ablation Electrospray Ionization for Atmospheric Pressure, in Vivo, and Imaging Mass Spectrometry. Anal. Chem. 2007, 79, 8098–8106. [Google Scholar] [CrossRef] [PubMed]
- Nemes, P.; Barton, A.A.; Li, Y.; Vertes, A. Ambient Molecular Imaging and Depth Profiling of Live Tissue by Infrared Laser Ablation Electrospray Ionization Mass Spectrometry. Anal. Chem. 2008, 80, 4575–4582. [Google Scholar] [CrossRef] [PubMed]
- Nemes, P.; Barton, A.A.; Vertes, A. Three-dimensional imaging of metabolites in tissues under ambient conditions by laser ablation electrospray ionization mass spectrometry. Anal. Chem. 2009, 81, 6668–6675. [Google Scholar] [CrossRef]
- Nemes, P.; Vertes, A. Laser Ablation Electrospray Ionization for Atmospheric Pressure Molecular Imaging Mass Spectrometry. In Mass Spectrometry Imaging: Principles and Protocols; Rubakhin, S.S., Sweedler, J.V., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 159–171. [Google Scholar]
- Etalo, D.W.; De Vos, R.C.H.; Joosten, M.H.A.J.; Hall, R.D. Spatially Resolved Plant Metabolomics: Some Potentials and Limitations of Laser-Ablation Electrospray Ionization Mass Spectrometry Metabolite Imaging. Plant Physiol. 2015, 169, 1424–1435. [Google Scholar] [CrossRef]
- Stopka, S.A.; Agtuca, B.J.; Koppenaal, D.W.; Paša-Tolić, L.; Stacey, G.; Vertes, A.; Anderton, C.R. Laser-ablation electrospray ionization mass spectrometry with ion mobility separation reveals metabolites in the symbiotic interactions of soybean roots and rhizobia. Plant J. 2017, 91, 340–354. [Google Scholar] [CrossRef]
- Etalo, D.W.; Díez-Simón, C.; de Vos, R.C.H.; Hall, R.D. Laser Ablation Electrospray Ionization-Mass Spectrometry Imaging (LAESI-MS) for Spatially Resolved Plant Metabolomics. In Plant Metabolomics: Methods and Protocols; António, C., Ed.; Springer: New York, NY, USA, 2018; pp. 253–267. [Google Scholar]
- Agtuca, B.J.; Stopka, S.A.; Evans, S.; Samarah, L.; Liu, Y.; Xu, D.; Stacey, M.G.; Koppenaal, D.W.; Paša-Tolić, L.; Anderton, C.R.; et al. Metabolomic profiling of wild-type and mutant soybean root nodules using laser-ablation electrospray ionization mass spectrometry reveals altered metabolism. Plant J. 2020, 103, 1937–1958. [Google Scholar] [CrossRef]
- Nemes, P.; Woods, A.S.; Vertes, A. Simultaneous Imaging of Small Metabolites and Lipids in Rat Brain Tissues at Atmospheric Pressure by Laser Ablation Electrospray Ionization Mass Spectrometry. Anal. Chem. 2010, 82, 982–988. [Google Scholar] [CrossRef]
- Zhou, W.; Xia, L.; Huang, C.; Yang, J.; Shen, C.; Jiang, H.; Chu, Y. Rapid analysis and identification of meat species by laser-ablation electrospray mass spectrometry (LAESI-MS). Rapid Commun. Mass Spectrom. 2016, 30, 116–121. [Google Scholar] [CrossRef]
- da Silva Lima, G.; Franco dos Santos, G.; Ramalho, R.R.F.; de Aguiar, D.V.A.; Roque, J.V.; Maciel, L.I.L.; Simas, R.C.; Pereira, I.; Vaz, B.G. Laser ablation electrospray ionization mass spectrometry imaging as a new tool for accessing patulin diffusion in mold-infected fruits. Food Chem. 2022, 373, 131490. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Javonillo, R.; Burns, J.R.; Pirger, Z.; Vertes, A. Comparative local analysis of metabolites, lipids and proteins in intact fish tissues by LAESI mass spectrometry. Analyst 2013, 138, 3444–3449. [Google Scholar] [CrossRef]
- Deimler, R.E.; Razunguzwa, T.T.; Reschke, B.R.; Walsh, C.M.; Powell, M.J.; Jackson, G.P. Direct analysis of drugs in forensic applications using laser ablation electrospray ionization-tandem mass spectrometry (LAESI-MS/MS). Anal. Methods 2014, 6, 4810–4817. [Google Scholar] [CrossRef]
- Dean, S.N.; Walsh, C.; Goodman, H.; van Hoek, M.L. Analysis of mixed biofilm (Staphylococcus aureus and Pseudomonas aeruginosa) by laser ablation electrospray ionization mass spectrometry. Biofouling 2015, 31, 151–161. [Google Scholar] [CrossRef]
- Li, H.; Balan, P.; Vertes, A. Molecular Imaging of Growth, Metabolism, and Antibiotic Inhibition in Bacterial Colonies by Laser Ablation Electrospray Ionization Mass Spectrometry. Angew. Chem. Int. Ed. 2016, 55, 15035–15039. [Google Scholar] [CrossRef]
- Shrestha, B.; Vertes, A. In Situ Metabolic Profiling of Single Cells by Laser Ablation Electrospray Ionization Mass Spectrometry. Anal. Chem. 2009, 81, 8265–8271. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Patt, J.M.; Vertes, A. In Situ Cell-by-Cell Imaging and Analysis of Small Cell Populations by Mass Spectrometry. Anal. Chem. 2011, 83, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Stolee, J.A.; Shrestha, B.; Mengistu, G.; Vertes, A. Observation of Subcellular Metabolite Gradients in Single Cells by Laser Ablation Electrospray Ionization Mass Spectrometry. Angew. Chem. Int. Ed. 2012, 51, 10386–10389. [Google Scholar] [CrossRef]
- Samarah, L.Z.; Khattar, R.; Tran, T.H.; Stopka, S.A.; Brantner, C.A.; Parlanti, P.; Veličković, D.; Shaw, J.B.; Agtuca, B.J.; Stacey, G.; et al. Single-Cell Metabolic Profiling: Metabolite Formulas from Isotopic Fine Structures in Heterogeneous Plant Cell Populations. Anal. Chem. 2020, 92, 7289–7298. [Google Scholar] [CrossRef]
- Jacobson, R.S.; Thurston, R.L.; Shrestha, B.; Vertes, A. In Situ Analysis of Small Populations of Adherent Mammalian Cells Using Laser Ablation Electrospray Ionization Mass Spectrometry in Transmission Geometry. Anal. Chem. 2015, 87, 12130–12136. [Google Scholar] [CrossRef]
- Taylor, M.J.; Mattson, S.; Liyu, A.; Stopka, S.A.; Ibrahim, Y.M.; Vertes, A.; Anderton, C.R. Optical Microscopy-Guided Laser Ablation Electrospray Ionization Ion Mobility Mass Spectrometry: Ambient Single Cell Metabolomics with Increased Confidence in Molecular Identification. Metabolites 2021, 11, 200. [Google Scholar] [CrossRef]
- Shrestha, B.; Vertes, A. High-Throughput Cell and Tissue Analysis with Enhanced Molecular Coverage by Laser Ablation Electrospray Ionization Mass Spectrometry Using Ion Mobility Separation. Anal. Chem. 2014, 86, 4308–4315. [Google Scholar] [CrossRef] [PubMed]
- Hillenkamp, F.; Karas, M.; Beavis, R.C.; Chait, B.T. Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry of Biopolymers. Anal. Chem. 1991, 63, 1193A–1203A. [Google Scholar] [CrossRef] [PubMed]
- Jurinke, C.; Oeth, P.; Van Den Boom, D. MALDI-TOF Mass Spectrometry: A Versatile Tool for High-Performance DNA Analysis. Mol. Biotechnol. 2004, 26, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [PubMed]
- Dreisewerd, K. Recent methodological advances in MALDI mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 2261–2278. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.; Popkova, Y.; Engel, K.M.; Schiller, J. Recent developments of useful MALDI matrices for the mass spectrometric characterization of lipids. Biomolecules 2018, 8, 173. [Google Scholar] [CrossRef]
- Zhou, Q.; Fülöp, A.; Hopf, C. Recent developments of novel matrices and on-tissue chemical derivatization reagents for MALDI-MSI. Anal. Bioanal. Chem. 2021, 413, 2599–2617. [Google Scholar] [CrossRef]
- Karas, M.; Krüger, R. Ion Formation in MALDI: The Cluster Ionization Mechanism. Chem. Rev. 2003, 103, 427–440. [Google Scholar] [CrossRef]
- Trimpin, S.; Inutan, E.D.; Herath, T.N.; McEwen, C.N. Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Method for Selectively Producing Either Singly or Multiply Charged Molecular Ions. Anal. Chem. 2010, 82, 11–15. [Google Scholar] [CrossRef]
- Bungert, D.; Heinzle, E.; Tholey, A. Quantitative matrix-assisted laser desorption/ionization mass spectrometry for the determination of enzyme activities. Anal. Biochem. 2004, 326, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, R.; Tabet, J.C.; Ducoroy, P.; Hendra, J.B.; Salzet, M.; Fournier, I. Solid Ionic Matrixes for Direct Tissue Analysis and MALDI Imaging. Anal. Chem. 2006, 78, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Chaurand, P.; Schwartz, S.A.; Caprioli, R.M. Imaging mass spectrometry: A new tool to investigate the spatial organization of peptides and proteins in mammalian tissue sections. Curr. Opin. Chem. Biol. 2002, 6, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Dekker, L.J.; Dalebout, J.C.; Siccama, I.; Jenster, G.; Sillevis Smitt, P.A.; Luider, T.M. A new method to analyze matrix-assisted laser desorption/ionization time-of-flight peptide profiling mass spectra. Rapid Commun. Mass Spectrom. 2005, 19, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Karpova, M.A.; Moshkovskii, S.A.; Toropygin, I.Y.; Archakov, A.I. Cancer-specific MALDI-TOF profiles of blood serum and plasma: Biological meaning and perspectives. J. Proteom. 2010, 73, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Serna, J.; García-Seisdedos, D.; Alcázar, A.; Lasunción, M.Á.; Busto, R.; Pastor, Ó. Quantitative lipidomic analysis of plasma and plasma lipoproteins using MALDI-TOF mass spectrometry. Chem. Phys. Lipids 2015, 189, 7–18. [Google Scholar] [CrossRef]
- Yan, L.; Yi, J.; Huang, C.; Zhang, J.; Fu, S.; Li, Z.; Lyu, Q.; Xu, Y.; Wang, K.; Yang, H.; et al. Rapid Detection of COVID-19 Using MALDI-TOF-Based Serum Peptidome Profiling. Anal. Chem. 2021, 93, 4782–4787. [Google Scholar] [CrossRef]
- Boggio, K.J.; Obasuyi, E.; Sugino, K.; Nelson, S.B.; Agar, N.Y.; Agar, J.N. Recent advances in single-cell MALDI mass spectrometry imaging and potential clinical impact. Expert Rev. Proteom. 2011, 8, 591–604. [Google Scholar] [CrossRef]
- Dai, Y.; Li, C.; Yi, J.; Qin, Q.; Liu, B.; Qiao, L. Plasmonic Colloidosome-Coupled MALDI-TOF MS for Bacterial Heteroresistance Study at Single-Cell Level. Anal. Chem. 2020, 92, 8051–8057. [Google Scholar] [CrossRef]
- Neumann, E.K.; Comi, T.J.; Rubakhin, S.S.; Sweedler, J.V. Lipid Heterogeneity between Astrocytes and Neurons Revealed by Single-Cell MALDI-MS Combined with Immunocytochemical Classification. Angew. Chem. 2019, 131, 5971–5975. [Google Scholar] [CrossRef]
- Amantonico, A.; Urban, P.L.; Fagerer, S.R.; Balabin, R.M.; Zenobi, R. Single-Cell MALDI-MS as an Analytical Tool for Studying Intrapopulation Metabolic Heterogeneity of Unicellular Organisms. Anal. Chem. 2010, 82, 7394–7400. [Google Scholar] [CrossRef] [PubMed]
- Weigt, D.; Sammour, D.A.; Ulrich, T.; Munteanu, B.; Hopf, C. Automated analysis of lipid drug-response markers by combined fast and high-resolution whole cell MALDI mass spectrometry biotyping. Sci. Rep. 2018, 8, 11260. [Google Scholar] [CrossRef] [PubMed]
- Papagiannopoulou, C.; Parchen, R.; Rubbens, P.; Waegeman, W. Fast Pathogen Identification Using Single-Cell Matrix-Assisted Laser Desorption/Ionization-Aerosol Time-of-Flight Mass Spectrometry Data and Deep Learning Methods. Anal. Chem. 2020, 92, 7523–7531. [Google Scholar] [CrossRef] [PubMed]
- Allam, M.; Cai, S.; Coskun, A.F. Multiplex bioimaging of single-cell spatial profiles for precision cancer diagnostics and therapeutics. NPJ Precis. Oncol. 2020, 4, 11. [Google Scholar] [CrossRef]
- Qin, J.; Chait, B.T. Identification and Characterization of Posttranslational Modifications of Proteins by MALDI Ion Trap Mass Spectrometry. Anal. Chem. 1997, 69, 4002–4009. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Gooley, A.A.; Herbert, B.R.; Molloy, M.P.; Binz, P.-A.; Ou, K.; Sanchez, J.-C.; Bairoch, A.; Williams, K.L.; et al. High-throughput mass spectrometric discovery of protein post-translational modifications. J. Mol. Biol. 1999, 289, 645–657. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, T.; Peng, C.; Wu, S. Advances in MALDI mass spectrometry imaging single cell and tissues. Front. Chem. 2022, 1076. [Google Scholar] [CrossRef]
- Mandal, A.; Singha, M.; Addy, P.S.; Basak, A. Laser desorption ionization mass spectrometry: Recent progress in matrix-free and label-assisted techniques. Mass Spectrom. Rev. 2019, 38, 3–21. [Google Scholar] [CrossRef]
- Peterson, D.S. Matrix-free methods for laser desorption/ionization mass spectrometry. Mass Spectrom. Rev. 2007, 26, 19–34. [Google Scholar] [CrossRef]
- Hölscher, D.; Shroff, R.; Knop, K.; Gottschaldt, M.; Crecelius, A.; Schneider, B.; Heckel, D.G.; Schubert, U.S.; Svatoš, A. Matrix-free UV-laser desorption/ionization (LDI) mass spectrometric imaging at the single-cell level: Distribution of secondary metabolites of Arabidopsis thaliana and Hypericum species. Plant J. 2009, 60, 907–918. [Google Scholar] [CrossRef]
- Le Pogam, P.; Schinkovitz, A.; Legouin, B.; Le Lamer, A.-C.; Boustie, J.; Richomme, P. Matrix-Free UV-Laser Desorption Ionization Mass Spectrometry as a Versatile Approach for Accelerating Dereplication Studies on Lichens. Anal. Chem. 2015, 87, 10421–10428. [Google Scholar] [CrossRef]
- Baumeister, T.U.H.; Vallet, M.; Kaftan, F.; Svatoš, A.; Pohnert, G. Live Single-Cell Metabolomics With Matrix-Free Laser/Desorption Ionization Mass Spectrometry to Address Microalgal Physiology. Front. Plant Sci. 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, T.U.H.; Vallet, M.; Kaftan, F.; Guillou, L.; Svatoš, A.; Pohnert, G. Identification to species level of live single microalgal cells from plankton samples with matrix-free laser/desorption ionization mass spectrometry. Metabolomics 2020, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- Proserpio, V.; Lönnberg, T. Single-cell technologies are revolutionizing the approach to rare cells. Immunol. Cell Biol. 2016, 94, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Malhotra, R.; Mayer, G.; Gorochov, G.; Miyara, M. Human FOXP3+ T regulatory cell heterogeneity. Clin. Transl. Immunol. 2018, 7, e1005. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guo, S.; Schrodi, S.J.; He, D. Molecular and Cellular Heterogeneity in Rheumatoid Arthritis: Mechanisms and Clinical Implications. Front. Immunol. 2021, 12, 790122. [Google Scholar] [CrossRef]
- Tracey, L.J.; An, Y.; Justice, M.J. CyTOF: An Emerging Technology for Single-Cell Proteomics in the Mouse. Curr. Protoc. 2021, 1, e118. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e386. [Google Scholar] [CrossRef]
- Cheung, R.K.; Utz, P.J. CyTOF—the next generation of cell detection. Nat. Rev. Rheumatol. 2011, 7, 502–503. [Google Scholar] [CrossRef]
- Anchang, B.; Davis, K.L.; Fienberg, H.G.; Williamson, B.D.; Bendall, S.C.; Karacosta, L.G.; Tibshirani, R.; Nolan, G.P.; Plevritis, S.K. DRUG-NEM: Optimizing drug combinations using single-cell perturbation response to account for intratumoral heterogeneity. Proc. Natl. Acad. Sci. USA 2018, 115, E4294–E4303. [Google Scholar] [CrossRef]
- Markowetz, F.; Kostka, D.; Troyanskaya, O.G.; Spang, R. Nested effects models for high-dimensional phenotyping screens. Bioinformatics 2007, 23, i305–i312. [Google Scholar] [CrossRef]
- Schielzeth, H.; Nakagawa, S. Nested by design: Model fitting and interpretation in a mixed model era. Methods Ecol. Evol. 2013, 4, 14–24. [Google Scholar] [CrossRef]
- Teh, C.E.; Gong, J.-N.; Segal, D.; Tan, T.; Vandenberg, C.J.; Fedele, P.L.; Low, M.S.Y.; Grigoriadis, G.; Harrison, S.J.; Strasser, A.; et al. Deep profiling of apoptotic pathways with mass cytometry identifies a synergistic drug combination for killing myeloma cells. Cell Death Differ. 2020, 27, 2217–2233. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Mazumder, S.; Chakravarti, S.; Sharma, N.; Mukherjee, U.K.; Kumar, S.; Baughn, L.B.; Van Ness, B.G.; Mitra, A.K. secDrug: A pipeline to discover novel drug combinations to kill drug-resistant multiple myeloma cells using a greedy set cover algorithm and single-cell multi-omics. Blood Cancer J. 2022, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Taverna, J.A.; Hung, C.-N.; DeArmond, D.T.; Chen, M.; Lin, C.-L.; Osmulski, P.A.; Gaczynska, M.E.; Wang, C.-M.; Lucio, N.D.; Chou, C.-W.; et al. Single-Cell Proteomic Profiling Identifies Combined AXL and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res. 2020, 80, 1551–1563. [Google Scholar] [CrossRef]
- Anchang, B.; Davis, K.; Fienberg, H.; Bendall, S.; Karacosta, L.; Nolan, G.; Plevritis, S.K. Abstract 2275: Individualized drug combination based on single-cell drug perturbations. Cancer Res. 2018, 78, 2275. [Google Scholar] [CrossRef]
- Ganesh, S.; Hu, T.; Woods, E.; Allam, M.; Cai, S.; Henderson, W.; Coskun, A.F. Spatially resolved 3D metabolomic profiling in tissues. Sci. Adv. 2021, 7, eabd0957. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E.K.; Migas, L.G.; Allen, J.L.; Caprioli, R.M.; Van de Plas, R.; Spraggins, J.M. Spatial metabolomics of the human kidney using MALDI trapped ion mobility imaging mass spectrometry. Anal. Chem. 2020, 92, 13084–13091. [Google Scholar] [CrossRef] [PubMed]
- Stopka, S.A.; Vertes, A. Metabolomic profiling of adherent mammalian cells in situ by LAESI-MS with ion mobility separation. In Ion Mobility-Mass Spectrometry; Springer: Berlin/Heidelberg, Germany, 2020; pp. 235–244. [Google Scholar]
- Roca, M.; Alcoriza, M.I.; Garcia-Cañaveras, J.C.; Lahoz, A. Reviewing the metabolome coverage provided by LC-MS: Focus on sample preparation and chromatography-A tutorial. Anal. Chim. Acta 2021, 1147, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Li, Y.; Yang, Y.; Liu, M. Mass spectrometry-based strategies for single-cell metabolomics. Mass Spectrom. Rev. 2023, 42, 67–94. [Google Scholar] [CrossRef]
- Taylor, M.J.; Lukowski, J.K.; Anderton, C.R. Spatially resolved mass spectrometry at the single cell: Recent innovations in proteomics and metabolomics. J. Am. Soc. Mass Spectrom. 2021, 32, 872–894. [Google Scholar] [CrossRef]
- Ostrowski, S.G.; Van Bell, C.T.; Winograd, N.; Ewing, A.G. Mass spectrometric imaging of highly curved membranes during Tetrahymena mating. Science 2004, 305, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Kollmer, F.; Paul, W.; Krehl, M.; Niehuis, E. Ultra high spatial resolution SIMS with cluster ions—approaching the physical limits. Surf. Interface Anal. 2013, 45, 312–314. [Google Scholar] [CrossRef]
- Anderton, C.R.; Gamble, L.J. Secondary ion mass spectrometry imaging of tissues, cells, and microbial systems. Micros. Today 2016, 24, 24–31. [Google Scholar] [CrossRef]
- Angerer, T.B.; Magnusson, Y.; Landberg, G.r.; Fletcher, J.S. Lipid heterogeneity resulting from fatty acid processing in the human breast cancer microenvironment identified by GCIB-ToF-SIMS imaging. Anal. Chem. 2016, 88, 11946–11954. [Google Scholar] [CrossRef]
- Tian, H.; Sparvero, L.J.; Blenkinsopp, P.; Amoscato, A.A.; Watkins, S.C.; Bayır, H.; Kagan, V.E.; Winograd, N. Secondary-ion mass spectrometry images Cardiolipins and phosphatidylethanolamines at the subcellular level. Angew. Chem. Int. Ed. 2019, 58, 3156–3161. [Google Scholar] [CrossRef]
- Pareek, V.; Tian, H.; Winograd, N.; Benkovic, S.J. Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 2020, 368, 283–290. [Google Scholar] [CrossRef]
- Korte, A.R.; Yandeau-Nelson, M.D.; Nikolau, B.J.; Lee, Y.J. Subcellular-level resolution MALDI-MS imaging of maize leaf metabolites by MALDI-linear ion trap-Orbitrap mass spectrometer. Anal. Bioanal. Chem. 2015, 407, 2301–2309. [Google Scholar] [CrossRef]
- Thiery-Lavenant, G.; Zavalin, A.I.; Caprioli, R.M. Targeted multiplex imaging mass spectrometry in transmission geometry for subcellular spatial resolution. J. Am. Soc. Mass Spectrom. 2013, 24, 609–614. [Google Scholar] [CrossRef]
- Niehaus, M.; Soltwisch, J.; Belov, M.; Dreisewerd, K. Transmission-mode MALDI-2 mass spectrometry imaging of cells and tissues at subcellular resolution. Nat. Methods 2019, 16, 925–931. [Google Scholar] [CrossRef]
- Zavalin, A.; Todd, E.M.; Rawhouser, P.D.; Yang, J.; Norris, J.L.; Caprioli, R.M. Direct imaging of single cells and tissue at sub-cellular spatial resolution using transmission geometry MALDI MS. J. Mass Spectrom. 2012, 47, 1473–1481. [Google Scholar] [CrossRef]
- Soltwisch, J.; Kettling, H.; Vens-Cappell, S.; Wiegelmann, M.; Müthing, J.; Dreisewerd, K. Mass spectrometry imaging with laser-induced postionization. Science 2015, 348, 211–215. [Google Scholar] [CrossRef]
- Rappez, L.; Stadler, M.; Triana, S.; Gathungu, R.M.; Ovchinnikova, K.; Phapale, P.; Heikenwalder, M.; Alexandrov, T. SpaceM reveals metabolic states of single cells. Nat. Methods 2021, 18, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, M.E.; Essner, J.J.; Lee, Y.J. 3D MALDI Mass Spectrometry Imaging of a Single Cell: Spatial Mapping of Lipids in the Embryonic Development of Zebrafish. Sci. Rep. 2017, 7, 14946. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.A.; Nagata, Y.; Waki, M.; Ide, Y.; Takei, S.; Fukano, H.; Romero-Perez, G.A.; Tajima, S.; Yao, I.; Ohnishi, K.; et al. Decreased level of phosphatidylcholine (16:0/20:4) in multiple myeloma cells compared to plasma cells: A single-cell MALDI-IMS approach. Anal. Bioanal. Chem. 2015, 407, 5273–5280. [Google Scholar] [CrossRef]
- Altschuler, S.J.; Wu, L.F. Cellular heterogeneity: Do differences make a difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Eldar, A.; Elowitz, M.B. Functional roles for noise in genetic circuits. Nature 2010, 467, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Gefen, O.; Balaban, N.Q. The importance of being persistent: Heterogeneity of bacterial populations under antibiotic stress. FEMS Microbiol. Rev. 2009, 33, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Srivastava, A.; Johri, S.; Gupta, I.; Karmodiya, K. Single-Cell RNA Sequencing Reveals Cellular Heterogeneity and Stage Transition under Temperature Stress in Synchronized Plasmodium falciparum Cells. Microbiol Spectr 2021, 9, e0000821. [Google Scholar] [CrossRef]
- Gasch, A.P.; Yu, F.B.; Hose, J.; Escalante, L.E.; Place, M.; Bacher, R.; Kanbar, J.; Ciobanu, D.; Sandor, L.; Grigoriev, I.V.; et al. Single-cell RNA sequencing reveals intrinsic and extrinsic regulatory heterogeneity in yeast responding to stress. PLoS Biol. 2017, 15, e2004050. [Google Scholar] [CrossRef] [PubMed]
- Ctortecka, C.; Mechtler, K. The rise of single-cell proteomics. Anal. Sci. Adv. 2021, 2, 84–94. [Google Scholar] [CrossRef]
- Gawel, D.R.; Serra-Musach, J.; Lilja, S.; Aagesen, J.; Arenas, A.; Asking, B.; Bengnér, M.; Björkander, J.; Biggs, S.; Ernerudh, J.; et al. A validated single-cell-based strategy to identify diagnostic and therapeutic targets in complex diseases. Genome Med. 2019, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.; Nestor, C.E.; Zhang, H.; Barabási, A.-L.; Baranzini, S.; Brunak, S.; Chung, K.F.; Federoff, H.J.; Gavin, A.-C.; Meehan, R.R.; et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014, 6, 82. [Google Scholar] [CrossRef]
- Sun, X.-x.; Yu, Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol. Sin. 2015, 36, 1219–1227. [Google Scholar] [CrossRef]
- Melo, F.D.S.E.; Vermeulen, L.; Fessler, E.; Medema, J.P. Cancer heterogeneity—A multifaceted view. EMBO Rep. 2013, 14, 686–695. [Google Scholar] [CrossRef]
- Chan, L.-K.; Tsui, Y.-M.; Ho, D.W.-H.; Ng, I.O.-L. Cellular heterogeneity and plasticity in liver cancer. Semin. Cancer Biol. 2022, 82, 134–149. [Google Scholar] [CrossRef]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell 2015, 17, 260–271. [Google Scholar] [CrossRef]
- Stewart, E.L.; Tan, S.Z.; Liu, G.; Tsao, M.S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations—A review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Chmielik, E.; Rusinek, D.; Oczko-Wojciechowska, M.; Jarzab, M.; Krajewska, J.; Czarniecka, A.; Jarzab, B. Heterogeneity of Thyroid Cancer. Pathobiology 2018, 85, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Montagnani, I.; Montironi, R.; Castiglione, F.; Martignoni, G.; Cheng, L.; Lopez-Beltran, A. Intratumoural heterogeneity may hinder precision medicine strategies in patients with clear cell renal cell carcinoma. J. Clin. Pathol. 2018, 71, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Capaldo, B.D.; Vaillant, F.; Pal, B.; van Ineveld, R.; Dawson, C.A.; Chen, Y.; Nolan, E.; Fu, N.Y.; Jackling, F.C.; et al. Intraclonal Plasticity in Mammary Tumors Revealed through Large-Scale Single-Cell Resolution 3D Imaging. Cancer Cell 2019, 35, 618–632.e616. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target /Non-Target | Spot Size | Molecular Coverage | Single-Cell Throughput | Ambient /Vacuum | Limitations | ||
|---|---|---|---|---|---|---|---|
| Metabolites | Proteins | ||||||
| MS-based scProteomics | |||||||
| Label-free | non-target | NA | NA | >1000 | very low | ambient | low data completeness in DDA mode |
| TMT labeling | non-target | NA | NA | >1000 | low | ambient | inaccurate quantification |
| CyTOF | target | NA | NA | ~100 (metal-tagged) | very high | vacuum | availability of antibodies biased data acquisition |
| Single-cell MSI | |||||||
| SIMS | non-target (TOF-SIMS) target (MIBI) | 50 nm–50 μm | tens | MIBI: ~100 (metal-tagged) | high | vacuum | TOF-SIMS: low identification/quantification MIBI: Availability of antibodies |
| DESI | non-target | >50 μm | tens | limited | very low | ambient | low spatial resolution |
| nanoDESI | non-target | >10 μm | hundreds | limited | medium | ambient | delicate fabrication of probes |
| LAESI | non-target | >30 μm | tens | limited | medium | ambient | environment-induced noises |
| MALDI | non-target | 1–25 μm | hundreds | tens | medium | vacuum | low detection of small moleculeslow quantification accuracy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Vu, H.M.; Lee, J.-H.; Lim, H.; Kim, M.-S. Advances in Mass Spectrometry-Based Single Cell Analysis. Biology 2023, 12, 395. https://doi.org/10.3390/biology12030395
Lee S, Vu HM, Lee J-H, Lim H, Kim M-S. Advances in Mass Spectrometry-Based Single Cell Analysis. Biology. 2023; 12(3):395. https://doi.org/10.3390/biology12030395
Chicago/Turabian StyleLee, Siheun, Hung M. Vu, Jung-Hyun Lee, Heejin Lim, and Min-Sik Kim. 2023. "Advances in Mass Spectrometry-Based Single Cell Analysis" Biology 12, no. 3: 395. https://doi.org/10.3390/biology12030395
APA StyleLee, S., Vu, H. M., Lee, J.-H., Lim, H., & Kim, M.-S. (2023). Advances in Mass Spectrometry-Based Single Cell Analysis. Biology, 12(3), 395. https://doi.org/10.3390/biology12030395