
On the Advent of Super-Resolution Microscopy in the Realm of Polycomb Proteins

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
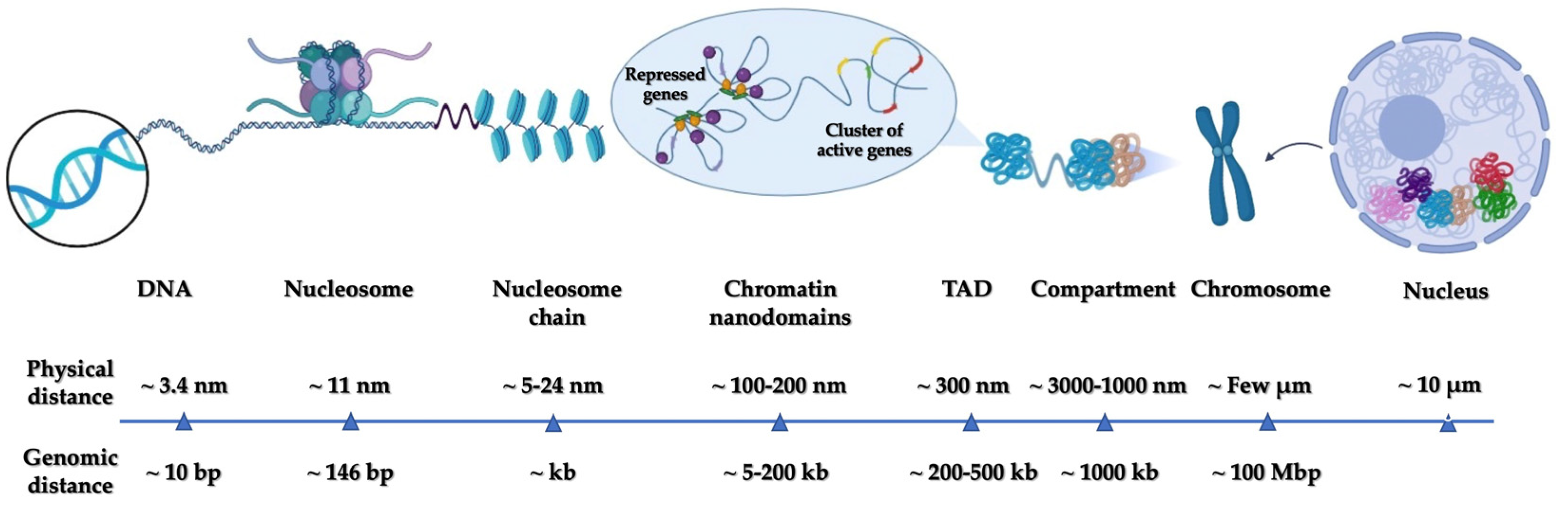
1. Introduction
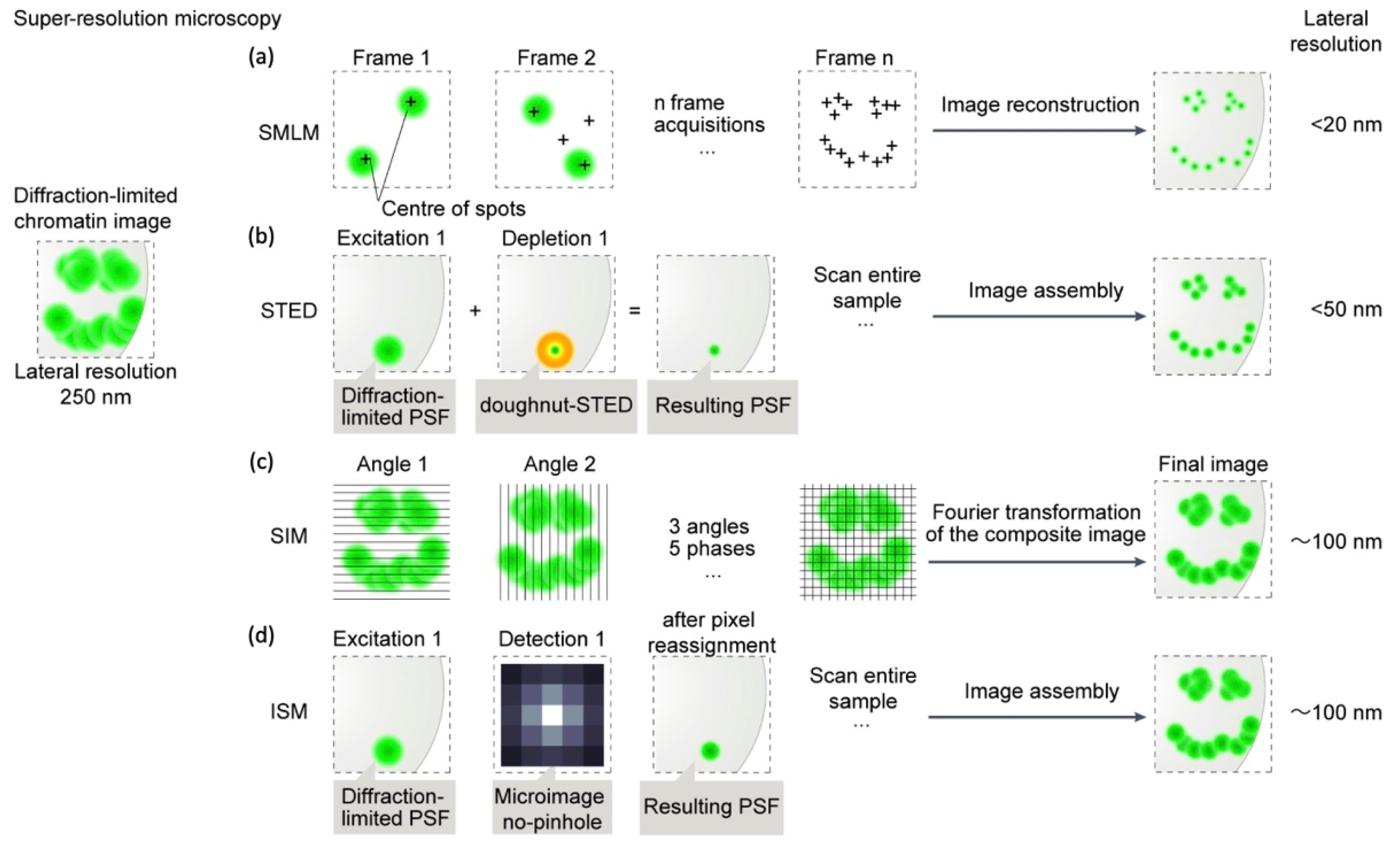
2. Super-Resolution Microscopy (SRM)
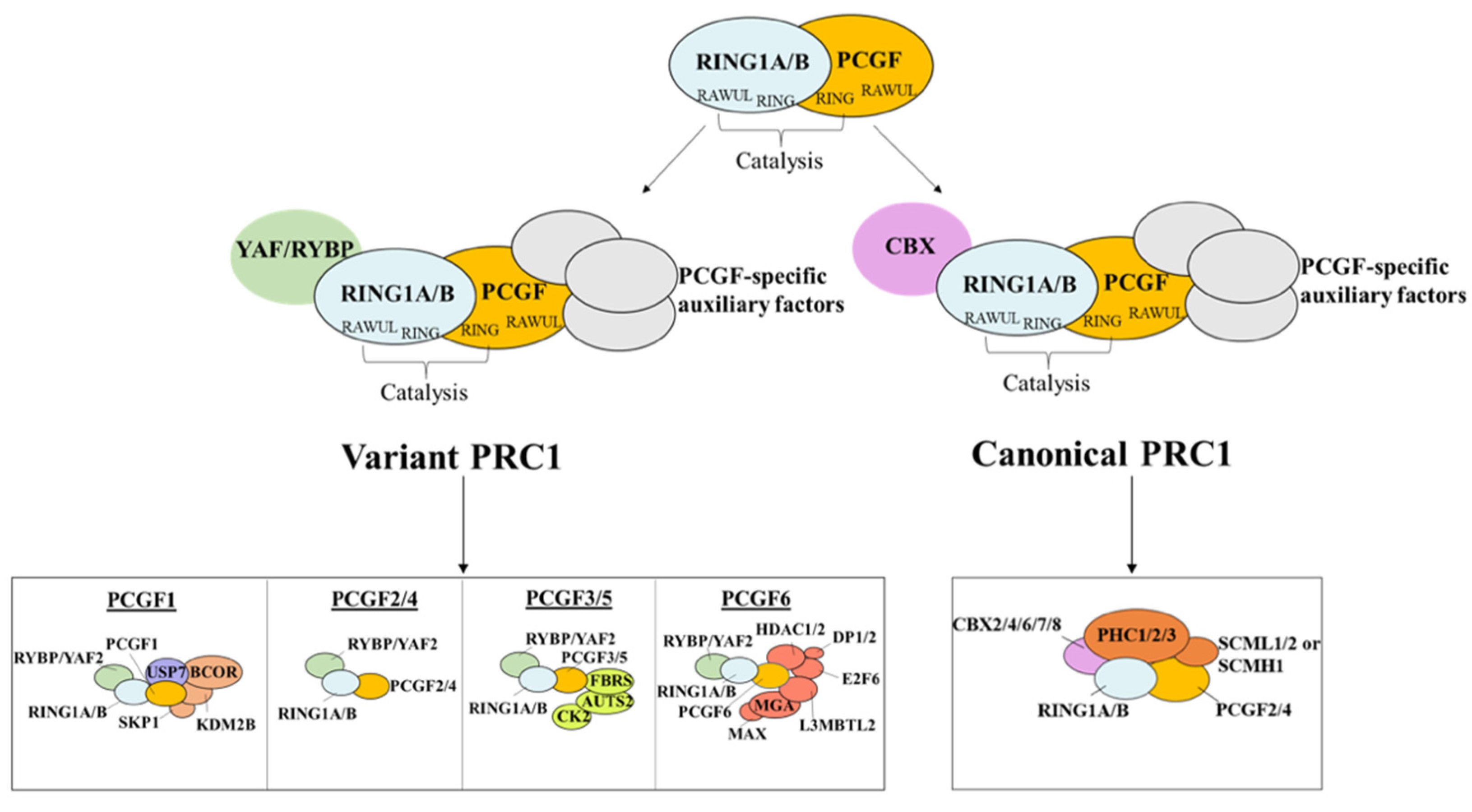
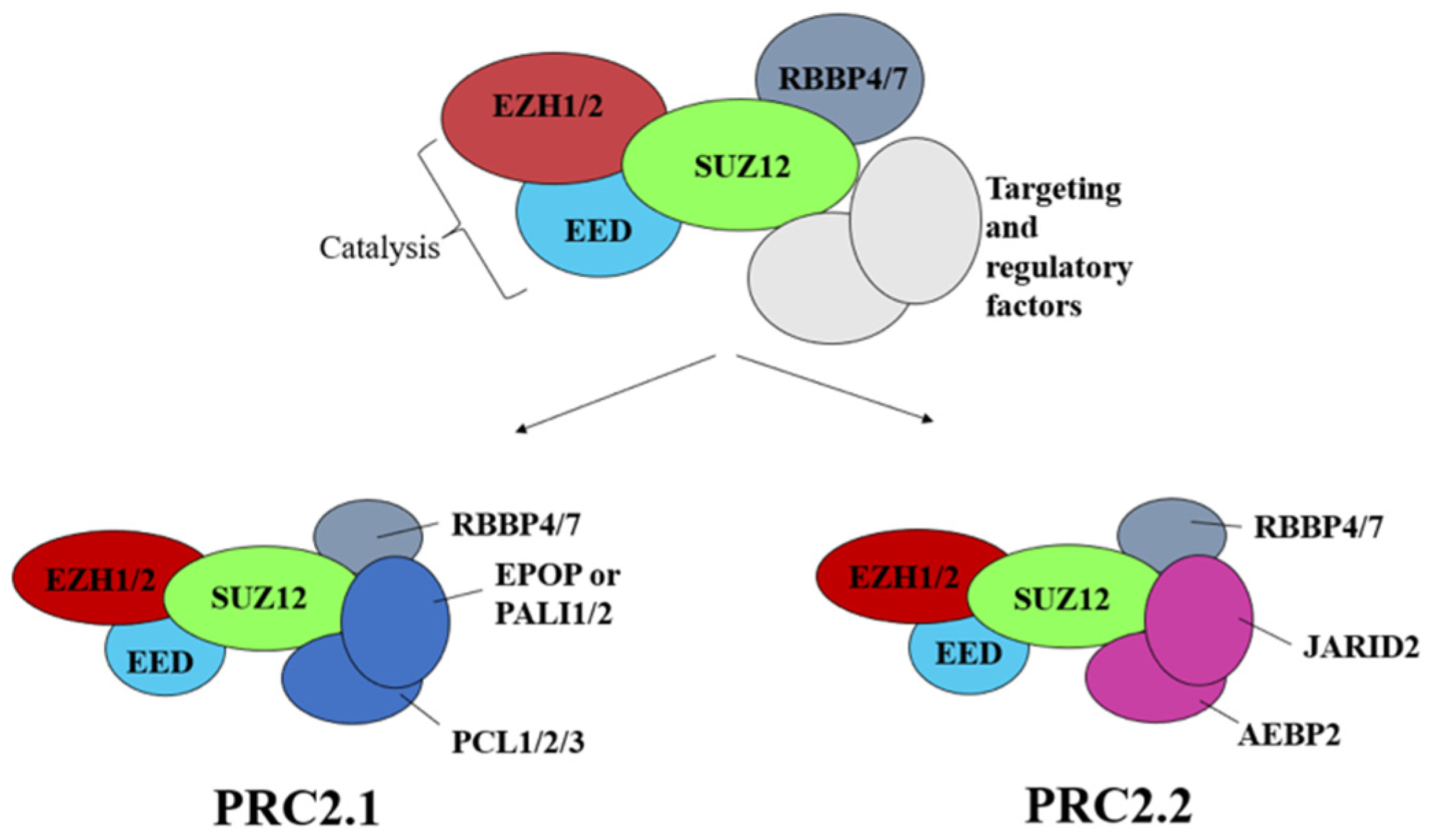
3. Structure of Polycomb Repressor Complexes PRC1 and PRC2
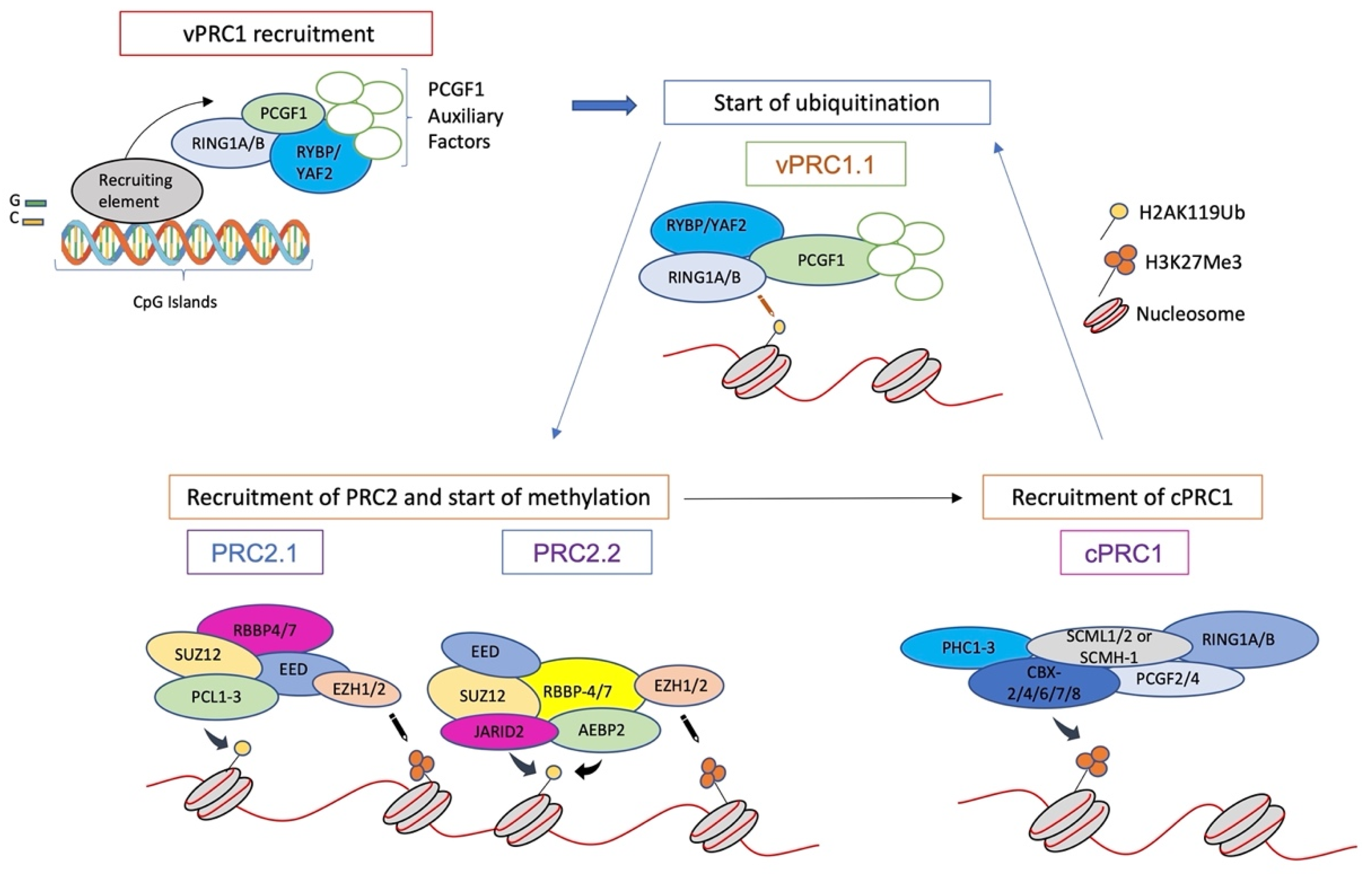
4. Formation and Spread of Polycomb Domains onto Chromatin
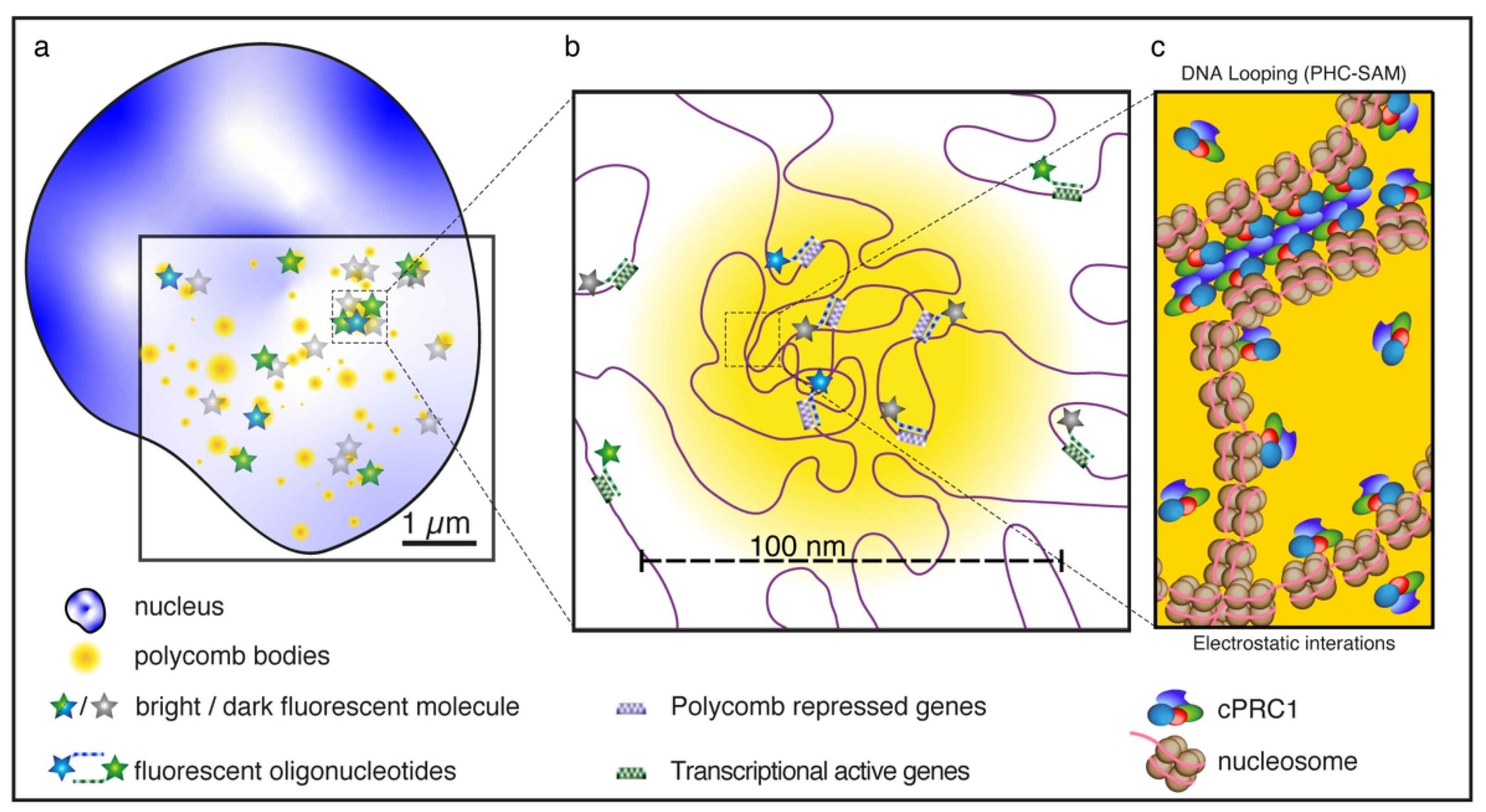
5. The Role of PRC1 in Shaping Chromatin Topology
6. Polycomb Complexes and Xist Regulation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, E.G.; Ha, H.; Lee, D.H.; Kim, W.R.; Lee, Y.J.; Bae, W.H.; Kim, H.S. Genomic Analyses of Non-Coding RNAs Overlapping Transposable Elements and Its Implication to Human Diseases. Int. J. Mol. Sci. 2022, 23, 8950. [Google Scholar] [CrossRef] [PubMed]
- Atlasi, Y.; Stunnenberg, H.G. The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 2017, 18, 643–658. [Google Scholar] [CrossRef]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.Q.; Liu, Z. Single-cell imaging of genome organization and dynamics. Mol. Syst. Biol. 2021, 17, e9653. [Google Scholar] [CrossRef] [PubMed]
- Jerkovic, I.; Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Bio. 2021, 22, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Klose, R.J. The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Bio. 2021, 22, 815–833. [Google Scholar] [CrossRef]
- Elsherbiny, A.; Dobreva, G. Epigenetic memory of cell fate commitment. Curr. Opin. Cell Biol. 2021, 69, 80–87. [Google Scholar] [CrossRef]
- Koppens, M.; van Lohuizen, M. Context-dependent actions of Polycomb repressors in cancer. Oncogene 2016, 35, 1341–1352. [Google Scholar] [CrossRef]
- Diaspro, A.; Bianchini, P. Optical nanoscopy. Riv. Nuovo Cimento 2020, 43, 385–455. [Google Scholar] [CrossRef]
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K.P.F. An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluores 2018, 6, 022003. [Google Scholar] [CrossRef]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Bio. 2017, 18, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Granboulan, N. The Fine Structure of the Cancer Cell Nucleus. Exp. Cell Res. 1963, 24 (Suppl. S9), 19–53. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 2017, 357, eaag0025. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Olins, A.L.; Olins, D.E. Chromatin structure in situ: The contribution of DNA ultrastructural cytochemistry. Eur. J. Histochem. 2014, 58, 2307. [Google Scholar] [CrossRef]
- Vicidomini, G. Image Formation in Fluorescence Microscopy; Springer: Dordrecht, The Netherlands, 2005; pp. 371–393. [Google Scholar]
- Masters, B.R. Abbe’s Theory of Image Formation in the Microscope. In Superresolution Optical Microscopy; Rhodes, W.T., Ed.; Springer Series in Optical Sciences; Springer Nature Switzerland AG: Cham, Switzerland, 2020; pp. 65–108. [Google Scholar]
- Toraldo di Francia, G. Resolving Power and Information. J. Opt. Soc. Am. 1955, 45, 497–501. [Google Scholar] [CrossRef]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffié, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat. Rev. Methods Prim. 2021, 1, 39. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.; Heilemann, M. Single-Molecule Localization Microscopy in Eukaryotes. Chem. Rev. 2017, 117, 7478–7509. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Moscatelli, J.; Smith, E.M.; Banerjee, C.; Puchner, E.M. Single-molecule localization microscopy and tracking with red-shifted states of conventional BODIPY conjugates in living cells. Nat. Comm. 2019, 10, 3400. [Google Scholar] [CrossRef]
- Klein, T.; Loschberger, A.; Proppert, S.; Wolter, S.; van de Linde, S.V.; Sauer, M. Live-cell dSTORM with SNAP-tag fusion proteins. Nat. Methods 2011, 8, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. STED super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef]
- Storti, B.; Carlotti, B.; Chiellini, G.; Ruglioni, M.; Salvadori, T.; Scotto, M.; Elisei, F.; Diaspro, A.; Bianchini, P.; Bizzarri, R. An Efficient Aequorea victoria Green Fluorescent Protein for Stimulated Emission Depletion Super-Resolution Microscopy. Int. J. Mol. Sci. 2022, 23, 2482. [Google Scholar] [CrossRef] [PubMed]
- Lanzano, L.; Hernandez, I.C.; Castello, M.; Gratton, E.; Diaspro, A.; Vicidomini, G. Encoding and decoding spatio-temporal information for super-resolution microscopy. Nat. Comm. 2015, 6, 6701. [Google Scholar] [CrossRef] [PubMed]
- Grotjohann, T.; Testa, I.; Reuss, M.; Brakemann, T.; Eggeling, C.; Hell, S.W.; Jakobs, S. rsEGFP2 enables fast RESOLFT nanoscopy of living cells. Elife 2012, 1, e00248. [Google Scholar] [CrossRef] [PubMed]
- Kostiuk, G.; Bucevicius, J.; Gerasimaite, R.; Lukinavicius, G. Application of STED imaging for chromatin studies. J. Phys. D Appl. Phys. 2019, 52, 504003. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynna, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef]
- Gwosch, K.C.; Pape, J.K.; Balzarotti, F.; Hoess, P.; Ellenberg, J.; Ries, J.; Hell, S.W. MINFLUX nanoscopy delivers 3D multicolor nanometer resolution in cells. Nat. Methods 2020, 17, 217–224. [Google Scholar] [CrossRef]
- Alvelid, J.; Damenti, M.; Sgattoni, C.; Testa, I. Event-triggered STED imaging. Nat. Methods 2022, 19, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Wegner, W.; Ilgen, P.; Gregor, C.; van Dort, J.; Mott, A.C.; Steffens, H.; Willig, K.I. In vivo mouse and live cell STED microscopy of neuronal actin plasticity using far-red emitting fluorescent proteins. Sci. Rep. 2017, 7, 11781. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, P.; Cardarelli, F.; Di Luca, M.; Diaspro, A.; Bizzarri, R. Nanoscale Protein Diffusion by STED-Based Pair Correlation Analysis. PLoS ONE 2014, 9, e99619. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef]
- Markaki, Y.; Smeets, D.; Fiedler, S.; Schmid, V.J.; Schermelleh, L.; Cremer, T.; Cremer, M. The potential of 3D-FISH and super-resolution structured illumination microscopy for studies of 3D nuclear architecture. BioEssays News Rev. Mol. Cell. Dev. Biol. 2012, 34, 412–426. [Google Scholar] [CrossRef]
- Miron, E.; Oldenkamp, R.; Brown, J.M.; Pinto, D.M.S.; Xu, C.S.; Faria, A.R.; Shaban, H.A.; Rhodes, J.D.P.; Innocent, C.; de Ornellas, S.; et al. Chromatin arranges in chains of mesoscale domains with nanoscale functional topography independent of cohesin. Sci. Adv. 2020, 6, eaba8811. [Google Scholar] [CrossRef]
- Szabo, Q.; Jost, D.; Chang, J.M.; Cattoni, D.I.; Papadopoulos, G.L.; Bonev, B.; Sexton, T.; Gurgo, J.; Jacquier, C.; Nollmann, M.; et al. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 2018, 4, eaar8082. [Google Scholar] [CrossRef]
- Castello, M.; Tortarolo, G.; Buttafava, M.; Deguchi, T.; Villa, F.; Koho, S.; Pesce, L.; Oneto, M.; Pelicci, S.; Lanzano, L.; et al. A robust and versatile platform for image scanning microscopy enabling super-resolution FLIM. Nat. Methods 2019, 16, 175–178. [Google Scholar] [CrossRef]
- Sheppard, C.J.R. Super-Resolution in Confocal Imaging. Optik 1988, 80, 53–54. [Google Scholar]
- Tortarolo, G.; Castello, M.; Diaspro, A.; Koho, S.; Vicidomini, G. Evaluating image resolution in stimulated emission depletion microscopy. Optica 2018, 5, 32–35. [Google Scholar] [CrossRef]
- Huff, J. The Airyscan detector from ZEISS: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 2015, 12, i–ii. [Google Scholar] [CrossRef]
- Sheppard, C.J.R.; Mehta, S.B.; Heintzmann, R. Superresolution by image scanning microscopy using pixel reassignment. Opt. Lett. 2013, 38, 2889–2892. [Google Scholar] [CrossRef] [PubMed]
- Nepita, I.; Piazza, S.; Ruglioni, M.; Cristiani, S.; Bosurgi, E.; Salvadori, T.; Vicidomini, G.; Diaspro, A.; Castello, M.; Bianchini, P.; et al. Image Scanning Microscopy to Investigate Polycomb Protein Colocalization onto Chromatin. Appl. Sci. 2023, 13, 1556. [Google Scholar] [CrossRef]
- Jurgens, G. A Group of Genes-Controlling the Spatial Expression of the Bithorax Complex in Drosophila. Nature 1985, 316, 153–155. [Google Scholar] [CrossRef]
- Loubiere, V.; Martinez, A.M.; Cavalli, G. Cell Fate and Developmental Regulation Dynamics by Polycomb Proteins and 3D Genome Architecture. BioEssays News Rev. Mol. Cell. Dev. Biol. 2019, 41, 15. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Occupying Chromatin: Polycomb Mechanisms for Getting to Genomic Targets, Stopping Transcriptional Traffic, and Staying Put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.H.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef]
- Rose, N.R.; King, H.W.; Blckledge, N.P.; Fursova, N.A.; Ember, K.J.I.; Fischer, R.; Kessler, B.M.; Klose, R.J. RYBP stimulates PRC1 to shape chromatin-based communication between Polycomb repressive complexes. Elife 2016, 5, e18591. [Google Scholar] [CrossRef] [PubMed]
- Kaustov, L.; Hui, O.Y.; Amaya, M.; Lemak, A.; Nady, N.; Duan, S.L.; Wasney, G.A.; Li, Z.H.; Vedadi, M.; Schapira, M.; et al. Recognition and Specificity Determinants of the Human Cbx Chromodomains. J. Biol. Chem. 2011, 286, 521–529. [Google Scholar] [CrossRef]
- Illingworth, R.S. Chromatin folding and nuclear architecture: PRC1 function in 3D. Curr. Opin. Genet. Dev. 2019, 55, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Taherbhoy, A.M.; Huang, O.W.; Cochran, A.G. BMI1-RING1B is an autoinhibited RING E3 ubiquitin ligase. Nat. Comm. 2015, 6, 7621. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.; Flyamer, I.M.; Williamson, I.; Sengupta, D.; Bickmore, W.A.; Illingworth, R.S. A central role for canonical PRC1 in shaping the 3D nuclear landscape. Gen. Dev. 2020, 34, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Justin, N.; Ohno, K.; Sharpe, M.L.; Son, J.; Drury, W.J.; Voigt, P.; Martin, S.R.; Taylor, W.R.; De Marco, V.; et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009, 461, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, J.L.; Cao, R.; Zhang, Y.; Kassis, J.A.; Jones, R.S. Hierarchical recruitment of Polycomb group silencing complexes. Mol. Cell 2004, 14, 637–646. [Google Scholar] [CrossRef]
- Kahn, T.G.; Dorafshan, E.; Schultheis, D.; Zare, A.; Stenberg, P.; Reim, I.; Pirrotta, V.; Schwartz, Y.B. Interdependence of PRC1 and PRC2 for recruitment to Polycomb Response Elements. Nucleic Acids Res. 2016, 44, 10132–10149. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Rose, N.R.; Klose, R.J. Targeting Polycomb systems to regulate gene expression: Modifications to a complex story. Nat. Rev. Mol. Cell Bio. 2015, 16, 643–649. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Gen. Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Farcas, A.M.; Blackledge, N.P.; Sudbery, I.; Long, H.K.; McGouran, J.F.; Rose, N.R.; Lee, S.; Sims, D.; Cerase, A.; Sheahan, T.W.; et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. Elife 2012, 1, e00205. [Google Scholar] [CrossRef]
- He, J.; Shen, L.; Wan, M.; Taranova, O.; Wu, H.; Zhang, Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat. Cell Biol. 2013, 15, 373–384. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.Y.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Comm. 2016, 7, 13661. [Google Scholar] [CrossRef]
- Perino, M.; van Mierlo, G.; Karemaker, I.D.; van Genesen, S.; Vermeulen, M.; Marks, H.; van Heeringen, S.J.; Veenstra, G.J.C. MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat. Genet. 2018, 50, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Liefke, R.; Jiang, J.Y.; Kurland, J.V.; Tian, W.; Deng, P.J.; Zhang, W.D.; He, Q.; Patel, D.J.; Bulyk, M.L.; et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 2017, 549, 287–291. [Google Scholar] [CrossRef]
- Lavarone, E.; Barbieri, C.M.; Pasini, D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat. Comm. 2019, 10, 1679. [Google Scholar] [CrossRef] [PubMed]
- Zhen, C.Y.; Tatavosian, R.; Huynh, T.N.; Duc, H.N.; Das, R.; Kokotovic, M.; Grimm, J.B.; Lavis, L.D.; Lee, J.; Mejia, F.J.; et al. Live-cell single-molecule tracking reveals co-recognition of H3K27me3 and DNA targets polycomb Cbx7-PRC1 to chromatin. Elife 2016, 5, 36. [Google Scholar] [CrossRef]
- Tamburri, S.; Lavarone, E.; Fernandez-Perez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856. [Google Scholar] [CrossRef]
- Oksuz, O.; Narendra, V.; Lee, C.H.; Descostes, N.; Leroy, G.; Raviram, R.; Blumenberg, L.; Karch, K.; Rocha, P.P.; Garcia, B.A.; et al. Capturing the Onset of PRC2-Mediated Repressive Domain Formation. Mol. Cell 2018, 70, 1149–1162. [Google Scholar] [CrossRef]
- Zhao, J.C.; Wang, M.; Chang, L.Y.; Yu, J.; Song, A.Q.; Liu, C.F.; Huang, W.J.; Zhang, T.T.; Wu, X.D.; Shen, X.H.; et al. RYBP/YAF2-PRC1 complexes and histone H1-dependent chromatin compaction mediate propagation of H2AK119ub1 during cell division. Nat. Cell Biol. 2020, 22, 439–452. [Google Scholar] [CrossRef]
- Alabert, C.; Barth, T.K.; Reveron-Gomez, N.; Sidoli, S.; Schmidt, A.; Jensen, O.N.; Imhof, A.; Groth, A. Two distinct modes for propagation of histone PTMs across the cell cycle. Gen. Dev. 2015, 29, 585–590. [Google Scholar] [CrossRef]
- Guo, Y.R.; Wang, G.G. Modulation of the high-order chromatin structure by Polycomb complexes. Front. Cell Dev. Biol. 2022, 10, 1021658. [Google Scholar] [CrossRef]
- Grau, D.J.; Chapman, B.A.; Garlick, J.D.; Borowsky, M.; Francis, N.J.; Kingston, R.E. Compaction of chromatin by diverse Polycomb group proteins requires localized regions of high charge. Gen. Dev. 2011, 25, 2210–2221. [Google Scholar] [CrossRef]
- Plys, A.J.; Davis, C.P.; Kim, J.; Rizki, G.; Keenen, M.M.; Marr, S.K.; Kingston, R.E. Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Gen. Dev. 2019, 33, 799–813. [Google Scholar] [CrossRef]
- Tatavosian, R.; Kent, S.; Brown, K.; Yao, T.T.; Huy, N.D.; Huynh, T.N.; Zhen, C.Y.; Ma, B.; Wang, H.B.; Ren, X.J. Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem. 2019, 294, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Isono, K.; Endo, T.A.; Ku, M.C.; Yamada, D.; Suzuki, R.; Sharif, J.; Ishikura, T.; Toyoda, T.; Bernstein, B.E.; Koseki, H. SAM Domain Polymerization Links Subnuclear Clustering of PRC1 to Gene Silencing. Dev. Cell 2013, 26, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Ji, F.; Sunwoo, H.; Jain, G.; Lee, J.T.; Sadreyev, R.I.; Dekker, J.; Kingston, R.E. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol. Cell 2017, 65, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Seif, E.; Kang, J.J.; Sasseville, C.; Senkovich, O.; Kaltashov, A.; Boulier, E.L.; Kapur, I.; Kim, C.A.; Francis, N.J. Phase separation by the polyhomeotic sterile alpha motif compartmentalizes Polycomb Group proteins and enhances their activity. Nat. Comm. 2020, 11, 5609. [Google Scholar] [CrossRef] [PubMed]
- Buchenau, P.; Hodgson, J.; Strutt, H.; Arndt-Jovin, D.J. The distribution of polycomb-group proteins during cell division and development in Drosophila embryos: Impact on models for silencing. J. Cell Biol. 1998, 141, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Pirrotta, V.; Li, H.B. A view of nuclear Polycomb bodies. Curr. Opin. Genet. Dev. 2012, 22, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Saurin, A.J.; Shiels, C.; Williamson, J.; Satijn, D.P.E.; Otte, A.P.; Sheer, D.; Freemont, P.S. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J. Cell Biol. 1998, 142, 887–898. [Google Scholar] [CrossRef]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.Y.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X.W. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.H.; Boettiger, A.N.; Schorderet, P.; Ergun, A.; Munger, C.; Sadreyev, R.I.; Zhuang, X.; Kingston, R.E.; Francis, N.J. Chromatin topology is coupled to Polycomb group protein subnuclear organization. Nat. Comm. 2016, 7, 10291. [Google Scholar] [CrossRef]
- Huang, B.; Wang, W.Q.; Bates, M.; Zhuang, X.W. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef]
- Szabo, Q.; Donjon, A.; Jerkovic, I.; Papadopoulos, G.L.; Cheutin, T.; Bonev, B.; Nora, E.P.; Bruneau, B.G.; Bantignies, F.; Cavalli, G. Regulation of single-cell genome organization into TADs and chromatin nanodomains. Nat. Genet. 2020, 52, 1151–1157. [Google Scholar] [CrossRef]
- Rivera, C.M.; Ren, B. Mapping Human Epigenomes. Cell 2013, 155, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Beliveau, B.J.; Joyce, E.F.; Apostolopoulos, N.; Yilmaz, F.; Fonseka, C.Y.; McCole, R.B.; Chang, Y.M.; Li, J.B.; Senaratne, T.N.; Williams, B.R.; et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. USA 2012, 109, 21301–21306. [Google Scholar] [CrossRef] [PubMed]
- Williamson, I.; Berlivet, S.; Eskeland, R.; Boyle, S.; Illingworth, R.S.; Paquette, D.; Dostie, J.; Bickmore, W.A. Spatial genome organization: Contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Gen. Dev. 2014, 28, 2778–2791. [Google Scholar] [CrossRef]
- Williamson, I.; Bickmore, W.A.; Illingworth, R.S. Polycomb-mediated chromatin compaction weathers the STORM. Genome Biol. 2016, 17, 35. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Sugar, R.; Dimond, A.; Javierre, B.M.; Armstrong, H.; Mifsud, B.; Dimitrova, E.; Matheson, L.; Tavares-Cadete, F.; Furlan-Magaril, M.; et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 2015, 47, 1179–1186. [Google Scholar] [CrossRef]
- Eskeland, R.; Leeb, M.; Grimes, G.R.; Kress, C.; Boyle, S.; Sproul, D.; Gilbert, N.; Fan, Y.H.; Skoultchi, A.I.; Wutz, A.; et al. Ring1B Compacts Chromatin Structure and Represses Gene Expression Independent of Histone Ubiquitination. Mol. Cell 2010, 38, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hyeon, C.B. Revisiting the organization of Polycomb-repressed domains: 3D chromatin models from Hi-C compared with super-resolution imaging. Nucleic Acids Res. 2020, 48, 11486–11494. [Google Scholar] [CrossRef]
- Liu, L.; Kim, M.H.; Hyeon, C. Heterogeneous Loop Model to Infer 3D Chromosome Structures from Hi-C. Biophys. J. 2019, 117, 613–625. [Google Scholar] [CrossRef]
- Cattoni, D.I.; Gizzi, A.M.C.; Georgieva, M.; Di Stefano, M.; Valeri, A.; Chamousset, D.; Houbron, C.; Dejardin, S.; Fiche, J.B.; Gonzalez, I.; et al. Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions. Nat. Comm. 2017, 8, 1753. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Thirumalai, D. Conformational heterogeneity in human interphase chromosome organization reconciles the FISH and Hi-C paradox. Nat. Comm. 2019, 10, 3894. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Bowness, J.S.; Wei, G.F. Progress toward understanding chromosome silencing by Xist RNA. Gen. Dev. 2020, 34, 733–744. [Google Scholar] [CrossRef]
- Loda, A.; Heard, E. Xist RNA in action: Past, present, and future. PLoS Genet. 2019, 15, e1008333. [Google Scholar] [CrossRef]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef]
- Markaki, Y.; Chong, J.G.; Wang, Y.Y.; Jacobson, E.C.; Luong, C.; Tan, S.Y.X.; Jachowicz, J.W.; Strehle, M.; Maestrini, D.; Banerjee, A.K.; et al. Xist nucleates local protein gradients to propagate silencing across the X chromosome (vol 184, pg 6174, 2021). Cell 2021, 184, 6212. [Google Scholar] [CrossRef]
- Simon, M.D.; Pinter, S.F.; Fang, R.; Sarma, K.; Rutenberg-Schoenberg, M.; Bowman, S.K.; Kesner, B.A.; Maier, V.K.; Kingston, R.E.; Lee, J.T. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 2013, 504, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Masui, O.; Corbel, C.; Nagao, K.; Endo, T.A.; Kezuka, F.; Diabangouaya, P.; Nakayama, M.; Kumon, M.; Koseki, Y.; Obuse, C.; et al. Polycomb repressive complexes 1 and 2 are each essential for maintenance of X inactivation in extra-embryonic lineages. Nat. Cell Biol. 2023, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.B.; de la Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003, 300, 131–135. [Google Scholar] [CrossRef]
- Cerase, A.; Smeets, D.; Tang, Y.A.; Gdula, M.; Kraus, F.; Spivakov, M.; Moindrot, B.; Leleu, M.; Tattermusch, A.; Demmerie, J.; et al. Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 2235–2240. [Google Scholar] [CrossRef] [PubMed]
- Smeets, D.; Markaki, Y.; Schmid, V.J.; Kraus, F.; Tattermusch, A.; Cerase, A.; Sterr, M.; Fiedler, S.; Demmerle, J.; Popken, J.; et al. Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenet Chromatin 2014, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, H.; Wu, J.Y.; Lee, J.T. The Xist RNA-PRC2 complex at 20-nm resolution reveals a low Xist stoichiometry and suggests a hit-and-run mechanism in mouse cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4216–E4225. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.K.; Deaton, A.M.; Lee, J.T. A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol. Cell 2006, 21, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Pandya-Jones, A.; Markaki, Y.; Serizay, J.; Chitiashvili, T.; Leon, W.R.M.; Damianov, A.; Chronis, C.; Papp, B.; Chen, C.K.; McKee, R.; et al. A protein assembly mediates Xist localization and gene silencing. Nature 2020, 587, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Jachowicz, J.W.; Strehle, M.; Banerjee, A.K.; Blanco, M.R.; Thai, J.; Guttman, M. Xist spatially amplifies SHARP/SPEN recruitment to balance chromosome-wide silencing and specificity to the X chromosome. Nat. Struct. Mol. Biol. 2022, 29, 239–249. [Google Scholar] [CrossRef]
- Cerase, A.; Calabrese, J.M.; Tartaglia, G.G. Phase separation drives X-chromosome inactivation. Nat. Struct. Mol. Biol. 2022, 29, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Pintacuda, G.; Wei, G.F.; Roustan, C.; Kirmizitas, B.A.; Solcan, N.; Cerase, A.; Castello, A.; Mohammed, S.; Moindrot, B.; Nesterova, T.B.; et al. hnRNPK Recruits PCGF3/5-PRC1 to the Xist RNA B-Repeat to Establish Polycomb-Mediated Chromosomal Silencing. Mol. Cell 2017, 68, 955–969.e10. [Google Scholar] [CrossRef] [PubMed]
- Bousard, A.; Raposo, A.C.; Zylicz, J.J.; Picard, C.; Pires, V.B.; Qi, Y.Y.; Gil, C.; Syx, L.; Chang, H.Y.; Heard, E.; et al. The role of Xist-mediated Polycomb recruitment in the initiation of X-chromosome inactivation. Embo Rep. 2019, 20, e48019. [Google Scholar] [CrossRef] [PubMed]
- Cerase, A.; Tartaglia, G.G. Long non-coding RNA-polycomb intimate rendezvous. Open Biol. 2020, 10, 200126. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nepita, I.; Piazza, S.; Ruglioni, M.; Cristiani, S.; Bosurgi, E.; Salvadori, T.; Vicidomini, G.; Diaspro, A.; Castello, M.; Cerase, A.; et al. On the Advent of Super-Resolution Microscopy in the Realm of Polycomb Proteins. Biology 2023, 12, 374. https://doi.org/10.3390/biology12030374
Nepita I, Piazza S, Ruglioni M, Cristiani S, Bosurgi E, Salvadori T, Vicidomini G, Diaspro A, Castello M, Cerase A, et al. On the Advent of Super-Resolution Microscopy in the Realm of Polycomb Proteins. Biology. 2023; 12(3):374. https://doi.org/10.3390/biology12030374
Chicago/Turabian StyleNepita, Irene, Simonluca Piazza, Martina Ruglioni, Sofia Cristiani, Emanuele Bosurgi, Tiziano Salvadori, Giuseppe Vicidomini, Alberto Diaspro, Marco Castello, Andrea Cerase, and et al. 2023. "On the Advent of Super-Resolution Microscopy in the Realm of Polycomb Proteins" Biology 12, no. 3: 374. https://doi.org/10.3390/biology12030374
APA StyleNepita, I., Piazza, S., Ruglioni, M., Cristiani, S., Bosurgi, E., Salvadori, T., Vicidomini, G., Diaspro, A., Castello, M., Cerase, A., Bianchini, P., Storti, B., & Bizzarri, R. (2023). On the Advent of Super-Resolution Microscopy in the Realm of Polycomb Proteins. Biology, 12(3), 374. https://doi.org/10.3390/biology12030374