
Immunopeptidomics in the Era of Single-Cell Proteomics

{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Miniaturization and Acceleration of Immunopeptidomics Sample Preparation
3. Improving Peptide Separation and Ionization for Increased Sensitivity
4. Evolution of Mass Spectrometers for SCP and Immunopeptidomics
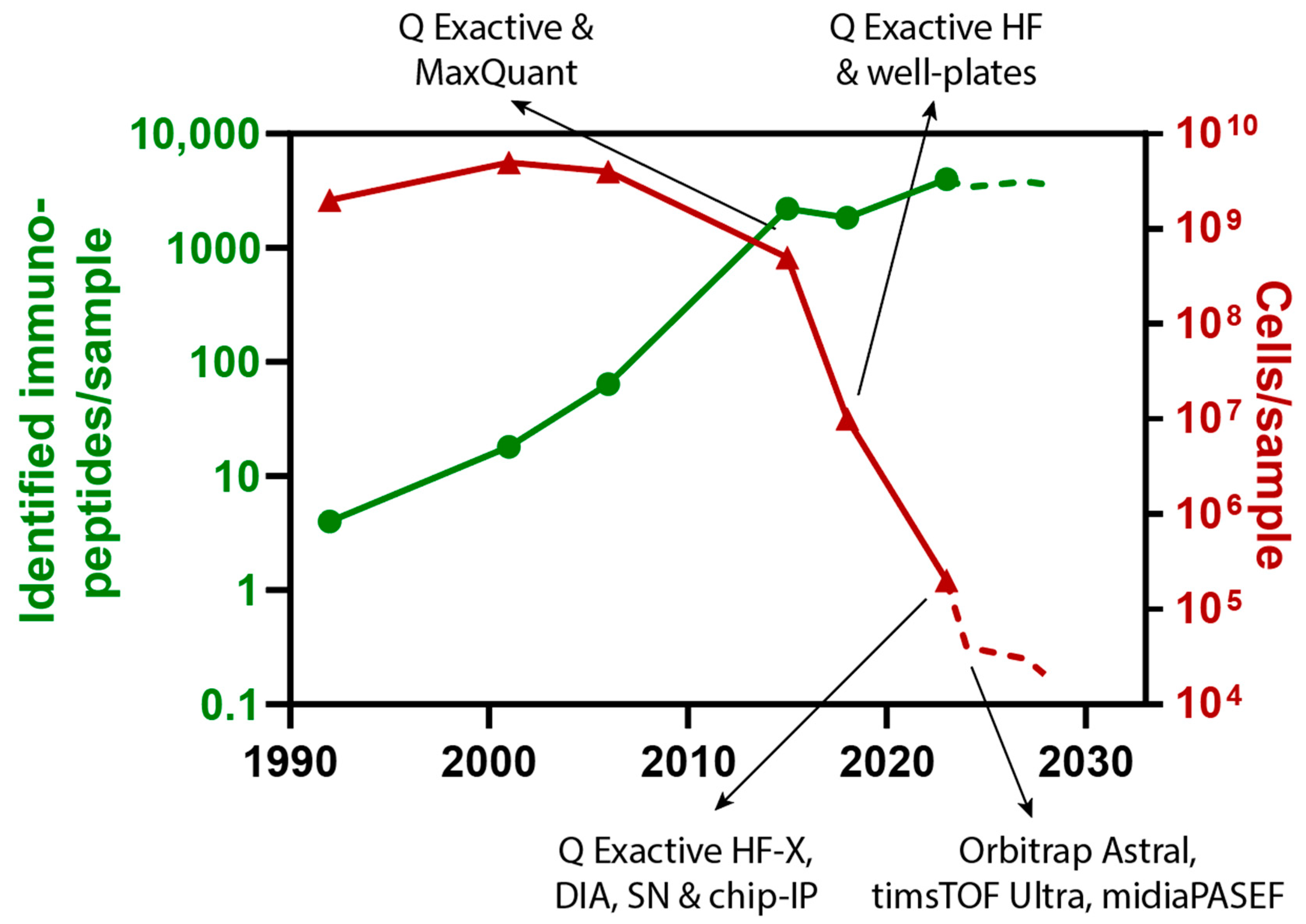
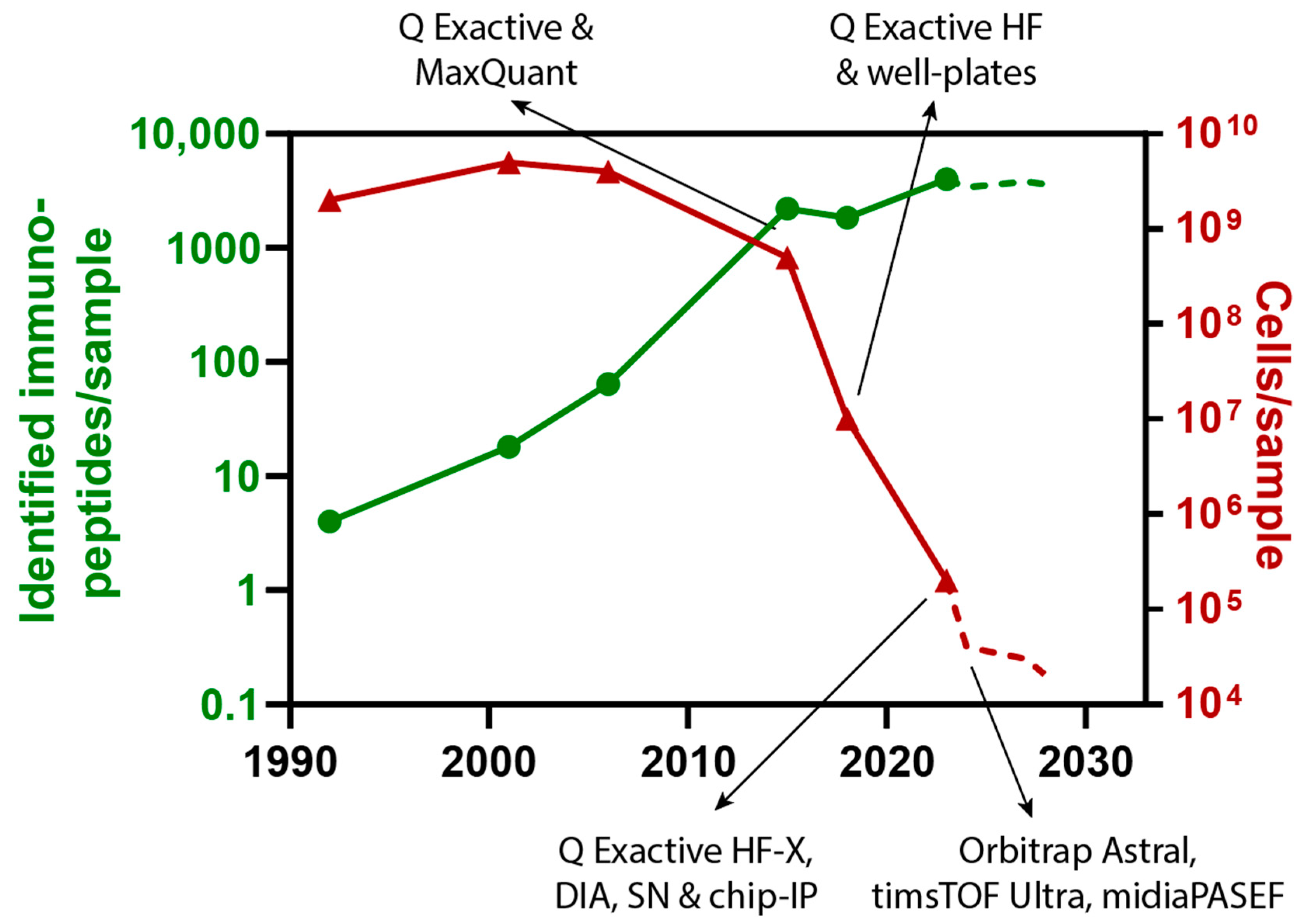
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunt, D.F.; Henderson, R.A.; Shabanowitz, J.; Sakaguchi, K.; Michel, H.; Sevilir, N.; Cox, A.L.; Appella, E.; Engelhard, V.H. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 1992, 255, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.L.; Verbeke, R.; Asselman, C.; Aernout, I.; Gul, A.; Eggermont, D.; Boucher, K.; Thery, F.; Maia, T.M.; Demol, H.; et al. Immunopeptidomics-based design of mRNA vaccine formulations against Listeria monocytogenes. Nat. Commun. 2022, 13, 6075. [Google Scholar] [CrossRef] [PubMed]
- Ingels, J.; De Cock, L.; Mayer, R.L.; Devreker, P.; Weening, K.; Heyns, K.; Lootens, N.; De Smet, S.; Brusseel, M.; De Munter, S.; et al. Small-scale manufacturing of neoantigen-encoding messenger RNA for early-phase clinical trials. Cytotherapy 2022, 24, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Klaeger, S.; Sarkizova, S.; Pearlman, L.R.; Chen, D.Y.; Gallagher, K.M.E.; Bauer, M.R.; Taylor, H.B.; Dunn, W.A.; Tarr, C.; et al. Profiling SARS-CoV-2 HLA-I peptidome reveals T cell epitopes from out-of-frame ORFs. Cell 2021, 184, 3962–3980.e17. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, P.; Muller, J.; Nicastri, A.; Cantillon, D.; Madhavan, M.; Charles, P.D.; Fotso, C.B.; Wittenberg, R.; Bull, N.; Pinpathomrat, N.; et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Bassani-Sternberg, M. Predicting Antigen Presentation-What Could We Learn from a Million Peptides? Front. Immunol. 2018, 9, 1716. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Karpinski, T.M.; Ozarowski, M.; Seremak-Mrozikiewicz, A.; Wolski, H.; Wlodkowic, D. The 2020 race towards SARS-CoV-2 specific vaccines. Theranostics 2021, 11, 1690–1702. [Google Scholar] [CrossRef]
- Poolman, J.T. Expanding the role of bacterial vaccines into life-course vaccination strategies and prevention of antimicrobial-resistant infections. NPJ Vaccines 2020, 5, 84. [Google Scholar] [CrossRef]
- Rosini, R.; Nicchi, S.; Pizza, M.; Rappuoli, R. Vaccines against Antimicrobial Resistance. Front. Immunol. 2020, 11, 1048. [Google Scholar] [CrossRef]
- Mayer, R.L.; Impens, F. Immunopeptidomics for next-generation bacterial vaccine development. Trends Microbiol. 2021, 29, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.E.; Bassani-Sternberg, M. The impact of immunopeptidomics: From basic research to clinical implementation. Semin. Immunol. 2023, 66, 101727. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, A.I.; Chong, C.; Huber, F.; Pak, H.; Stevenson, B.J.; Muller, M.; Michaux, J.; Altimiras, E.R.; Rusakiewicz, S.; Simo-Riudalbas, L.; et al. The immunopeptidome landscape associated with T cell infiltration, inflammation and immune editing in lung cancer. Nat. Cancer 2023, 4, 608–628. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, M.; Mayer, R.L.; Mechtler, K. Label-free single cell proteomics utilizing ultrafast LC and MS instrumentation: A valuable complementary technique to multiplexing. Proteomics 2023, 23, e2200162. [Google Scholar] [CrossRef]
- Matzinger, M.; Muller, E.; Durnberger, G.; Pichler, P.; Mechtler, K. Robust and Easy-to-Use One-Pot Workflow for Label-Free Single-Cell Proteomics. Anal. Chem. 2023, 95, 4435–4445. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.D.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol. 2022, 18, e10798. [Google Scholar] [CrossRef]
- Ctortecka, C.; Hartlmayr, D.; Seth, A.; Mendjan, S.; Tourniaire, G.; Udeshi, N.D.; Carr, S.A.; Mechtler, K. An automated nanowell-array workflow for quantitative multiplexed single-cell proteomics sample preparation at high sensitivity. Mol. Cell Proteom. 2023, 22, 100665. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwangler, B.; Uresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; Keller, U.A.D.; Dick, J.E.; Porse, B.T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 3341. [Google Scholar] [CrossRef]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22, 50. [Google Scholar] [CrossRef]
- Budnik, B.; Levy, E.; Harmange, G.; Slavov, N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19, 161. [Google Scholar] [CrossRef]
- Johnston, S.M.; Webber, K.G.I.; Xie, X.; Truong, T.; Nydegger, A.; Lin, H.L.; Nwosu, A.; Zhu, Y.; Kelly, R.T. Rapid, One-Step Sample Processing for Label-Free Single-Cell Proteomics. J. Am. Soc. Mass. Spectrom. 2023, 34, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Mun, D.G.; Bhat, F.A.; Ding, H.; Madden, B.J.; Natesampillai, S.; Badley, A.D.; Johnson, K.L.; Kelly, R.T.; Pandey, A. Optimizing single cell proteomics using trapped ion mobility spectrometry for label-free experiments. Analyst 2023, 148, 3466–3475. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Clair, G.; Chrisler, W.B.; Shen, Y.; Zhao, R.; Shukla, A.K.; Moore, R.J.; Misra, R.S.; Pryhuber, G.S.; Smith, R.D.; et al. Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angew. Chem. Int. Ed. Engl. 2018, 57, 12370–12374. [Google Scholar] [CrossRef] [PubMed]
- Virant-Klun, I.; Leicht, S.; Hughes, C.; Krijgsveld, J. Identification of Maturation-Specific Proteins by Single-Cell Proteomics of Human Oocytes. Mol. Cell Proteom. 2016, 15, 2616–2627. [Google Scholar] [CrossRef]
- Petrosius, V.; Aragon-Fernandez, P.; Uresin, N.; Kovacs, G.; Phlairaharn, T.; Furtwangler, B.; Op De Beeck, J.; Skovbakke, S.L.; Goletz, S.; Thomsen, S.F.; et al. Exploration of cell state heterogeneity using single-cell proteomics through sensitivity-tailored data-independent acquisition. Nat. Commun. 2023, 14, 5910. [Google Scholar] [CrossRef]
- Chang, Y.H.; Gregorich, Z.R.; Chen, A.J.; Hwang, L.; Guner, H.; Yu, D.; Zhang, J.; Ge, Y. New mass-spectrometry-compatible degradable surfactant for tissue proteomics. J. Proteome Res. 2015, 14, 1587–1599. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F.; Mao, J.; Zhang, Z.; Liu, Z.; Huang, G.; Cheng, K.; Zou, H. High-sensitivity N-glycoproteomic analysis of mouse brain tissue by protein extraction with a mild detergent of N-dodecyl beta-D-maltoside. Anal. Chem. 2015, 87, 2054–2057. [Google Scholar] [CrossRef]
- Liang, Y.; Acor, H.; McCown, M.A.; Nwosu, A.J.; Boekweg, H.; Axtell, N.B.; Truong, T.; Cong, Y.; Payne, S.H.; Kelly, R.T. Fully Automated Sample Processing and Analysis Workflow for Low-Input Proteome Profiling. Anal. Chem. 2021, 93, 1658–1666. [Google Scholar] [CrossRef]
- Chong, C.; Marino, F.; Pak, H.; Racle, J.; Daniel, R.T.; Muller, M.; Gfeller, D.; Coukos, G.; Bassani-Sternberg, M. High-throughput and Sensitive Immunopeptidomics Platform Reveals Profound Interferongamma-Mediated Remodeling of the Human Leukocyte Antigen (HLA) Ligandome. Mol. Cell Proteom. 2018, 17, 533–548. [Google Scholar] [CrossRef]
- Phulphagar, K.M.; Ctortecka, C.; Jacome, A.S.V.; Klaeger, S.; Verzani, E.K.; Hernandez, G.M.; Udeshi, N.D.; Clauser, K.R.; Abelin, J.G.; Carr, S.A. Sensitive, High-Throughput HLA-I and HLA-II Immunopeptidomics Using Parallel Accumulation-Serial Fragmentation Mass Spectrometry. Mol. Cell Proteom. 2023, 22, 100563. [Google Scholar] [CrossRef]
- Klaeger, S.; Apffel, A.; Clauser, K.R.; Sarkizova, S.; Oliveira, G.; Rachimi, S.; Le, P.M.; Tarren, A.; Chea, V.; Abelin, J.G.; et al. Optimized Liquid and Gas Phase Fractionation Increases HLA-Peptidome Coverage for Primary Cell and Tissue Samples. Mol. Cell Proteom. 2021, 20, 100133. [Google Scholar] [CrossRef] [PubMed]
- Lim Kam Sian, T.C.C.; Goncalves, G.; Steele, J.R.; Shamekhi, T.; Bramberger, L.; Jin, D.; Shahbazy, M.; Purcell, A.W.; Ramarathinam, S.; Stoychev, S.; et al. SAPrIm, a semi-automated protocol for mid-throughput immunopeptidomics. Front. Immunol. 2023, 14, 1107576. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Huang, M.; Wang, X.K.; Zhu, Y.; Li, J.S.; Wong, C.C.L.; Fang, Q. Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem. 2018, 90, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Motamedchaboki, K.; Misal, S.A.; Liang, Y.; Guise, A.J.; Truong, T.; Huguet, R.; Plowey, E.D.; Zhu, Y.; Lopez-Ferrer, D.; et al. Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 2020, 12, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Furlan, C.; Dirks, R.A.M.; Thomas, P.C.; Jones, R.C.; Wang, J.; Lynch, M.; Marks, H.; Vermeulen, M. Miniaturised interaction proteomics on a microfluidic platform with ultra-low input requirements. Nat. Commun. 2019, 10, 1525. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pak, H.S.; Huber, F.; Michaux, J.; Taillandier-Coindard, M.; Altimiras, E.R.; Bassani-Sternberg, M. A microfluidics-enabled automated workflow of sample preparation for MS-based immunopeptidomics. Cell Rep. Methods 2023, 3, 100479. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; O'Brien Johnson, R.; Livson, Y.; Greer, T.; Zheng, X.; Li, N. Maximizing hydrophobic peptide recovery in proteomics and antibody development using a mass spectrometry compatible surfactant. Anal. Biochem. 2022, 658, 114924. [Google Scholar] [CrossRef]
- Krisp, C.L.M.; Almeida, A.; Sandow, J.; Hartlmayr, D.; Seth, A.; Kruppa, G. Robust and High-Throughput Single Cell Proteomics with the Evosep One. Available online: https://www.evosep.com/wp-content/uploads/2022/06/AN-018B-Whisper40SPD_SingleCell.pdf (accessed on 30 September 2023).
- Webber, K.G.I.; Truong, T.; Johnston, S.M.; Zapata, S.E.; Liang, Y.; Davis, J.M.; Buttars, A.D.; Smith, F.B.; Jones, H.E.; Mahoney, A.C.; et al. Label-Free Profiling of up to 200 Single-Cell Proteomes per Day Using a Dual-Column Nanoflow Liquid Chromatography Platform. Anal. Chem. 2022, 94, 6017–6025. [Google Scholar] [CrossRef]
- Zheng, R.; Matzinger, M.; Mayer, R.; Valenta, A.; Sun, X.; Mechtler, K. A high-sensitivity low-nanoflow LC-MS configuration for high-throughput sample-limited proteomics. bioRxiv 2023. [Google Scholar] [CrossRef]
- Xiang, P.; Zhu, Y.; Yang, Y.; Zhao, Z.; Williams, S.M.; Moore, R.J.; Kelly, R.T.; Smith, R.D.; Liu, S. Picoflow Liquid Chromatography-Mass Spectrometry for Ultrasensitive Bottom-Up Proteomics Using 2-mum-i.d. Open Tubular Columns. Anal. Chem. 2020, 92, 4711–4715. [Google Scholar] [CrossRef]
- Kreimer, S.; Binek, A.; Chazarin, B.; Cho, J.H.; Haghani, A.; Hutton, A.; Marban, E.; Mastali, M.; Meyer, J.G.; Mesquita, T.; et al. High-Throughput Single-Cell Proteomic Analysis of Organ-Derived Heterogeneous Cell Populations by Nanoflow Dual-Trap Single-Column Liquid Chromatography. Anal. Chem. 2023, 95, 9145–9150. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.L.; Matzinger, M.; Schmücker, A.; Stejskal, K.; Krššáková, G.; Berger, F.; Mechtler, K. Wide Window Acquisition and AI-based data analysis to reach deep proteome coverage for a wide sample range, including single cell proteomic inputs. bioRxiv 2022. [Google Scholar] [CrossRef]
- Stewart, H.I.; Grinfeld, D.; Giannakopulos, A.; Petzoldt, J.; Shanley, T.; Garland, M.; Denisov, E.; Peterson, A.C.; Damoc, E.; Zeller, M.; et al. Parallelized Acquisition of Orbitrap and Astral Analyzers Enables High-Throughput Quantitative Analysis. Anal. Chem. 2023, 95, 15656–15664. [Google Scholar] [CrossRef] [PubMed]
- Battellino, T.; Ogata, K.; Spicer, V.; Ishihama, Y.; Krokhin, O. Acetic Acid Ion Pairing Additive for Reversed-Phase HPLC Improves Detection Sensitivity in Bottom-up Proteomics Compared to Formic Acid. J. Proteome Res. 2023, 22, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Orsburn, B.C. Acetic acid is a superior acidifier for sub-nanogram and single cell proteomic studies. bioRxiv 2023. [Google Scholar] [CrossRef]
- Olsen, J.V.; de Godoy, L.M.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell Proteom. 2005, 4, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Damoc, E.; Hauschild, J.P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell Proteom. 2011, 10, M111-011015. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Szyrwiel, L.; Sinn, L.; Ralser, M.; Demichev, V. Slice-PASEF: Fragmenting all ions for maximum sensitivity in proteomics. bioRxiv 2022. [Google Scholar] [CrossRef]
- Skowronek, P.; Krohs, F.; Lubeck, M.; Wallmann, G.; Itang, E.C.M.; Koval, P.; Wahle, M.; Thielert, M.; Meier, F.; Willems, S.; et al. Synchro-PASEF Allows Precursor-Specific Fragment Ion Extraction and Interference Removal in Data-Independent Acquisition. Mol. Cell Proteom. 2023, 22, 100489. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Łącki, M.K.; Startek, M.P.; Teschner, D.; Brehmer, S.; Decker, J.; Schild, T.; Krieger, J.; Krohs, F.; Raether, O.; et al. midiaPASEF maximizes information content in data-independent acquisition proteomics. bioRxiv 2023. [Google Scholar] [CrossRef]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, Z.; Bomgarden, R.D.; Pike, I.; Kuhn, K.; Rogers, J.C.; Roberts, T.M.; Gygi, S.P.; Paulo, J.A. TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J. Proteome Res. 2021, 20, 2964–2972. [Google Scholar] [CrossRef] [PubMed]
- Petelski, A.A.; Emmott, E.; Leduc, A.; Huffman, R.G.; Specht, H.; Perlman, D.H.; Slavov, N. Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc. 2021, 16, 5398–5425. [Google Scholar] [CrossRef]
- Vegvari, A.; Zhang, X.; Zubarev, R.A. Toward Single Bacterium Proteomics. J. Am. Soc. Mass. Spectrom. 2023, 34, 2098–2106. [Google Scholar] [CrossRef]
- Pfammatter, S.; Bonneil, E.; Lanoix, J.; Vincent, K.; Hardy, M.P.; Courcelles, M.; Perreault, C.; Thibault, P. Extending the Comprehensiveness of Immunopeptidome Analyses Using Isobaric Peptide Labeling. Anal. Chem. 2020, 92, 9194–9204. [Google Scholar] [CrossRef]
- Ye, Z.; Sabatier, P.; van der Hoeven, L.; Phlairaharn, T.; Hartlmayr, D.; Izaguirre, F.; Seth, A.; Joshi, H.J.; Bekker-Jensen, D.B.; Bache, N.; et al. High-throughput and scalable single cell proteomics identifies over 5000 proteins per cell. bioRxiv 2023. [Google Scholar] [CrossRef]
- Purcell, A.W.; Gorman, J.J.; Garcia-Peydro, M.; Paradela, A.; Burrows, S.R.; Talbo, G.H.; Laham, N.; Peh, C.A.; Reynolds, E.C.; Lopez De Castro, J.A.; et al. Quantitative and qualitative influences of tapasin on the class I peptide repertoire. J. Immunol. 2001, 166, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Gebreselassie, D.; Spiegel, H.; Vukmanovic, S. Sampling of major histocompatibility complex class I-associated peptidome suggests relatively looser global association of HLA-B*5101 with peptides. Hum. Immunol. 2006, 67, 894–906. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayer, R.L.; Mechtler, K. Immunopeptidomics in the Era of Single-Cell Proteomics. Biology 2023, 12, 1514. https://doi.org/10.3390/biology12121514
Mayer RL, Mechtler K. Immunopeptidomics in the Era of Single-Cell Proteomics. Biology. 2023; 12(12):1514. https://doi.org/10.3390/biology12121514
Chicago/Turabian StyleMayer, Rupert L., and Karl Mechtler. 2023. "Immunopeptidomics in the Era of Single-Cell Proteomics" Biology 12, no. 12: 1514. https://doi.org/10.3390/biology12121514
APA StyleMayer, R. L., & Mechtler, K. (2023). Immunopeptidomics in the Era of Single-Cell Proteomics. Biology, 12(12), 1514. https://doi.org/10.3390/biology12121514