
Types of Cell Death from a Molecular Perspective



Abstract
Simple Summary
Abstract
1. Introduction
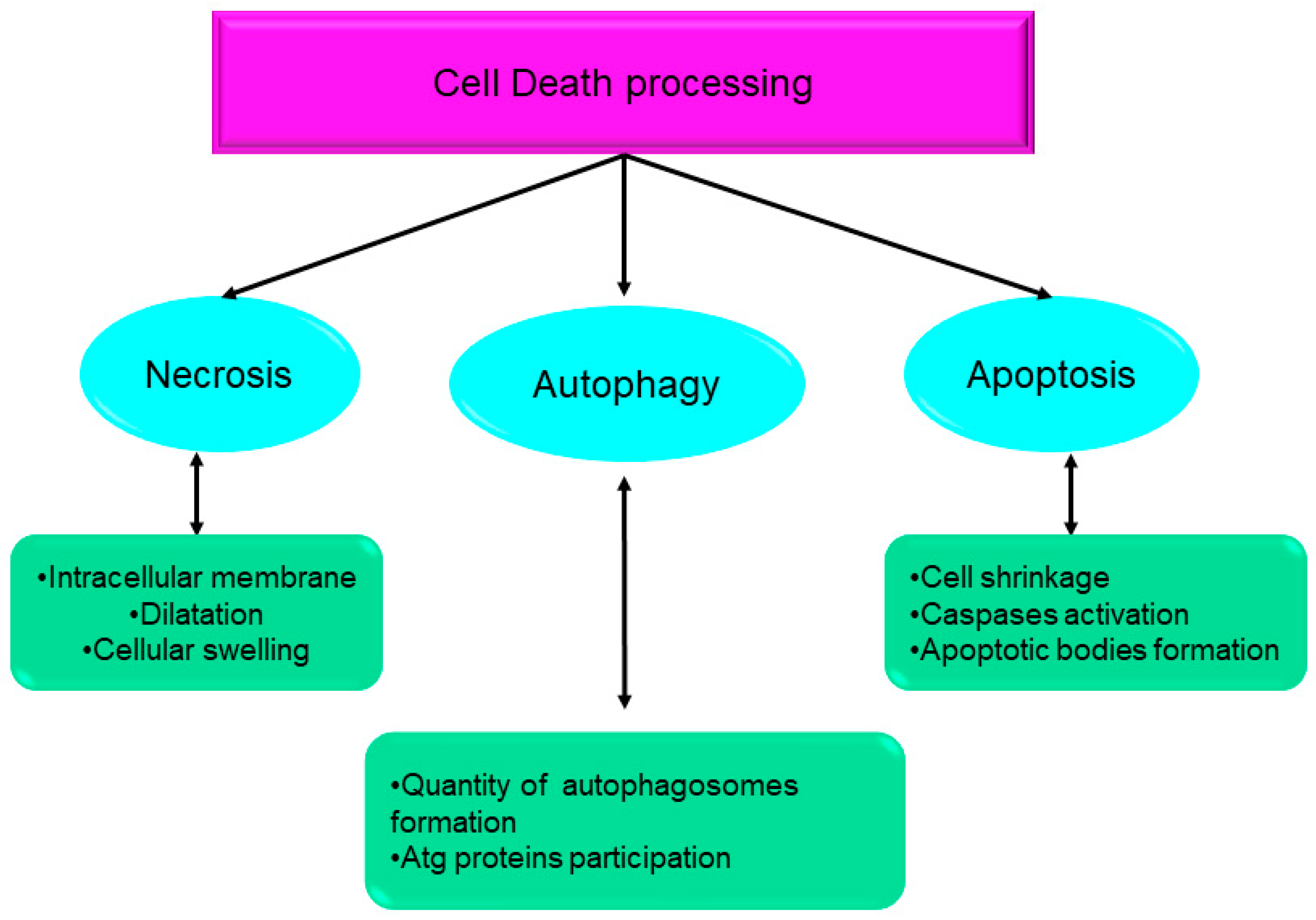
- Type I cell death (apoptosis);
- Type II cell death (autophagy);
- Type III cell death (necrosis).
2. Types of Cell Death
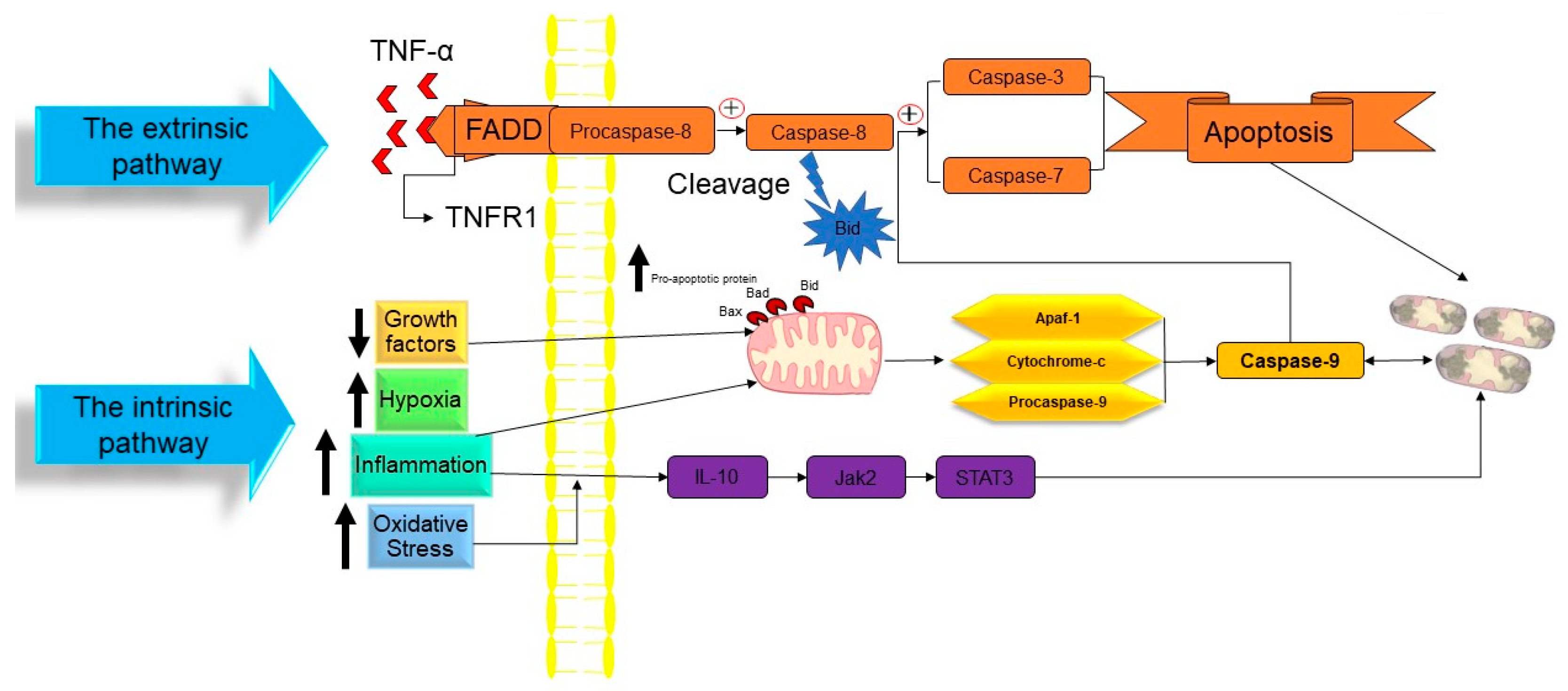
2.1. Apoptosis
- Cytoplasmic shrinkage;
- The irreversible condensation of chromatin in the nucleus (pyknosis);
- The destructive fragmentation of the nucleus (karyorrhexis);
- The formation of apoptotic bodies based on the establishment of intact small vesicles;
- The phagocytosis and decomposition of apoptotic bodies in neighboring cells’ lysosomes [34].
- The intrinsic pathway based on intracellular damage sensors’ detection.
- The extrinsic pathway based on immune cell and damaged cell attachment.
- The intrinsic pathway of apoptosis
- Extrinsic pathway of apoptosis
2.2. Anoikis
- A.
- Myeloid cell leukemia sequence 1, as well as the anti-apoptotic proteins Bcl-2 and B-cell lymphoma-extra large, Bcl-XL (Mcl-1).
- B.
- Pro-apoptotic proteins Bax, Bcl-2 homologous antagonist/killer (Bak), and Bcl-2 related ovarian killer (Bok), all with several domains.
- C.
- BH3 interacting domain death agonist (Bid), BCL2-associated agonist of cell death (Bad), Bcl-2 interacting mediator of cell death (Bim), BCL-2 interacting killer (Bik), BCL-2 modifying factor (Bmf), Noxa, Puma, and Harakiri (Hrk) are all pro-apoptotic BH3-only proteins [66].
- The intrinsic pathway of Anoikis
- The extrinsic pathway of Anoikis
2.3. Pyroptosis
2.4. NETosis: Neutrophil Extracellular Trap-Associated Cell Death
2.5. Ferroptosis: Iron-Dependent Cell Death
2.6. Autophagy
2.7. Entosis
2.8. Methuosis
- Class I is activated by Ras oncogenes, which induce vacuole formation by multiple sequential processes. The activation of Rac1 induces the process of macro-pinocytosis. Furthermore, the activated version of Rac1 interacts with G-protein-coupled receptor kinase-interacting protein 1 (GIT-1) to deactivate ADP-ribosylation factor 6 (Arf-6), thereby impeding the recycling of macro-pinosomes back to the plasma membrane. Consequently, the accumulating macro-pinosomes exhibit certain characteristics of late endosomes and subsequently merge together to form vacuoles [192].
- Vacuole development in class II methuosis inducer including mitogen-activated protein kinase kinase 4 (MKK-4), casein kinase 1 (CK1), nucleolin (Nuc), Arf6, GIT1, nerve growth factor (NGF), and early endosome antigen 1 (EEA1) [192].
2.9. Paraptosis
2.10. Mitoptosis
2.11. Parthanatos
2.12. Necroptosis
2.13. Necrosis
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Yan, G.E.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features. World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Srinivasan, M.; Clarke, R.; Kraikivski, P. Mathematical Models of Death Signaling Networks. Entropy 2022, 24, 1402. [Google Scholar] [CrossRef]
- Tyson, J.J.; Novak, B. A dynamical paradigm for molecular cell biology. Trends Cell Biol. 2020, 30, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Kraikivski, P.; Chen, K.C.; Laomettachit, T.; Murali, T.M.; Tyson, J.J. From START to FINISH: Computational analysis of cell cycle control in budding yeast. NPJ Syst. Biol. Appl. 2015, 1, 15016. [Google Scholar] [CrossRef] [PubMed]
- Shafiekhani, S.; Kraikivski, P.; Gheibi, N.; Ahmadian, M.; Jafari, A.H. Dynamical analysis of the fission yeast cell cycle via Markov chain. Curr. Genet. 2021, 67, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Jalihal, A.P.; Kraikivski, P.; Murali, T.M.; Tyson, J.J. Modeling and analysis of the macronutrient signaling network in budding yeast. Mol. Biol. Cell 2021, 32, ar20. [Google Scholar] [CrossRef]
- Jung, Y.; Kraikivski, P.; Shafiekhani, S.; Terhune, S.S.; Dash, R.K. Crosstalk between Plk1, p53, cell cycle, and G2/M DNA damage checkpoint regulation in cancer: Computational modeling and analysis. NPJ Syst. Biol. Appl. 2021, 7, 46. [Google Scholar] [CrossRef]
- Clarke, R.; Kraikivski, P.; Jones, B.C.; Sevigny, C.M.; Sengupta, S.; Wang, Y. A systems biology approach to discovering pathway signaling dysregulation in metastasis. Cancer Metastasis Rev. 2020, 39, 903–918. [Google Scholar] [CrossRef]
- Xu, F.; Yin, Z.; Zhu, L.; Jin, J.; He, Q.; Li, X.; Shuai, J. Oscillations governed by the incoherent dynamics in necroptotic signaling. Front. Phys. 2021, 9, 726638. [Google Scholar] [CrossRef]
- Zhu, L.; Li, X.; Xu, F.; Yin, Z.; Jin, J.; Liu, Z.; Qi, H.; Shuai, J. Network modeling-based identification of the switching targets between pyroptosis and secondary pyroptosis. Chaos Solitons Fractals 2022, 155, 111724. [Google Scholar] [CrossRef] [PubMed]
- Konstorum, A.; Tesfay, L.; Paul, B.T.; Torti, F.M.; Laubenbacher, R.C.; Torti, S.V. Systems biology of ferroptosis: A modeling approach. J. Theor. Biol. 2020, 493, 110222. [Google Scholar] [CrossRef]
- Checcoli, A.; Pol, J.G.; Naldi, A.; Noel, V.; Barillot, E.; Kroemer, G.; Thieffry, D.; Calzone, L.; Stoll, G. Dynamical Boolean Modeling of Immunogenic Cell Death. Front. Physiol. 2020, 11, 590479. [Google Scholar] [CrossRef]
- D’arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell. Mol. Life Sci. 2016, 73, 2405–2410. [Google Scholar] [CrossRef]
- Galluzzi, L.; López-Soto, A.; Kumar, S.; Kroemer, G. Caspases connect cell-death signaling to organismal homeostasis. Immunity 2016, 44, 221–231. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, B.-S.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.M.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kutscher, L.M.; Shaham, S. Non-apoptotic cell death in animal development. Cell Death Differ. 2017, 24, 1326–1336. [Google Scholar] [CrossRef]
- Cooper, J.P.; Youle, R.J. Balancing cell growth and death. Curr. Opin. Cell Biol. 2012, 24, 802. [Google Scholar] [CrossRef]
- Liao, M.; Qin, R.; Huang, W.; Zhu, H.-P.; Peng, F.; Han, B.; Liu, B. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: A revisited perspective from molecular mechanisms to targeted therapies. J. Hematol. Oncol. 2022, 15, 44. [Google Scholar] [CrossRef]
- Jellinger, K.A. Cell death mechanisms in neurodegeneration. J. Cell. Mol. Med. 2001, 5, 1–17. [Google Scholar] [CrossRef]
- Agid, Y.; Blin, J. Nerve cell death in degenerative diseases of the central nervous system: Clinical aspects. Ciba Found. Symp. 1987, 126, 3–29. [Google Scholar]
- Moore, M.J.; Wang, Q.; Kennedy, C.J.; Silver, P.A. An alternative splicing network links cell-cycle control to apoptosis. Cell 2010, 142, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Van Cruchten, S.; Van Den Broeck, W. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol. 2002, 31, 214–223. [Google Scholar] [CrossRef]
- Yadav, A.B.; Angadi, P.V.; Kale, A.D.; Yadav, S.K. Histological assessment of cellular changes in postmortem gingival specimens for estimation of time since death. J. Forensic Odonto-Stomatol. 2015, 33, 19. [Google Scholar]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. Clearance of apoptotic cells: Implications in health and disease. J. Cell Biol. 2010, 189, 1059–1070. [Google Scholar] [CrossRef]
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-only proteins noxa and puma are key regulators of induced apoptosis. Life 2022, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Song, C.-H. Effect of reactive oxygen species on the endoplasmic reticulum and mitochondria during intracellular pathogen infection of mammalian cells. Antioxidants 2021, 10, 872. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L. Apoptotic Cell Death in Disease—Current Understanding of the Nccd 2023. Cell Death Differ. 2023, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. Bcl-2 Family Proteins: Changing Partners in the Dance Towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Kanai, M.; Inoue-Yamauchi, A.; Tu, H.-C.; Huang, Y.; Ren, D.; Kim, H.; Takeda, S.; Reyna, D.E.; Chan, P.M. An Interconnected Hierarchical Model of Cell Death Regulation by the Bcl-2 Family. Nat. Cell Biol. 2015, 17, 1270–1281. [Google Scholar] [CrossRef]
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef]
- Cain, K.; Bratton, S.B.; Cohen, G.M. The Apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214. [Google Scholar] [CrossRef]
- Green, D.R. The mitochondrial pathway of apoptosis Part I: MOMP and beyond. Cold Spring Harb. Perspect. Biol. 2022, 14, a041038. [Google Scholar] [CrossRef]
- Ferraro, E.; Fuoco, C.; Strappazzon, F.; Cecconi, F. Apoptosome structure and regulation. In Apoptosome: Up-and-Coming Therapeutical Tool; Springer: Berlin/Heidelberg, Germany, 2010; pp. 27–39. [Google Scholar]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Ekert, P.G.; Vaux, D.L. The mitochondrial death squad: Hardened killers or innocent bystanders? Curr. Opin. Cell Biol. 2005, 17, 626–630. [Google Scholar] [CrossRef]
- Shliapina, V.L.; Yurtaeva, S.V.; Rubtsova, M.P.; Dontsova, O.A. At the crossroads: Mechanisms of apoptosis and autophagy in cell life and death. Acta Naturae 2021, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.R.; Harringto, W.J., Jr. Apoptosis: Programmed cell death at a molecular level. In Seminars in Arthritis and Rheumatism; WB Saunders: Philadelphia, PA, USA, 2003; pp. 345–369. [Google Scholar]
- Raducka-Jaszul, O.; Bogusławska, D.M.; Jędruchniewicz, N.; Sikorski, A.F. Role of extrinsic apoptotic signaling pathway during definitive erythropoiesis in normal patients and in patients with β-thalassemia. Int. J. Mol. Sci. 2020, 21, 3325. [Google Scholar] [CrossRef] [PubMed]
- Sessler, T.; Healy, S.; Samali, A.; Szegezdi, E. Structural determinants of DISC function: New insights into death receptor-mediated apoptosis signalling. Pharmacol. Ther. 2013, 140, 186–199. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef]
- Tan, C.-P.; Lu, Y.-Y.; Ji, L.-N.; Mao, Z.-W. Metallomics insights into the programmed cell death induced by metal-based anticancer compounds. Metallomics 2014, 6, 978–995. [Google Scholar] [CrossRef]
- Samraj, A.K.; Keil, E.; Ueffing, N.; Schulze-Osthoff, K.; Schmitz, I. Loss of caspase-9 provides genetic evidence for the type I/II concept of CD95-mediated apoptosis. J. Biol. Chem. 2006, 281, 29652–29659. [Google Scholar] [CrossRef]
- Engels, I.H.; Stepczynska, A.; Stroh, C.; Lauber, K.; Berg, C.; Schwenzer, R.; Wajant, H.; Jänicke, R.U.; Porter, A.G.; Belka, C. Caspase-8/FLICE functions as an executioner caspase in anticancer drug-induced apoptosis. Oncogene 2000, 19, 4563–4573. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 147–171. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Orrenius, S. Apoptosis: A basic biological phenomenon with wide-ranging implications in human disease. J. Intern. Med. 2005, 258, 479–517. [Google Scholar] [CrossRef]
- Long, J.S.; Ryan, K.M. New frontiers in promoting tumour cell death: Targeting apoptosis, necroptosis and autophagy. Oncogene 2012, 31, 5045–5060. [Google Scholar] [CrossRef]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed]
- Reddig, P.J.; Juliano, R.L. Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005, 24, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. Complexity and specificity of integrin signalling. Nat. Cell Biol. 2000, 2, E13–E14. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364. [Google Scholar] [CrossRef]
- Frisch, S.M.; Screaton, R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001, 13, 555–562. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [CrossRef]
- Li, S.; Chen, Y.; Zhang, Y.; Jiang, X.; Jiang, Y.; Qin, X.; Yang, H.; Wu, C.; Liu, Y. Shear stress promotes anoikis resistance of cancer cells via caveolin-1-dependent extrinsic and intrinsic apoptotic pathways. J. Cell. Physiol. 2019, 234, 3730–3743. [Google Scholar] [CrossRef]
- Grossmann, J. Molecular mechanisms of “detachment-induced apoptosis—Anoikis”. Apoptosis 2002, 7, 247–260. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Bras, M.; Queenan, B.; Susin, S.A. Programmed cell death via mitochondria: Different modes of dying. Biochemistry 2005, 70, 231–239. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Puthalakath, H.; Villunger, A.; O’Reilly, L.A.; Beaumont, J.G.; Coultas, L.; Cheney, R.E.; Huang, D.C.S.; Strasser, A. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 2001, 293, 1829–1832. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Z.; Ouyang, Z.; Ren, Y.; Cheng, Y.; Liu, P.; Wen, Y.; Shao, Y. Non-canonical phosphorylation of Bmf by p38 MAPK promotes its apoptotic activity in anoikis. Cell Death Differ. 2022, 29, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.-J.; Wildey, G.M.; Howe, P.H. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J. Biol. Chem. 2006, 281, 813–823. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef]
- McConkey, D.J.; Bondar, V. Regulation and function of detachment-induced cell death (Anoikis) in cancer progression and metastasis. Apoptosis Senescence Cancer 2007, 109–122. [Google Scholar] [CrossRef]
- Bunek, J.; Kamarajan, P.; Kapila, Y.L. Anoikis mediators in oral squamous cell carcinoma. Oral Dis. 2011, 17, 355–361. [Google Scholar] [CrossRef]
- Rosen, K.; Shi, W.; Calabretta, B.; Filmus, J. Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of fas ligand: A novel mechanism of anoikis of intestinal epithelial cells. J. Biol. Chem. 2002, 277, 46123–46130. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Mühlethaler-Mottet, A.; Bourloud, K.B.; Auderset, K.; Joseph, J.-M.; Gross, N. Drug-mediated sensitization to TRAIL-induced apoptosis in caspase-8-complemented neuroblastoma cells proceeds via activation of intrinsic and extrinsic pathways and caspase-dependent cleavage of XIAP, Bcl-xL and RIP. Oncogene 2004, 23, 5415–5425. [Google Scholar] [CrossRef][Green Version]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Aoudjit, F.; Vuori, K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: A role for c-flip and implications for anoikis. J. Cell Biol. 2001, 152, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Alahari, S.K.; Reddig, P.J.; Juliano, R.L. Biological aspects of signal transduction by cell adhesion receptors. Int. Rev. Cytol. 2002, 220, 145–184. [Google Scholar]
- Shim, S.R.; Kook, S.; Kim, J.I.; Song, W.K. Degradation of focal adhesion proteins paxillin and p130cas by caspases or calpains in apoptotic rat-1 and L929 cells. Biochem. Biophys. Res. Commun. 2001, 286, 601–608. [Google Scholar] [CrossRef]
- Kim, W.; Kook, S.; Kim, D.J.; Teodorof, C.; Song, W.K. The 31-kDa caspase-generated cleavage product of p130cas functions as a transcriptional repressor of E2A in apoptotic cells. J. Biol. Chem. 2004, 279, 8333–8342. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Labbe, K.; Saleh, M. Cell death in the host response to infection. Cell Death Differ. 2008, 15, 1339–1349. [Google Scholar] [CrossRef]
- Fink, S.L.; Bergsbaken, T.; Cookson, B.T. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 4312–4317. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Narula, J.; Burke, A.P.; Haider, N.; Farb, A.; Hui-Liong, Y.; Smialek, J.; Virmani, R. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am. J. Pathol. 2000, 157, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Khalid, N.; Azimpouran, M. Necrosis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Wang, L.; Du, F.; Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Feltham, R.; Yabal, M.; Conos, S.A.; Chen, K.W.; Ziehe, S.; Graß, C.; Zhan, Y.; Nguyen, T.A.; Hall, C. XIAP loss triggers RIPK3-and caspase-8-driven IL-1β activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 2017, 20, 668–682. [Google Scholar] [CrossRef]
- Demarco, B.; Grayczyk, J.P.; Bjanes, E.; Le Roy, D.; Tonnus, W.; Assenmacher, C.-A.; Radaelli, E.; Fettrelet, T.; Mack, V.; Linkermann, A. Caspase-8–dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 2020, 6, eabc3465. [Google Scholar] [CrossRef] [PubMed]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; De Nardo, D.; Gao, W.; Vince, A.J.; Hall, C.; McArthur, K.; Simpson, D.; Vijayaraj, S.; Lindqvist, L.M.; Bouillet, P. The mitochondrial apoptotic effectors BAX/BAK activate caspase-3 and-7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1β activation. Cell Rep. 2018, 25, 2339–2353. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Thi Nguyen, D.; Hattori, T.; Manh Le, T.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef]
- Cui, J.; Zhou, Z.; Yang, H.; Jiao, F.; Li, N.; Gao, Y.; Wang, L.; Chen, J.; Quan, M. MST1 suppresses pancreatic cancer progression via ROS-induced pyroptosis. Mol. Cancer Res. 2019, 17, 1316–1325. [Google Scholar] [CrossRef]
- Sreevalsan, S.; Safe, S. Reactive oxygen species and colorectal cancer. Curr. Color. Cancer Rep. 2013, 9, 350–357. [Google Scholar] [CrossRef]
- Hajibabaie, F.; Abedpoor, N.; Javanmard, S.H.; Hasan, A.; Sharifi, M.; Rahimmanesh, I.; Shariati, L.; Makvandi, P. The molecular perspective on the development of melanoma and genome engineering of T-cells in targeting therapy. Environ. Res. 2023, 237, 116980. [Google Scholar] [CrossRef] [PubMed]
- Larocque, K.; Ovadje, P.; Djurdjevic, S.; Mehdi, M.; Green, J.; Pandey, S. Novel analogue of colchicine induces selective pro-death autophagy and necrosis in human cancer cells. PLoS ONE 2014, 9, e87064. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.D.; Kang, K.A.; Kim, H.S.; Kim, D.H.; Choi, Y.H.; Lee, S.J.; Hyun, J.W. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013, 4, e750. [Google Scholar] [CrossRef]
- Kim, J.Y.; Yu, S.-J.; Oh, H.J.; Lee, J.Y.; Kim, Y.; Sohn, J. Panaxydol induces apoptosis through an increased intracellular calcium level, activation of JNK and p38 MAPK and NADPH oxidase-dependent generation of reactive oxygen species. Apoptosis 2011, 16, 347–358. [Google Scholar] [CrossRef]
- Hsieh, C.-J.; Kuo, P.-L.; Hsu, Y.-C.; Huang, Y.-F.; Tsai, E.-M.; Hsu, Y.-L. Arctigenin, a dietary phytoestrogen, induces apoptosis of estrogen receptor-negative breast cancer cells through the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic. Biol. Med. 2014, 67, 159–170. [Google Scholar] [CrossRef]
- Sun, W.; Zeng, C.; Liu, S.; Fu, J.; Hu, L.; Shi, Z.; Yue, D.; Ren, Z.; Zhong, Z.; Zuo, Z. Ageratina adenophora induces mice hepatotoxicity via ROS-NLRP3-mediated pyroptosis. Sci. Rep. 2018, 8, 16032. [Google Scholar] [CrossRef]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.-F.; Mei, Q.-B.; Zhou, X.-G.; Tang, Y.; Xiong, R.; Qiu, W.-Q.; Pan, R.; Law, B.Y.-K.; Wong, V.K.-W.; Yu, C.-L. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers 2020, 12, 193. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef]
- Remijsen, Q.; Kuijpers, T.W.; Wirawan, E.; Lippens, S.; Vandenabeele, P.; Vanden Berghe, T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011, 18, 581–588. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef]
- Csomós, K.; Kristóf, E.; Jakob, B.; Csomós, I.; Kovács, G.; Rotem, O.; Hodrea, J.; Bagoly, Z.; Muszbek, L.; Balajthy, Z. Protein cross-linking by chlorinated polyamines and transglutamylation stabilizes neutrophil extracellular traps. Cell Death Dis. 2016, 7, e2332. [Google Scholar] [CrossRef]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Berghe, T.V.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Chauhan, D.; Demon, D.; Vande Walle, L.; Paerewijck, O.; Zecchin, A.; Bosseler, L.; Santoni, K.; Planès, R.; Ribo, S.; Fossoul, A. GSDMD drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for NETosis. EMBO Rep. 2022, 23, e54277. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef]
- Sánchez, M.; Sabio, L.; Gálvez, N.; Capdevila, M.; Dominguez-Vera, J.M. Iron chemistry at the service of life. IUBMB Life 2017, 69, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Forciniti, S.; Greco, L.; Grizzi, F.; Malesci, A.; Laghi, L. Iron metabolism in cancer progression. Int. J. Mol. Sci. 2020, 21, 2257. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Foret, M.K.; Lincoln, R.; Do Carmo, S.; Cuello, A.C.; Cosa, G. Connecting the “Dots”: From free radical lipid autoxidation to cell pathology and disease. Chem. Rev. 2020, 120, 12757–12787. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta BBA-Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Chen, S.; Chen, Y.; Zhang, Y.; Kuang, X.; Liu, Y.; Guo, M.; Ma, L.; Zhang, D.; Li, Q. Iron metabolism and ferroptosis in epilepsy. Front. Neurosci. 2020, 14, 601193. [Google Scholar] [CrossRef]
- Zia, Q.; Azhar, A.; Hassan, N.; Jain, P.; Singh, M.; Mirza, M.A.; Ali, A.; Parveen, S.; Hasan, S.; Alothaim, A.S. Cell Death: A Molecular Perspective. Curr. Mol. Biol. Rep. 2021, 1–26. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and cancer: Mitochondria meet the “iron maiden” cell death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Zhou, R.-P.; Chen, Y.; Wei, X.; Yu, B.; Xiong, Z.-G.; Lu, C.; Hu, W. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics 2020, 10, 11976. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X. The chemistry and biology of ferroptosis. Cell Chem. Biol. 2020, 27, 365–375. [Google Scholar] [CrossRef]
- Hadian, K. Ferroptosis suppressor protein 1 (FSP1) and coenzyme Q10 cooperatively suppress ferroptosis. Biochemistry 2020, 59, 637–638. [Google Scholar] [CrossRef]
- Yan, H.-f.; Zou, T.; Tuo, Q.-z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Badadani, M. Autophagy mechanism, regulation, functions, and disorders. ISRN Cell Biol. 2012, 2012, 927064. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Autophagy: Dual roles in life and death? Nat. Rev. Mol. Cell Biol. 2005, 6, 505–510. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Zhu, L. Chaperone-mediated autophagy. Autophagy: Biol. Dis. Basic Sci. 2019, 435–452. [Google Scholar] [CrossRef]
- Andrade-Tomaz, M.; de Souza, I.; Ribeiro Reily Rocha, C.; Rodrigues Gomes, L. The role of chaperone-mediated autophagy in cell cycle control and its implications in cancer. Cells 2020, 9, 2140. [Google Scholar] [CrossRef]
- Suzuki, K.; Ohsumi, Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007, 581, 2156–2161. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467. [Google Scholar] [CrossRef]
- Miracco, C.; Cevenini, G.; Franchi, A.; Luzi, P.; Cosci, E.; Mourmouras, V.; Monciatti, I.; Mannucci, S.; Biagioli, M.; Toscano, M. Beclin 1 and LC3 autophagic gene expression in cutaneous melanocytic lesions. Hum. Pathol. 2010, 41, 503–512. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ohbayashi, S.; Sakoh-Nakatogawa, M.; Kakuta, S.; Suzuki, S.W.; Kirisako, H.; Kondo-Kakuta, C.; Noda, N.N.; Yamamoto, H.; Ohsumi, Y. The autophagy-related protein kinase Atg1 interacts with the ubiquitin-like protein Atg8 via the Atg8 family interacting motif to facilitate autophagosome formation. J. Biol. Chem. 2012, 287, 28503–28507. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Bo Otto, F.; Thumm, M. Nucleophagy—Implications for microautophagy and health. Int. J. Mol. Sci. 2020, 21, 4506. [Google Scholar] [CrossRef]
- Muciño-Hernández, G.; Acevo-Rodríguez, P.S.; Cabrera-Benitez, S.; Guerrero, A.O.; Merchant-Larios, H.; Castro-Obregón, S. Nucleophagy contributes to genome stability through degradation of type II topoisomerases A and B and nucleolar components. J. Cell Sci. 2023, 136, jcs260563. [Google Scholar] [CrossRef]
- Papandreou, M.-E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ivanov, A.; Adams, P.D.; Berger, S.L. Mammalian autophagy degrades nuclear constituents in response to tumorigenic stress. Autophagy 2016, 12, 1416–1417. [Google Scholar] [CrossRef] [PubMed]
- Arifur, M.R.; Golam, M.M.; Ushimaru, T. The Nem1/Spo7–Pah1/lipin axis is required for autophagy induction after TORC1 inactivation. FEBS J. 2018, 285, 1840–1860. [Google Scholar]
- Schäfer, J.A.; Schessner, J.P.; Bircham, P.W.; Tsuji, T.; Funaya, C.; Pajonk, O.; Schaeff, K.; Ruffini, G.; Papagiannidis, D.; Knop, M. ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J. 2020, 39, e102586. [Google Scholar] [CrossRef]
- Otto, F.B.; Thumm, M. Mechanistic dissection of macro-and micronucleophagy. Autophagy 2021, 17, 626–639. [Google Scholar] [CrossRef]
- Krick, R.; Muehe, Y.; Prick, T.; Bremer, S.; Schlotterhose, P.; Eskelinen, E.-L.; Millen, J.; Goldfarb, D.S.; Thumm, M. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol. Biol. Cell 2008, 19, 4492–4505. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef]
- Millen, J.I.; Krick, R.; Prick, T.; Thumm, M.; Goldfarb, D.S. Measuring piecemeal microautophagy of the nucleus in Saccharomyces cerevisiae. Autophagy 2009, 5, 75–81. [Google Scholar] [CrossRef]
- Dubots, E.; Cottier, S.; Péli-Gulli, M.-P.; Jaquenoud, M.; Bontron, S.; Schneiter, R.; De Virgilio, C. TORC1 regulates Pah1 phosphatidate phosphatase activity via the Nem1/Spo7 protein phosphatase complex. PLoS ONE 2014, 9, e104194. [Google Scholar] [CrossRef]
- Siniossoglou, S.; Santos-Rosa, H.; Rappsilber, J.; Mann, M.; Hurt, E. A novel complex of membrane proteins required for formation of a spherical nucleus. EMBO J. 1998, 17, 6449–6464. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Fujimoto, M.; Tatematsu, T.; Cheng, J.; Orii, M.; Takatori, S.; Fujimoto, T. Niemann-Pick type C proteins promote microautophagy by expanding raft-like membrane domains in the yeast vacuole. Elife 2017, 6, e25960. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, X.; Zhang, Y.; Gao, Z.; Liu, Y.; Hu, J.; Hu, X.; Li, L.; Shi, J.; Gao, N. Nuclear accumulation of UBC9 contributes to SUMOylation of lamin A/C and nucleophagy in response to DNA damage. J. Exp. Clin. Cancer Res. 2019, 38, 67. [Google Scholar] [CrossRef]
- Kim, Y.; Gentry, M.S.; Harris, T.E.; Wiley, S.E.; Lawrence, J.C., Jr.; Dixon, J.E. A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6596–6601. [Google Scholar] [CrossRef]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Overholtzer, M. Mechanisms and consequences of entosis. Cell. Mol. Life Sci. 2016, 73, 2379–2386. [Google Scholar] [CrossRef] [PubMed]
- White, E. Entosis: It’s a cell-eat-cell world. Cell 2007, 131, 840–842. [Google Scholar] [CrossRef]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007, 131, 966–979. [Google Scholar] [CrossRef]
- Sun, Q.; Cibas, E.S.; Huang, H.; Hodgson, L.; Overholtzer, M. Induction of entosis by epithelial cadherin expression. Cell Res. 2014, 24, 1288–1298. [Google Scholar] [CrossRef]
- Wan, Q.; Liu, J.; Zheng, Z.; Zhu, H.; Chu, X.; Dong, Z.; Huang, S.; Du, Q. Regulation of myosin activation during cell–cell contact formation by Par3-Lgl antagonism: Entosis without matrix detachment. Mol. Biol. Cell 2012, 23, 2076–2091. [Google Scholar] [CrossRef]
- Hamann, J.C.; Surcel, A.; Chen, R.; Teragawa, C.; Albeck, J.G.; Robinson, D.N.; Overholtzer, M. Entosis is induced by glucose starvation. Cell Rep. 2017, 20, 201–210. [Google Scholar] [CrossRef]
- Huang, R.; Liu, W. Identifying an essential role of nuclear LC3 for autophagy. Autophagy 2015, 11, 852–853. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2012, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, W.A. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [Google Scholar] [CrossRef]
- Kaul, A.; Overmeyer, J.H.; Maltese, W.A. Activated Ras induces cytoplasmic vacuolation and non-apoptotic death in glioblastoma cells via novel effector pathways. Cell. Signal. 2007, 19, 1034–1043. [Google Scholar] [CrossRef][Green Version]
- Bhanot, H.; Young, A.M.; Overmeyer, J.H.; Maltese, W.A. Induction of Nonapoptotic Cell Death by Activated Ras Requires Inverse Regulation of Rac1 and Arf6Roles of Rac1 and Arf6 in Ras-Induced Cell Death. Mol. Cancer Res. 2010, 8, 1358–1374. [Google Scholar] [CrossRef]
- Overmeyer, J.H.; Young, A.M.; Bhanot, H.; Maltese, W.A. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 2011, 10, 1–17. [Google Scholar] [CrossRef]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic vacuolization in cell death and survival. Oncotarget 2016, 7, 55863. [Google Scholar] [CrossRef]
- Kim, E.; Lee, D.M.; Seo, M.J.; Lee, H.J.; Choi, K.S. Intracellular Ca2+ imbalance critically contributes to paraptosis. Front. Cell Dev. Biol. 2021, 8, 607844. [Google Scholar] [CrossRef]
- Yokoi, K.; Balachandran, C.; Umezawa, M.; Tsuchiya, K.; Mitrić, A.; Aoki, S. Amphiphilic Cationic Triscyclometalated Iridium (III) Complex–Peptide Hybrids Induce Paraptosis-like Cell Death of Cancer Cells via an Intracellular Ca2+-Dependent Pathway. ACS Omega 2020, 5, 6983–7001. [Google Scholar] [CrossRef] [PubMed]
- Jangamreddy, J.R.; Los, M.J. Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat. Mon. 2012, 12, e6159. [Google Scholar] [CrossRef]
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Nepryakhina, O.K.; Saprunova, V.B.; Bakeeva, L.E.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): Formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta BBA-Bioenerg. 2008, 1777, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005, 15, 2112–2118. [Google Scholar] [CrossRef]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 2005, 14, R283–R289. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Ren, L.; Chen, X.; Chen, X.; Li, J.; Cheng, B.; Xia, J. Mitochondrial dynamics: Fission and fusion in fate determination of mesenchymal stem cells. Front. Cell Dev. Biol. 2020, 8, 580070. [Google Scholar] [CrossRef]
- Yu, R.; Liu, T.; Ning, C.; Tan, F.; Jin, S.-B.; Lendahl, U.; Zhao, J.; Nistér, M. The phosphorylation status of Ser-637 in dynamin-related protein 1 (Drp1) does not determine Drp1 recruitment to mitochondria. J. Biol. Chem. 2019, 294, 17262–17277. [Google Scholar] [CrossRef]
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014, 33, 418–436. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef]
- Prieto, J.; León, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; López-García, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. N. Y. Acad. Sci. 2008, 1147, 233–241. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.-W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C. Poly (ADP-ribose)(PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [PubMed]
- Teloni, F.; Altmeyer, M. Readers of poly (ADP-ribose): Designed to be fit for purpose. Nucleic Acids Res. 2015, 44, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Chang, P. Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat. Chem. Biol. 2018, 14, 236–243. [Google Scholar] [CrossRef]
- Wang, R.; Li, C.; Qiao, P.; Xue, Y.; Zheng, X.; Chen, H.; Zeng, X.; Liu, W.; Boldogh, I.; Ba, X. OGG1-initiated base excision repair exacerbates oxidative stress-induced parthanatos. Cell Death Dis. 2018, 9, 628. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Ke, Y.; Zeng, X.; Gao, J.; Ba, X.; Wang, R. The key players of parthanatos: Opportunities for targeting multiple levels in the therapy of parthanatos-based pathogenesis. Cell. Mol. Life Sci. 2022, 79, 60. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.-H.; Li, T.; Ding, W.-X.; Jaeschke, H. Receptor-interacting serine/threonine-protein kinase 3 (RIPK3)–mixed lineage kinase domain-like protein (MLKL)–mediated necroptosis contributes to ischemia-reperfusion injury of steatotic livers. Am. J. Pathol. 2019, 189, 1363–1374. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol. 2017, 27, 800–809. [Google Scholar] [CrossRef]
- Méry, B.; Guy, J.-B.; Vallard, A.; Espenel, S.; Ardail, D.; Rodriguez-Lafrasse, C.; Rancoule, C.; Magné, N. In vitro cell death determination for drug discovery: A landscape review of real issues. J. Cell Death 2017, 10, 1179670717691251. [Google Scholar] [CrossRef]
- Berghe, T.V.; Grootjans, S.; Goossens, V.; Dondelinger, Y.; Krysko, D.V.; Takahashi, N.; Vandenabeele, P. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 2013, 61, 117–129. [Google Scholar] [CrossRef]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef]
- Seehawer, M.; Heinzmann, F.; D’artista, L.; Harbig, J.; Roux, P.-F.; Hoenicke, L.; Dang, H.; Klotz, S.; Robinson, L.; Doré, G. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018, 562, 69–75. [Google Scholar] [CrossRef]
- Krysko, D.V.; Berghe, T.V.; Parthoens, E.; D’Herde, K.; Vandenabeele, P. Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol. 2008, 442, 307–341. [Google Scholar]
- Clarke, P.G.H. Developmental cell death: Morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Sandle, T.; Chesca, A.; Akhayeva, A.S.; Marchenko, A.B. Apoptosis versus necrosis. SFJ Chronic Dis. 2018, 1, 1–4. [Google Scholar]
- Nusratillo o’g’li, X.B.; Akmal o’g’li, E.D.; Ahmad o’g’li, A.A. The Concept of Necrosis, Causes, Consequences and Types of Origin. Etiology of Prevention and Treatment. Ta’lim Va Rivojlanish Tahlili Onlayn Ilmiy Jurnali 2023, 3, 329–331. [Google Scholar]
- Gramaglia, D.; Gentile, A.; Battaglia, M.; Ranzato, L.; Petronilli, V.; Fassetta, M.; Bernardi, P.; Rasola, A. Apoptosis to necrosis switching downstream of apoptosome formation requires inhibition of both glycolysis and oxidative phosphorylation in a BCL-XL-and PKB/AKT-independent fashion. Cell Death Differ. 2004, 11, 342–353. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wallig, M.A.; Janovitz, E.B. Morphologic manifestations of toxic cell Injury. In Haschek and Rousseaux’s Handbook of Toxicologic Pathology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 113–148. [Google Scholar]
- Nicotera, P.; Melino, G. Regulation of the apoptosis–necrosis switch. Oncogene 2004, 23, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anoikis | Apoptosis |
|---|---|
| Programmed cell death, occurring in cells, separated from the extracellular matrix. Induced upon the separation of a cell from the extracellular matrix. Prevents the adherent-independent cell growth and the attachment of cells to an improper matrix, thus preventing the colonization of distant organs. | Programmed cell death, occurring in cells that are redundant, functionally incomplete, or dangerous for an organism. Induced when a cell becomes redundant, functionally incomplete, or dangerous for an organism. Mostly removes useful cells during the fethal/larval development and also the potentially harmful cells. |
| Pyroptosis | Apoptosis | |
|---|---|---|
| Morphological Changes |
|
|
| Molecular Mechanism |
|
|
| Regulation | The apoptotic cell death occurs when the ubiquitination of RIPK1 is inhibited, leading to the formation of a complex between RIPK1, FADD, and pro-caspase-8. This complex activates caspase-8, which then cleaves RIPK1, ultimately resulting in the apoptotic cell death. | NLRP3 and other proteins are regulated by post-translational phosphorylation and ubiquitylation modifications. |
| Mitochondrial Participation | The release of Cytochrome-C from mitochondria and the subsequent generation of apoptotic bodies. | Mitochondria are engaged in the control of gasdermin D oligomerization and the consequent development of pores in the plasma membrane. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajibabaie, F.; Abedpoor, N.; Mohamadynejad, P. Types of Cell Death from a Molecular Perspective. Biology 2023, 12, 1426. https://doi.org/10.3390/biology12111426
Hajibabaie F, Abedpoor N, Mohamadynejad P. Types of Cell Death from a Molecular Perspective. Biology. 2023; 12(11):1426. https://doi.org/10.3390/biology12111426
Chicago/Turabian StyleHajibabaie, Fatemeh, Navid Abedpoor, and Parisa Mohamadynejad. 2023. "Types of Cell Death from a Molecular Perspective" Biology 12, no. 11: 1426. https://doi.org/10.3390/biology12111426
APA StyleHajibabaie, F., Abedpoor, N., & Mohamadynejad, P. (2023). Types of Cell Death from a Molecular Perspective. Biology, 12(11), 1426. https://doi.org/10.3390/biology12111426