
Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
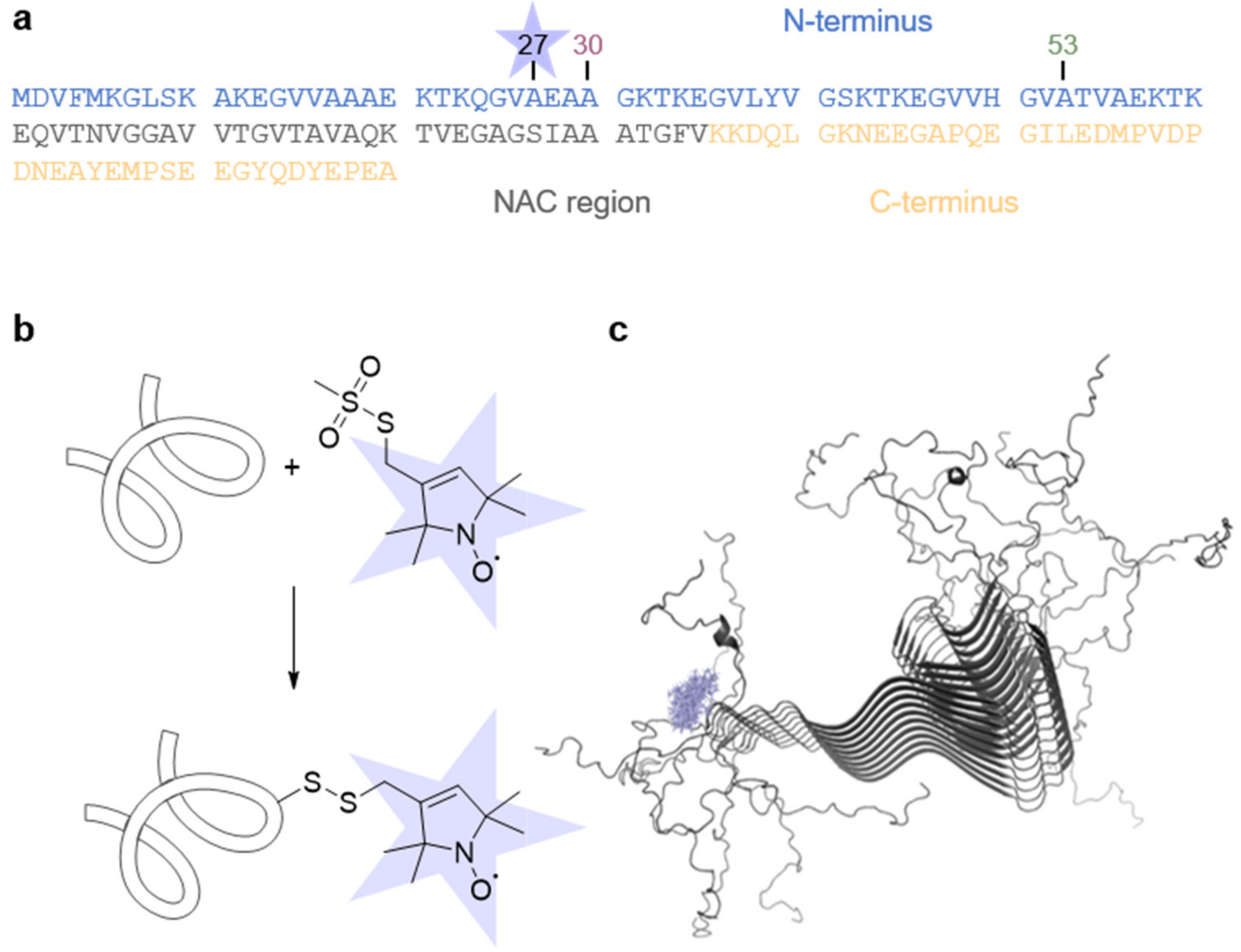
2.1. Protein Production and Spin Labeling
2.2. Aggregation Assay
2.3. Rapid Scan EPR Spectroscopy
3. Results
3.1. Spin-Labeled α-Synuclein Variants Aggregate in the Presence of Ethanol
3.2. Spin-Dilution Is Not a Prerequisite in Our Experimental Setup
3.3. Circular Dichroism Reveals the Global Aggregation Process of α-Synuclein
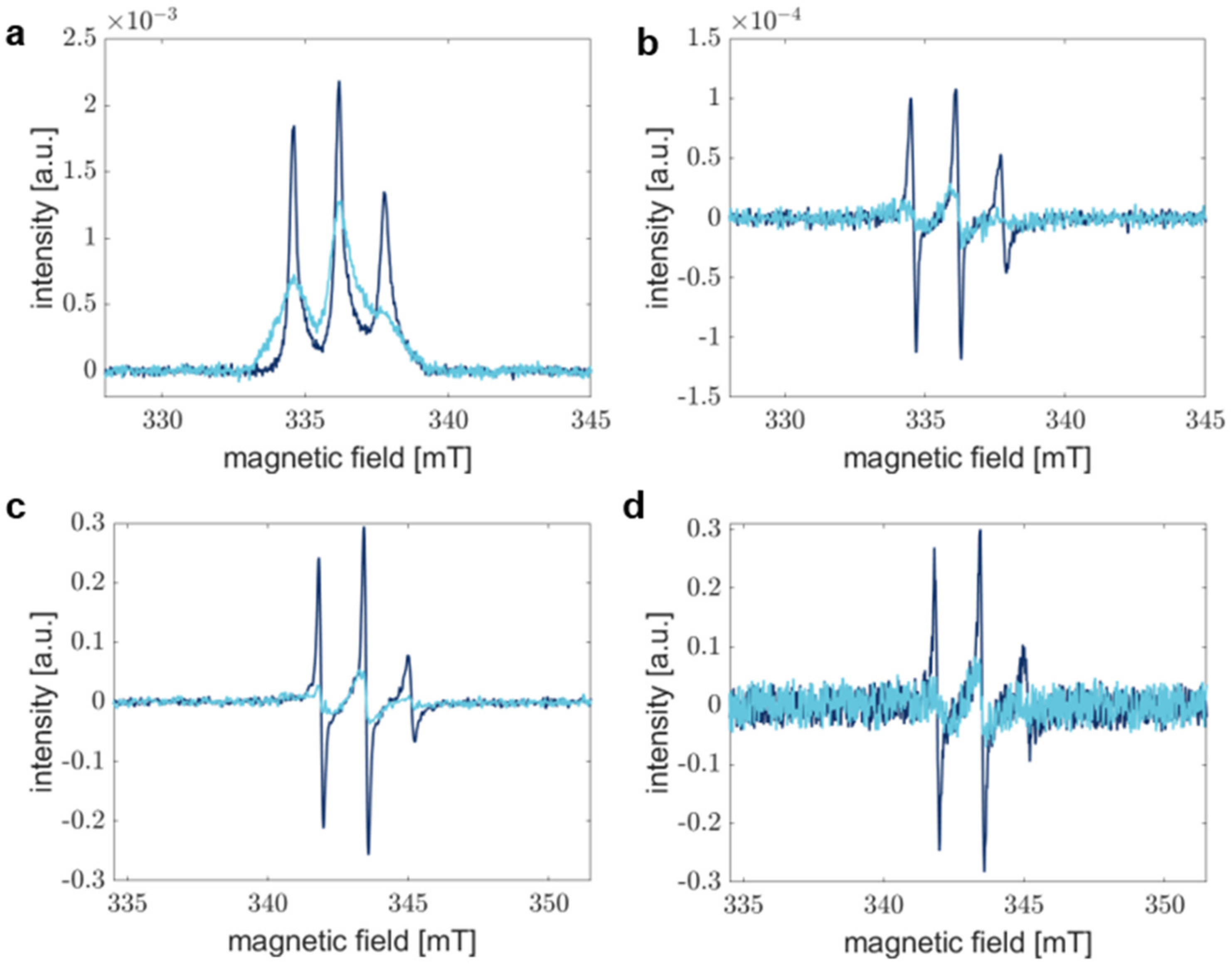
3.4. The Signal-to-Noise Ratio of Rapid Scan EPR Spectroscopy Outperforms Conventional Continuous Wave EPR Spectroscopy
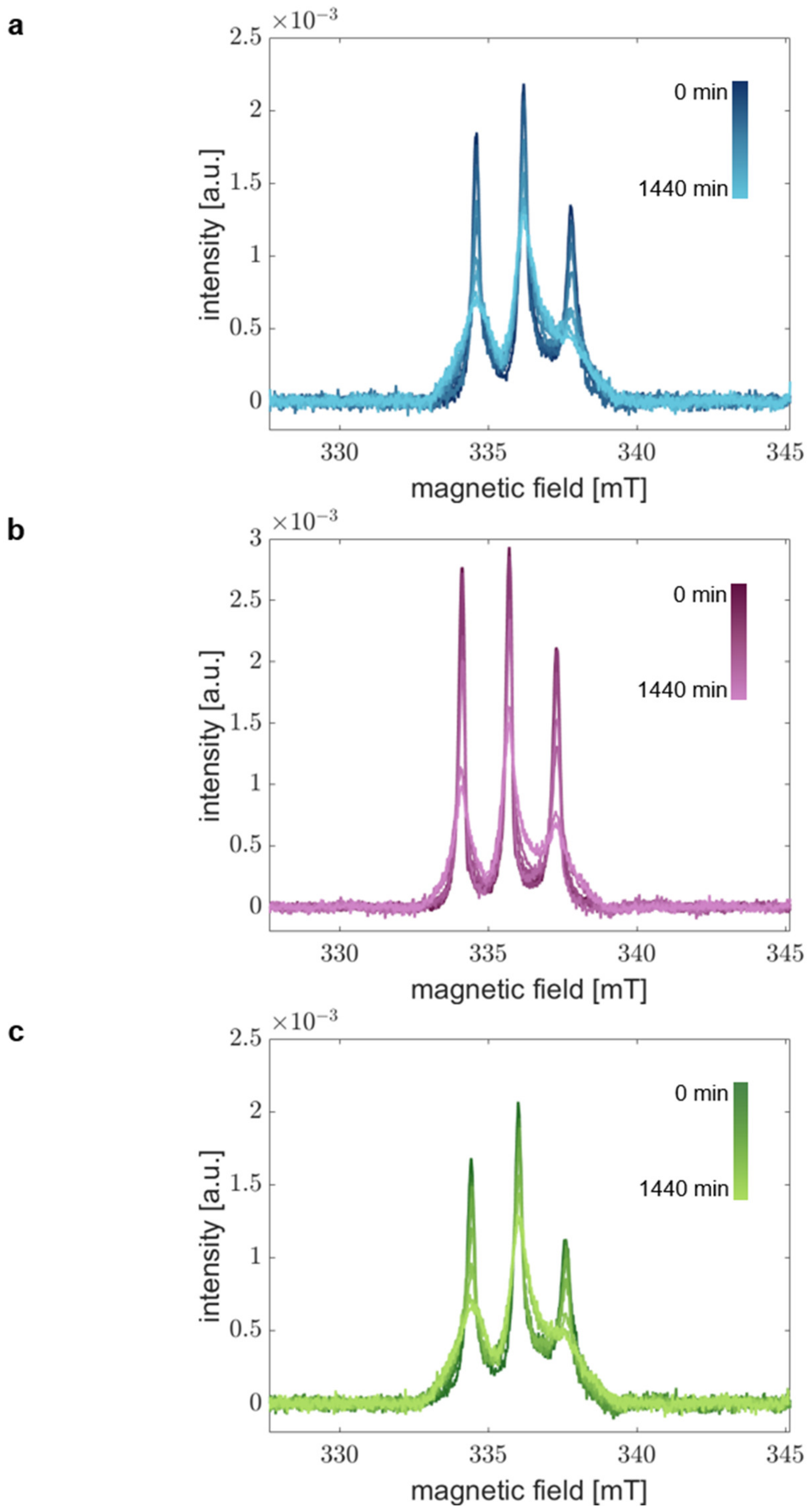
3.5. Rapid Scan Experiments Capture the Local Kinetics of α-Synuclein Aggregation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braun, P.; Gingras, A.C. History of Protein-Protein Interactions: From Egg-White to Complex Networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signaling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Coupling of Folding and Binding for Unstructured Proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of Alpha-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-Bound Human α-Synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- Li, X.; Dong, C.; Hoffmann, M.; Garen, C.R.; Cortez, L.M.; Petersen, N.O.; Woodside, M.T. Early Stages of Aggregation of Engineered α-Synuclein Monomers and Oligomers in Solution. Sci. Rep. 2019, 9, 1734. [Google Scholar] [CrossRef]
- Villar-Piqué, A.; Lopes da Fonseca, T.; Outeiro, T.F. Structure, Function and Toxicity of Alpha-Synuclein: The Bermuda Triangle in Synucleinopathies. J. Neurochem. 2016, 139, 240–255. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural Characterization of Toxic Oligomers That Are Kinetically Trapped during α-Synuclein Fibril Formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro Mutation in the Gene Encoding α-Synuclein in Parkinson’s Disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the Alpha-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in Vitro Fibril Formation by a Mutant α-Synuclein Linked to Early-Onset Parkinson Disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both α-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Lemkau, L.R.; Comellas, G.; Kloepper, K.D.; Woods, W.S.; George, J.M.; Rienstra, C.M. Mutant Protein A30P α-Synuclein Adopts Wild-Type Fibril Structure, despite Slower Fibrillation Kinetics. J. Biol. Chem. 2012, 287, P11526–P11532. [Google Scholar] [CrossRef]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both Familial Parkinson’s Disease Mutations Accelerate Alpha-Synuclein Aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of Familial Parkinson’s Disease Point Mutations A30P and A53T on the Structural Properties, Aggregation, and Fibrillation of Human α-Synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of A-Synuclein Aggregate Morphology on Solution Conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations Associated with Familial Parkinson’s Disease Alter the Initiation and Amplification Steps of α-Synuclein Aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef]
- Fallah, M.A.; Gerding, H.R.; Scheibe, C.; Drescher, M.; Karreman, C.; Schildknecht, S.; Leist, M.; Hauser, K. Simultaneous IR-Spectroscopic Observation of α-Synuclein, Lipids, and Solvent Reveals an Alternative Membrane-Induced Oligomerization Pathway. ChemBioChem 2017, 18, 2312–2316. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Karmakar, S.; Bose, A.; Chowdhury, P.K. β-Cyclodextrin and Curcumin, a Potent Cocktail for Disaggregating and/or Inhibiting Amyloids: A Case Study with α-Synuclein. Biochemistry 2014, 53, 4081–4083. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Karmakar, S.; Batra, R.; Sharma, P.; Pradhan, P.; Singh, J.; Kundu, B.; Chowdhury, P.K. Polyphenols in Combination with β-Cyclodextrin Can Inhibit and Disaggregate α-Synuclein Amyloids under Cell Mimicking Conditions: A Promising Therapeutic Alternative. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational Behavior and Aggregation of α-Synuclein in Organic Solvents: Modeling the Effects of Membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef] [PubMed]
- Altenbach, C.; Flitsch, S.L.; Khorana, H.G.; Hubbell, W.L. Structural Studies on Transmembrane Proteins. 2. Spin Labeling of Bacteriorhodopsin Mutants at Unique Cysteines. Biochemistry 1989, 28, 7806–7812. [Google Scholar] [CrossRef]
- Hubbell, W.L.; Altenbach, C. Investigation of Structure and Dynamics in Membrane Proteins Using Site-Directed Spin Labeling. Curr. Opin. Struct. Biol. 1994, 4, 566–573. [Google Scholar] [CrossRef]
- Chen, M.; Margittai, M.; Chen, J.; Langen, R. Investigation of Alpha-Synuclein Fibril Structure by Site-Directed Spin Labeling. J. Biol. Chem. 2007, 282, 24970–24979. [Google Scholar] [CrossRef]
- Der-Sarkissian, A.; Jao, C.C.; Chen, J.; Langen, R. Structural Organization of Alpha-Synuclein Fibrils Studied by Site-Directed Spin Labeling. J. Biol. Chem. 2003, 278, 37530–37535. [Google Scholar] [CrossRef]
- Karyagina, I.; Becker, S.; Giller, K.; Riedel, D.; Jovin, T.M.; Griesinger, C.; Bennati, M. Electron Paramagnetic Resonance Spectroscopy Measures the Distance between the External β-Strands of Folded α-Synuclein in Amyloid Fibrils. Biophys. J. 2011, 101, L1–L3. [Google Scholar] [CrossRef]
- Pornsuwan, S.; Giller, K.; Riedel, D.; Becker, S.; Griesinger, C.; Bennati, M. Long-Range Distances in Amyloid Fibrils of α-Synuclein from PELDOR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 10290–10294. [Google Scholar] [CrossRef]
- Strohäker, T.; Jung, B.C.; Liou, S.-H.; Fernandez, C.O.; Riedel, D.; Becker, S.; Halliday, G.M.; Bennati, M.; Kim, W.S.; Lee, S.-J.; et al. Structural Heterogeneity of α-Synuclein Fibrils Amplified from Patient Brain Extracts. Nat. Commun. 2019, 10, 5535. [Google Scholar] [CrossRef]
- Zurlo, E.; Kumar, P.; Meisl, G.; Dear, A.J.; Mondal, D.; Claessens, M.M.A.E.; Knowles, T.P.J.; Huber, M. In Situ Kinetic Measurements of α-Synuclein Aggregation Reveal Large Population of Short-Lived Oligomers. PLoS ONE 2021, 16, e0245548. [Google Scholar] [CrossRef]
- Zurlo, E.; Passerini, L.; Kumar, P.; Huber, M. In Situ Continuous Wave Electron Paramagnetic Resonance Investigation of the Amyloid Aggregation of Parkinson’s Protein Alpha-Synuclein—The Second Spin-Label Position. Appl. Magn. Reson. 2021, 53, 1133–1150. [Google Scholar] [CrossRef]
- Eaton, S.S.; Eaton, G.R. Rapid-Scan EPR of Nitroxide Spin Labels and Semiquinones. Methods Enzymol. 2015, 563, 3–21. [Google Scholar] [CrossRef]
- Eaton, G.R.; Eaton, S.S. Advances in Rapid Scan EPR Spectroscopy. Methods Enzymol. 2022, 666, 1–24. [Google Scholar] [CrossRef]
- Braun, T.S.; Stehle, J.; Kacprzak, S.; Carl, P.; Höfer, P.; Subramaniam, V.; Drescher, M. Intracellular Protein-Lipid Interactions Studied by Rapid-Scan Electron Paramagnetic Resonance Spectroscopy. J. Phys. Chem. Lett. 2021, 12, 2471–2475. [Google Scholar] [CrossRef]
- Tseitlin, M.; Eaton, S.S.; Eaton, G.R. Uncertainty Analysis for Absorption and First-Derivative EPR Spectra. Concepts Magn. Reson. Part A 2012, 40, 295–305. [Google Scholar] [CrossRef]
- Cattani, J.; Braun, T.; Drescher, M. Probing Alpha-Synuclein Conformations by Electron Paramagnetic Resonance (EPR) Spectroscopy. In Methods in Molecular Biology; Holtzbrinck Springer Nature Publishing Group: New York, NY, USA, 2019; Volume 1948, pp. 247–260. [Google Scholar]
- Eichhoff, U.; Höfer, P. 75 Years of EPR. EPR Milestones in 60 Years Bruker History. Appl. Magn. Reson. 2020, 51, 1723–1737. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of Two Distinct Synucleins from Human Brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The Synaptic Function of-Synuclein. J. Parkinsons. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct Observation of the Three Regions in α-Synuclein That Determine Its Membrane-Bound Behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-State NMR Structure of a Pathogenic Fibril of Full-Length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Chattopadhyay, K. Cryo-Electron Microscopy Uncovers Key Residues within the Core of Alpha-Synuclein Fibrils. ACS Chem. Neurosci. 2019, 10, 1135–1136. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM Structure of Alpha-Synuclein Fibrils. Elife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef]
- Mitchell, D.G.; Quine, R.W.; Tseitlin, M.; Eaton, S.S.; Eaton, G.R. X-Band Rapid-Scan EPR of Nitroxyl Radicals. J. Magn. Reson. 2012, 214, 221–226. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Snead, D.; Eliezer, D. Alpha-Synuclein Function and Dysfunction on Cellular Membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.Y. Synucleins Are Developmentally Expressed, and α-Synuclein Regulates the Size of the Presynaptic Vesicular Pool in Primary Hippocampal Neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Accumulation Time | RS | CW |
|---|---|---|
| monomer | 121 | 26 |
| fibril | 54 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dröden, J.; Drescher, M. Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein. Biology 2023, 12, 79. https://doi.org/10.3390/biology12010079
Dröden J, Drescher M. Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein. Biology. 2023; 12(1):79. https://doi.org/10.3390/biology12010079
Chicago/Turabian StyleDröden, Jessica, and Malte Drescher. 2023. "Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein" Biology 12, no. 1: 79. https://doi.org/10.3390/biology12010079
APA StyleDröden, J., & Drescher, M. (2023). Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein. Biology, 12(1), 79. https://doi.org/10.3390/biology12010079