
Macromolecular Crowding Is Surprisingly Unable to Deform the Structure of a Model Biomolecular Condensate

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Dissipative Particle Dynamics Simulations
2.2. POETS
- 1.
- How do we compute multiple simulations fast enough to support semi-interactive parameter space exploration?
- 2.
- What visualization and workflow support can be created to support a human who wishes to perform such an exploration?
- -
- It allowed the experiments to be completed in under two hours, which allowed them to be submitted to the “fast” low-latency queue in the Iridis job manager;
- -
- It means we do not need to use GPUs to achieve low latency, which is useful because GPUs are less common in many HPC clusters and they are heavily subscribed by chemists, physicists, and machine learning researchers. In our experience, it typically takes more than 2 hours for a GPU job to even start running, even though it may perform faster once it has started.
- 1.
- Manual: Define a model with multiple parameter dimensions to sweep;
- 2.
- Manual: Construct a parameterized scenario generator that can instantiate the model for specific parameter values;
- 3.
- Perform human–computer collaborative search:
- a.
- Manual: Identify two interesting parameter dimensions and ranges; pick X points for one parameter and Y points for the other parameter and generate the XxY concrete scenarios to be simulated;
- b.
- Automatic: Simulate the scenarios in parallel using multiple machines in a HPC system;
- c.
- Automatic: Collect the outputs and produce tiled XxY images and videos;
- d.
- Manual: Inspect the tiled images to understand the parameter response. If necessary, repeat step 3.a.
- 4.
- Manual: explore and analyze the results in more detail.
3. Results
3.1. Crowding Assists Phase Separation of IDPs with Sub-Critical Affinity
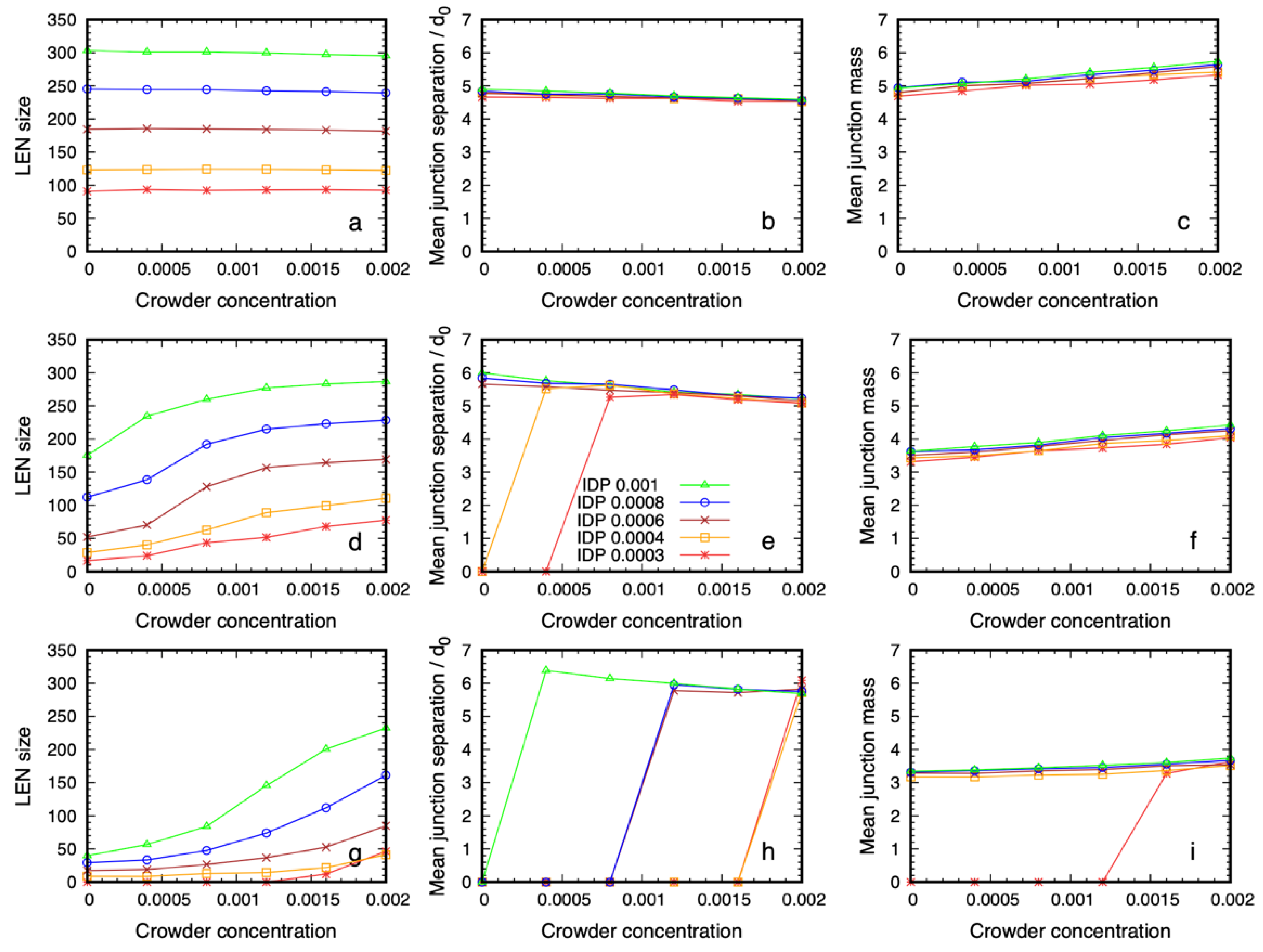
3.2. Quantitative Properties of Condensate Structure Are Insensitive to Crowder Concentration
3.3. Dense Phase Structure Is Partially Decoupled from the Crowder Molecular Weight and Enthalpic Repulsion from the IDPs
3.3.1. The Phase Boundary but Not the Dense Phase Structure Varies with the Crowder Volume Fraction
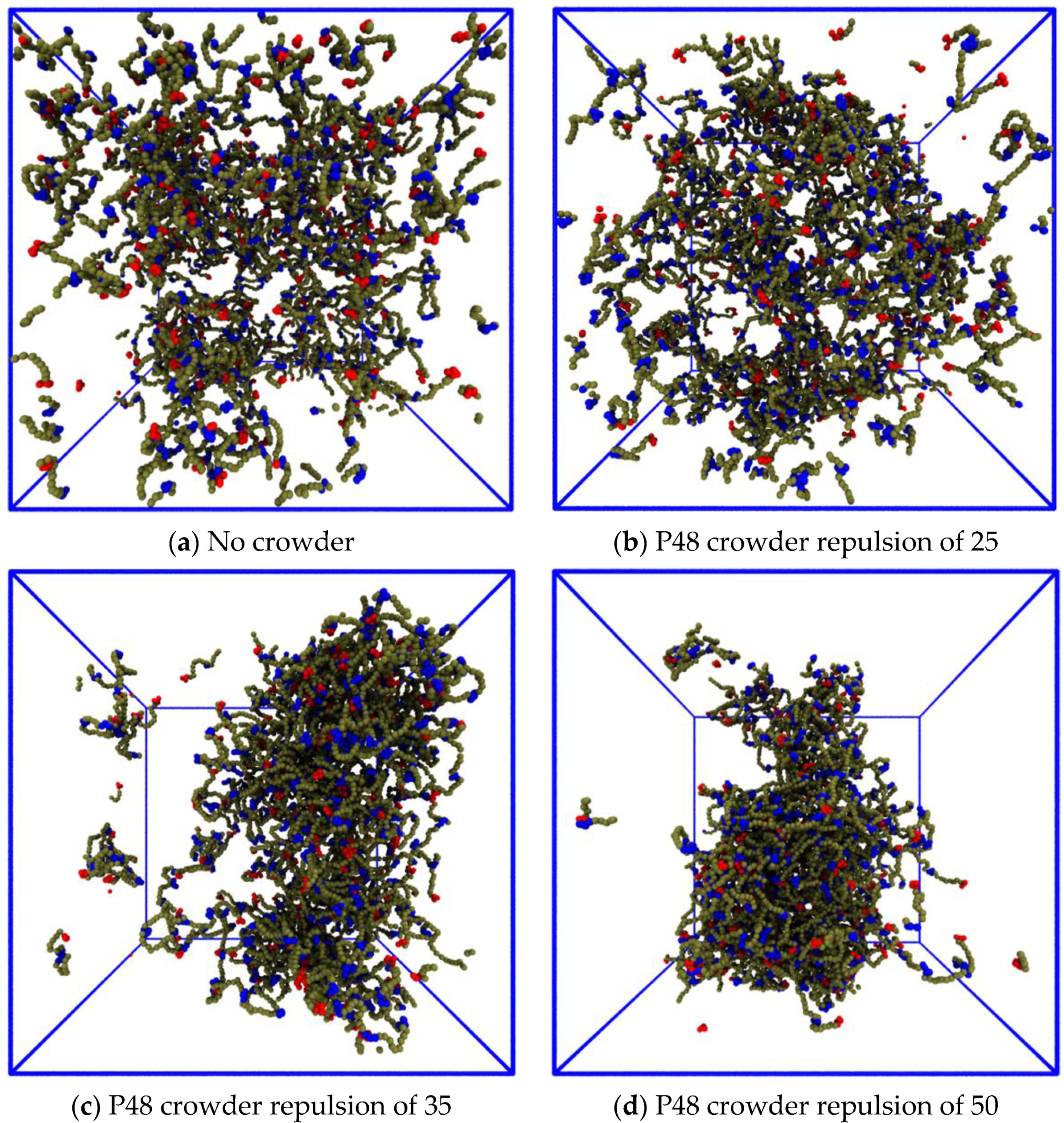
3.3.2. Reducing the Enthalpic Repulsion of the IDPs from the Crowder Molecules Leaves the Dense Phase Stable
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellis, R.J.; Minton, A.P. Join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Feig, M.; Yu, I.; Wang, P.-H.; Nawrocki, G.; Sugita, Y. Crowding in Cellular Environments at an Atomistic Level from Computer Simulations. J. Phys. Chem. B 2017, 121, 8009–8025. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, J.; Weeks, K.M.; Pielak, G.J. Challenge of Mimicking the Influences of the Cellular Environment on RNA Structure by PEG-Induced Macromolecular Crowding. Biochemistry 2015, 54, 6447–6453. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B. The Structure of Protoplasm. Science 1899, 10, 33–45. [Google Scholar] [CrossRef]
- Hyman, A.A.; Brangwynne, C.P. Beyond Stereospecificity: Liquids and Mesoscale Organization of Cytoplasm. Dev. Cell 2011, 21, 14–16. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal. 2016, 14, 1. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; van den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2020, 22, 215–235. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Pappu, R.V. Functional Implications of Intracellular Phase Transitions. Biochemistry 2018, 57, 2415–2423. [Google Scholar] [CrossRef]
- Bratek-Skicki, A.; Pancsa, R.; Meszaros, B.; Van Lindt, J.; Tompa, P. A guide to regulation of the formation of biomolecular condensates. FEBS J. 2020, 287, 1924–1935. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Mehta, S.; Zhang, J. Liquid–liquid phase separation: A principal organizer of the cell’s biochemical activity architecture. Trends Pharmacol. Sci. 2021, 42, 845–856. [Google Scholar] [CrossRef]
- Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Boyd-Shiwarski, C.R.; Shiwarski, D.J.; Griffiths, S.E.; Beacham, R.T.; Norrell, L.; Morrison, D.E.; Wang, J.; Mann, J.; Tennant, W.; Anderson, E.N.; et al. WNK kinases sense molecular crowding and rescue cell volume via phase separation. Cell 2022, 185, 4488–4506. [Google Scholar] [CrossRef]
- Pastore, A.; Temussi, P.A. Crowding revisited: Open questions and future perspectives. Trends Biochem. Sci. 2022, 47, 1048–1058. [Google Scholar] [CrossRef]
- Nedelsky, N.B.; Taylor, J.P. Bridging biophysics and neurology: Aberrant phase transitions in neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 272–286. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Are Aberrant Phase Transitions a Driver of Cellular Aging? Bioessays 2016, 38, 959–968. [Google Scholar] [CrossRef]
- Ranganathan, S.; Shakhnovich, E. The physics of liquid-to-solid transitions in multi-domain protein condensates. Biophys. J. 2022, 121, 2751–2766. [Google Scholar] [CrossRef]
- Agarwal, A.; Mukhopadhyay, S. Prion Protein Biology Through the Lens of Liquid-Liquid Phase Separation. J. Mol. Biol. 2021, 434, 167368. [Google Scholar] [CrossRef] [PubMed]
- Bracha, D.; Walls, M.T.; Brangwynne, C.P. Probing and engineering liquid-phase organelles. Nat. Biotechnol. 2019, 37, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Dzuricky, M.; Rogers, B.A.; Shahid, A.; Cremer, P.S.; Chilkoti, A. De novo engineering of intracellular condensates using artificial disordered proteins. Nat. Chem. 2020, 12, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, M.; Georgeson, J.M.; Locatelli, E.; Rovigatti, L.; Nandi, S.K.; Steinberg, A.; Nadav, Y.; Shimoni, E.; Safran, S.A.; Doye, J.P.K.; et al. Designer protein assemblies with tunable phase diagrams in living cells. Nat. Chem. Biol. 2020, 16, 939–945. [Google Scholar] [CrossRef]
- Garabedian, M.V.; Wang, W.; Dabdoub, J.B.; Tong, M.; Caldwell, R.M.; Benman, W.; Schuster, B.S.; Deiters, A.; Good, M.C. Designer membraneless organelles sequester native factors for control of cell behavior. Nat. Chem. Biol. 2021, 17, 998–1007. [Google Scholar] [CrossRef]
- Hastings, R.L.; Boeynaems, S. Designer Condensates: A Toolkit for the Biomolecular Architect. J. Mol. Biol. 2021, 433, 166837. [Google Scholar] [CrossRef]
- Mu, W.; Ji, Z.; Zhou, M.; Wu, J.; Lin, Y.; Qiao, Y. Membrane-confined liquid-liquid phase separation toward artificial organelles. Sci. Adv. 2021, 7, eabf9000. [Google Scholar] [CrossRef]
- Qian, Z.-G.; Huang, S.-C.; Xia, X.-X. Synthetic protein condensates for cellular and metabolic engineering. Nat. Chem. Biol. 2022, 18, 1330–1340. [Google Scholar] [CrossRef]
- Biesaga, M.; Frigolé-Vivas, M.; Salvatella, X. Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr. Opin. Chem. Biol. 2021, 62, 90–100. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Mittasch, M.; Gomes, B.F.; Klein, I.A.; Murcko, M.A. Modulating biomolecular condensates: A novel approach to drug discovery. Nat. Rev. Drug Discov. 2022, 21, 841–862. [Google Scholar] [CrossRef]
- Patel, A.; Mitrea, D.; Namasivayam, V.; Murcko, M.A.; Wagner, M.; Klein, I.A. Principles and functions of condensate modifying drugs. Front. Mol. Biosci. 2022, 9, 1007744. [Google Scholar] [CrossRef]
- Nott Timothy, J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forma-Kay, J.D.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membrane less Organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef]
- Shin, Y.; Chang, Y.-C.; Lee, D.S.; Berry, J.; Sanders, D.W.; Ronceray, P.; Wingreen, N.S.; Haataja, M.; Brangwynne, C.P. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell 2018, 175, 1481–1491.e13. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Phase Separation in Membrane Biology: The Interplay between Membrane-Bound Organelles and Membraneless Condensates. Dev. Cell 2020, 55, 30–44. [Google Scholar] [CrossRef]
- Gouveia, B.; Kim, Y.; Shaevitz, J.W.; Petry, S.; Stone, H.A.; Brangwynne, C.P. Capillary forces generated by biomolecular condensates. Nature 2022, 609, 255–264. [Google Scholar] [CrossRef]
- Lasker, K.; Boeynaems, S.; Lam, V.; Scholl, D.; Stainton, E.; Briner, A.; Jacquemyn, M.; Daelemans, D.; Deniz, A.; Villa, E.; et al. The material properties of a bacterial-derived biomolecular condensate tune biological function in natural and synthetic systems. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Chan, S.H.; Dey, S.; Castello-Serrano, I.; Ditlev, J.A.; Rosen, M.K. Coupling of protein condensates to ordered lipid domains determines functional membrane organization. Biorxiv 2022. [Google Scholar] [CrossRef]
- Musacchio, A. On the role of phase separation in the biogenesis of membrane less compartments. EMBO J. 2022, 41, e109952. [Google Scholar] [CrossRef]
- Mittag, T.; Pappu, R.V. A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol. Cell 2022, 82, 2201–2214. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell 2015, 60, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid−liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019, 26, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.C.; Tang, W.S.; Jovic, N.; Janke, A.M.; Seo, D.H.; Perdikari, T.M.; Mittal, J.; Fawzi, N.L. Molecular interactions contributing to FUS SYGQ LC-RGG phase separation and co-partitioning with RNA polymerase II heptads. Nat. Struct. Mol. Biol. 2021, 28, 923–935. [Google Scholar] [CrossRef]
- Harmon, T.S.; Holehouse, A.S.; Rosen, M.K.; Pappu, R.V. Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. eLife 2017, 6, e30294. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Kim, Y.C.; Best, R.B.; Mittal, J. Sequence determinants of protein phase behavior from a coarse-grained model. PLOS Comput. Biol. 2018, 14, e1005941. [Google Scholar] [CrossRef]
- Dignon, G.L.; Zheng, W.; Mittal, J. Simulation methods for liquid–liquid phase separation of disordered proteins. Curr. Opin. Chem. Eng. 2019, 23, 92–98. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V.; Holehouse, A.S. Conformational Preferences and Phase Behaviour of Intrinsically Disordered Low Complexity Sequences: Insights from Multiscale Simulations. Curr. Op. Struct. Biol. 2019, 56, 1–10. [Google Scholar] [CrossRef]
- Benayad, Z.; von Bülow, S.; Stelzl, L.S.; Hummer, G. Simulation of FUS Protein Condensates with an Adapted Coarse-Grained Model. J. Chem. Theory Comput. 2020, 17, 525–537. [Google Scholar] [CrossRef]
- Shillcock, J.C.; Lagisquet, C.; Alexandre, J.; Vuillon, L.; Ipsen, J.H. Model biomolecular condensates have heterogeneous structure quantitatively dependent on the interaction profile of their constituent macromolecules. Soft Matter 2022, 18, 6674–6693. [Google Scholar] [CrossRef]
- Shillcock, J.C.; Thomas, D.B.; Beaumont, J.R.; Bragg, G.M.; Vousden, M.L.; Brown, A.D. Coupling Bulk Phase Separation of Disordered Proteins to Membrane Domain Formation in Molecular Simulations on a Bespoke Compute Fabric. Membranes 2022, 12, 1–20. [Google Scholar] [CrossRef]
- Pyo, A.G.; Zhang, Y.; Wingreen, N.S. Surface tension and super-stoichiometric surface enrichment in two-component biomolecular condensates. Iscience 2022, 25, 103852. [Google Scholar] [CrossRef]
- Weiner, B.G.; Pyo, A.G.T.; Meir, Y.; Wingreen, N.S. Motif-pattern dependence of biomolecular phase separation driven by specific interactions. PLOS Comput. Biol. 2021, 17, e1009748. [Google Scholar] [CrossRef]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. EPL Europhys. Lett. 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Shillcock, J.C.; Brochut, M.; Chénais, E.; Ipsen, J.H. Phase behaviour and structure of a model biomolecular condensate. Soft Matter 2020, 16, 6413–6423. [Google Scholar] [CrossRef]
- Beaumont, J.R.; Brown, A.D.; Thomas, D.B.; Shillcock, J.C.; Naylor, M.F.; Bragg, G.M.; Vousden, M.; Moore, S.W.; Flemming, S. An event-driven approach to Dissipative Particle Dynamics. ACM Trans. Parallel Comput. 2023. (In Press)
- Brown, A.D.; Thomas, D.B.; Reeve, J.; Tarawneh, G.; De Gennaro, A.; Mokhov, A.; Naylor, M.; Kazmierski, T. Distributed event-based computing. In Proceedings of the ParCo 2017: Parallel Computing is Everywhere, Bologna, Italy, 12–15 September 2017; IOS Press: Amsterdam, the Netherlands, 2017. [Google Scholar]
- Español, P.; Warren, P. Statistical Mechanics of Dissipative Particle Dynamics. EPL Europhys. Lett. 1995, 30, 191–196. [Google Scholar] [CrossRef]
- Espagnol, P.; Warren, P.B. Perspective: Dissipative Particle Dynamics. J Chem Phys. 2017, 146, 150901-17. [Google Scholar] [CrossRef]
- Shillcock, J.C.; Lipowsky, R. Equilibrium structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations. J. Chem. Phys. 2002, 117, 5048–5061. [Google Scholar] [CrossRef]
- Shillcock, J.C. OSPREY-DPD. Open Source Polymer Research Engine-Dissipative Particle Dynamics. 2020. Available online: https://github.com/Osprey-DPD/osprey-dpd (accessed on 16 January 2023).
- Wang, J.; Choi, J.-M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 2018, 174, 688–699.e16. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: New York, NY, USA, 2003. [Google Scholar]
- Zilman, A.; Tlusty, T.; Safran, S.A. Entropic networks in colloidal, polymeric and amphiphilic systems. J. Phys. Condens. Matter. 2002, 15, S57–S64. [Google Scholar] [CrossRef]
- Dudowicz, J.; Freed, K.F. Lattice cluster theory of associating polymers. I. Solutions of linear telechelic polymer chains. J. Chem. Phys. 2012, 136, 064902. [Google Scholar] [CrossRef] [PubMed]
- Parada, G.A.; Zhao, X. Ideal reversible polymer networks. Soft Matter 2018, 14, 5186–5196. [Google Scholar] [CrossRef]
- Wen, J.; Hong, L.; Krainer, G.; Yao, Q.-Q.; Knowles, T.P.J.; Wu, S.; Perrett, S. Conformational Expansion of Tau in Condensates Promotes Irreversible Aggregation. J. Am. Chem. Soc. 2021, 143, 13056–13064. [Google Scholar] [CrossRef]
- Horvath, I.; Kumar, R.; Wittung-Stafshede, P. Macromolecular crowding modulates α-synuclein amyloid fiber growth. Biophys. J. 2021, 120, 3374–3381. [Google Scholar] [CrossRef]
- Linsenmeier, M.; Faltova, L.; Palmiero, U.C.; Seiffert, C.; Küffner, A.M.; Pinotsi, D. The interface of condensates of the hnRNPA1 low complexity domain promotes formation of amyloid fibrils. BioRxiv 2022. [Google Scholar] [CrossRef]
- Küffner, A.M.; Linsenmeier, M.; Grigolato, F.; Prodan, M.; Zuccarini, R.; Palmiero, U.C.; Faltova, L.; Arosio, P. Sequestration within biomolecular condensates inhibits Aβ-42 amyloid formation. Chem. Sci. 2021, 12, 4373–4382. [Google Scholar] [CrossRef]
- Houser, J.R.; Cho, H.W.; Hayden, C.C.; Yang, N.X.; Wang, L.; Lafer, E.M.; Thirumalai, D.; Stachowiak, J.C. Molecular mechanisms of steric pressure generation and membrane remodeling by disordered proteins. Biophys. J. 2022, 121, 3320–3333. [Google Scholar] [CrossRef]
- Boija, A.; Klein, I.A.; Young, R.A. Biomolecular Condensates and Cancer. Cancer Cell 2021, 39, 174–192. [Google Scholar] [CrossRef]
- Kar, M.; Dar, F.; Welsh, T.J.; Vogel, L.T.; Kühnemuth, R.; Majumdar, A.; Krainer, G.; Franzmann, T.M.; Alberti, S.; Seidel, C.A.M.; et al. Phase-separating RNA-binding proteins form heterogeneous distributions of clusters in subsaturated solutions. Proc. Natl. Acad. Sci. USA 2022, 119, e2202222119. [Google Scholar] [CrossRef]
- Tesei, G.; Schulze, T.K.; Crehuet, R.; Lindorff-Larsen, K. Accurate model of liquid–liquid phase behavior of intrinsically disordered proteins from optimization of single-chain properties. Proc. Natl. Acad. Sci. USA 2021, 118, 2111696118. [Google Scholar] [CrossRef]
- Farag, M.; Cohen, S.R.; Borcherds, W.M.; Bremer, A.; Mittag, T.; Pappu, R.V. Condensates formed by prion-like low-complexity domains have small-world network structures and interfaces defined by expanded conformations. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Arter, W.E.; Qi, R.; Erkamp, N.A.; Krainer, G.; Didi, K.; Welsh, T.J.; Acker, J.; Nixon-Abell, J.; Qamar, S.; Guillén-Boixet, J.; et al. Biomolecular condensate phase diagrams with a combinatorial microdroplet platform. Nat. Commun. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Bai, Q.; Liu, Z.; Chen, J.; Liang, D. Crowded Environment Regulates the Coacervation of Biopolymers via Nonspecific Interactions. Biomacromolecules 2023, 24, 283–293. [Google Scholar] [CrossRef]
- Harmon, T.S.; Holehouse, A.S.; Pappu, R.V. Differential Solvation of Intrinsically Disordered Linkers Drives the Formation of Spatially Organised Droplets in Ternary Systems of Linear Multivalent Proteins. New J. Physics. 2018, 20, 045002-16. [Google Scholar] [CrossRef]
- Alshareedah, I.; Moosa, M.M.; Pham, M.; Potoyan, D.A.; Banerjee, P.R. Programmable viscoelasticity in protein-RNA condensates with disordered sticker-spacer polypeptides. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Ginell, G.M.; Griffith, D.; Böke, E. Clustering of Aromatic Residues in Prion-like Domains Can Tune the Formation, State, and Organization of Biomolecular Condensates. Biochemistry 2021, 60, 3566–3581. [Google Scholar] [CrossRef]
- Das, S.; Muthukumar, M. Microstructural Organization in α-Synuclein Solutions. Macromolecules 2022, 55, 4228–4236. [Google Scholar] [CrossRef]
- André, A.A.; Yewdall, N.A.; Spruijt, E. Crowding-induced phase separation and gelling by co-condensation of PEG in NPM1-rRNA condensates. bioRxiv 2022. [Google Scholar] [CrossRef]
- Alshareedah, I.; Singh, A.; Quinn, A.; Banerjee, P.R. Determinants of Viscoelasticity and Flow Activation Energy in Biomolecular Condensates. bioRxiv 2022. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| W | E | B | F | P | |
|---|---|---|---|---|---|
| W | 25 | ||||
| E | 25 | aEE | |||
| B | 23 | 25 | 25 | ||
| F | 25 | aEE | 25 | aEE | |
| P | 25 | 80 | 80 | 80 | 80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shillcock, J.C.; Thomas, D.B.; Ipsen, J.H.; Brown, A.D. Macromolecular Crowding Is Surprisingly Unable to Deform the Structure of a Model Biomolecular Condensate. Biology 2023, 12, 181. https://doi.org/10.3390/biology12020181
Shillcock JC, Thomas DB, Ipsen JH, Brown AD. Macromolecular Crowding Is Surprisingly Unable to Deform the Structure of a Model Biomolecular Condensate. Biology. 2023; 12(2):181. https://doi.org/10.3390/biology12020181
Chicago/Turabian StyleShillcock, Julian C., David B. Thomas, John H. Ipsen, and Andrew D. Brown. 2023. "Macromolecular Crowding Is Surprisingly Unable to Deform the Structure of a Model Biomolecular Condensate" Biology 12, no. 2: 181. https://doi.org/10.3390/biology12020181
APA StyleShillcock, J. C., Thomas, D. B., Ipsen, J. H., & Brown, A. D. (2023). Macromolecular Crowding Is Surprisingly Unable to Deform the Structure of a Model Biomolecular Condensate. Biology, 12(2), 181. https://doi.org/10.3390/biology12020181