

Trait-Based Method of Quantitative Assessment of Ecological Functional Groups in the Human Intestinal Microbiome


 , and
, and
Abstract
Simple Summary
Abstract
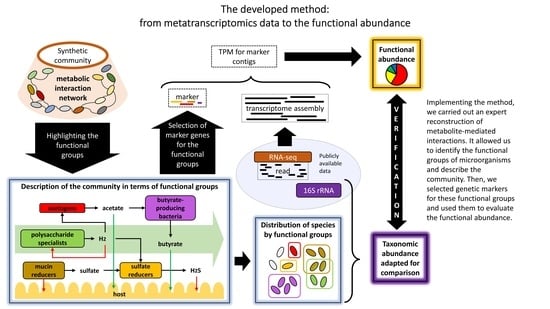
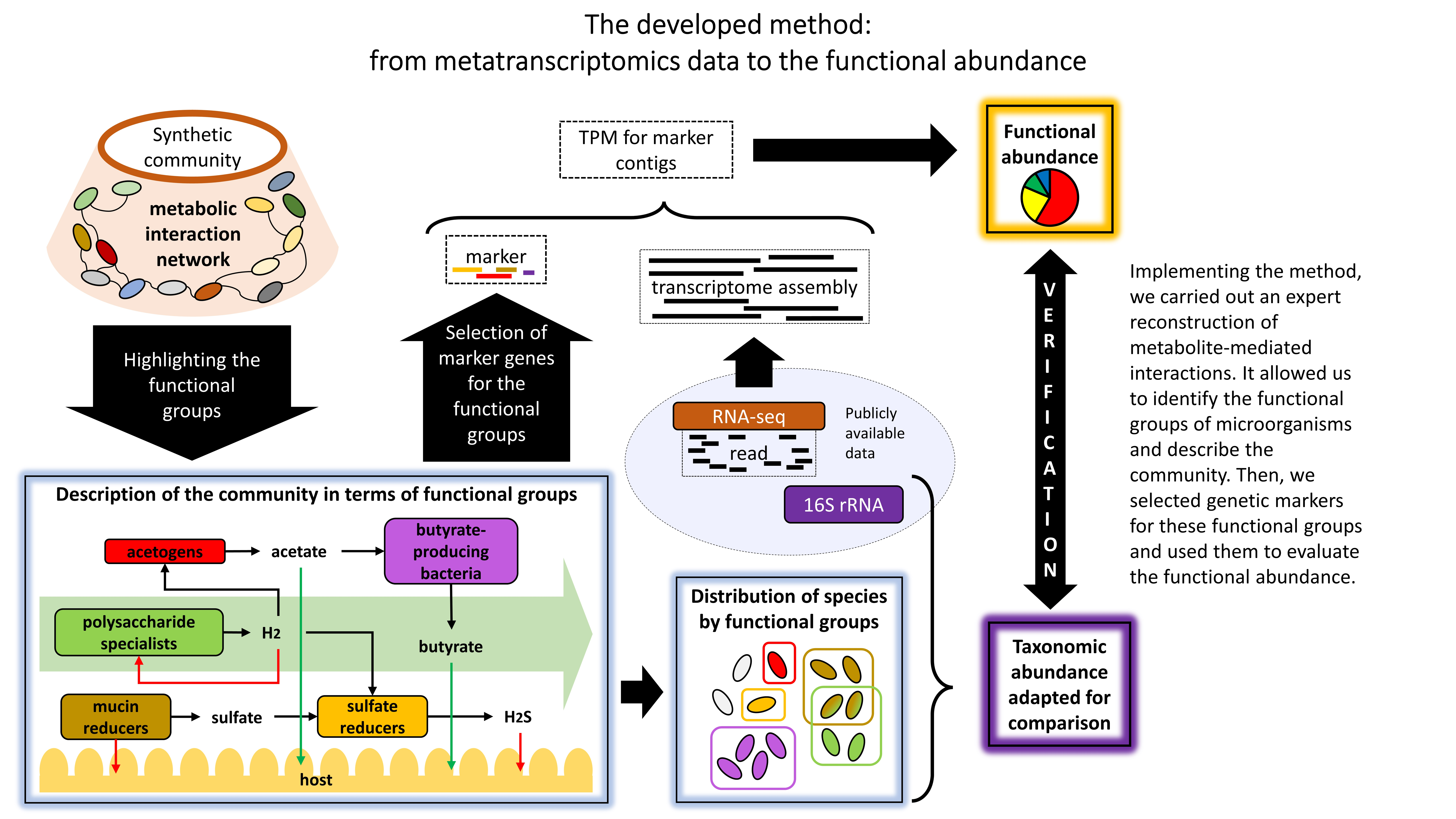
1. Introduction
2. Methods
2.1. Metagenomic and Metatranscriptomic Data from a Synthetic Community Modelling the Core Microbiome of the Human Gut
2.2. Methodology for Identification of the Key Functional Groups and Their Trait-Determining Genetic Features (TDGFs)
2.3. Implementation of the Method for Quantitative Assessment of Functional Groups in the Human Intestinal Microbiome
2.4. Comparing Functional Analysis Results with Humann
3. Results
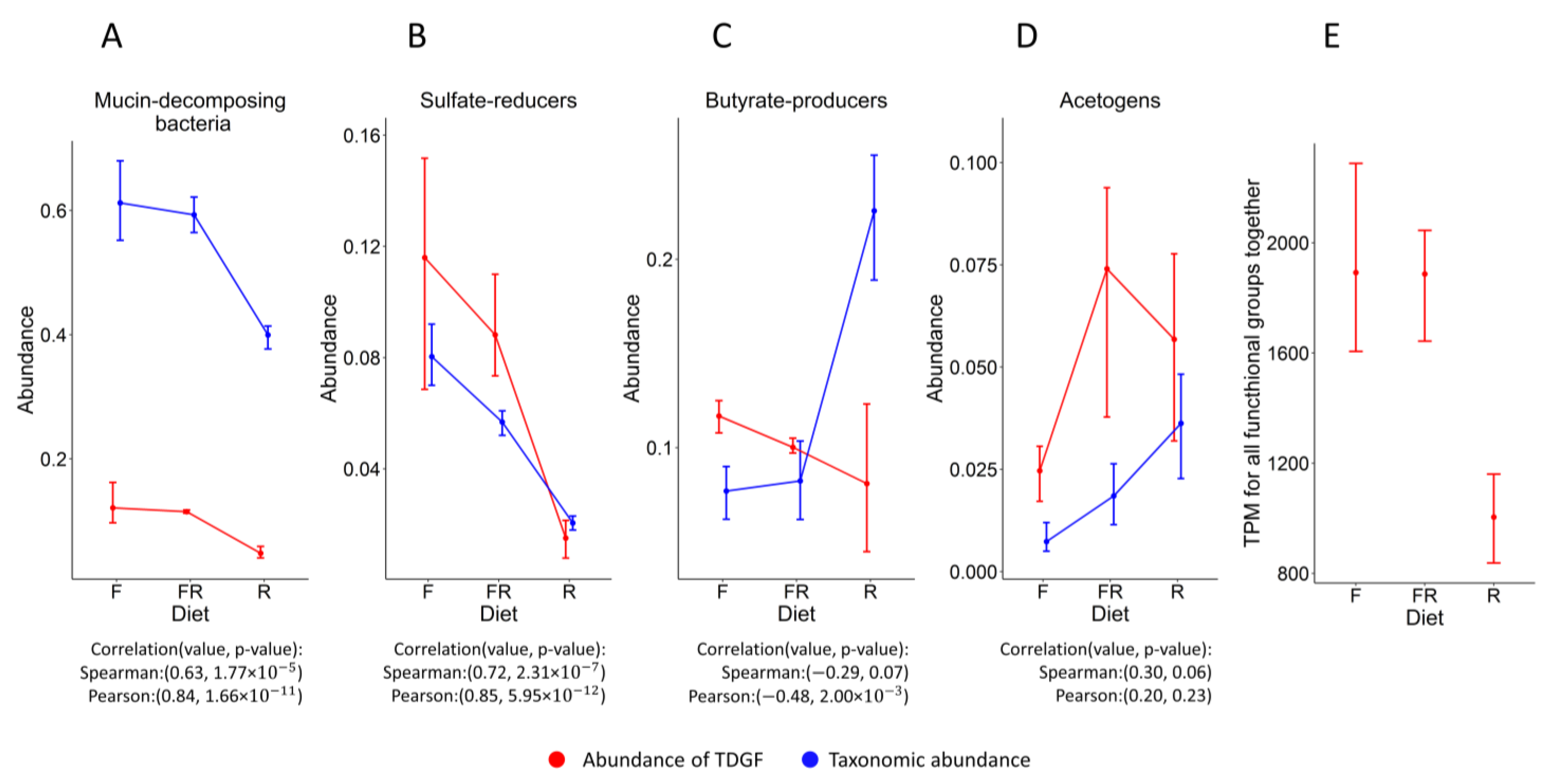
3.1. The Ecological Structure of the Human Intestinal Microbial Community in Terms of Functional Groups
3.2. The Similarity of the Patterns of Changes in the Abundance of Functional Groups When Evaluated Based on Different Data
3.3. Comparing Functional Analysis Results with HumanN and RSEM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TDGF | trait-determining genetic feature |
| R | fiber-rich diet |
| F | fiber-free diet |
| FR | alternation of a fiber-rich diet and fiber-free diet |
| P | prebiotic diet |
| FP | alternation of a prebiotic and fiber-free diet |
| TBQM | trait-based quantification method method |
| TPM | transcript per million [42] |
| CPM | copies per million [18] |
| EC | Enzyme Commission |
References
- Ottman, N.; Smidt, H.; de Vos, W.M.; Belzer, C. The Function of Our Microbiota: Who Is out There and What Do They Do? Front. Cell. Infect. Microbiol. 2012, 2, 104. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Round, J.L. Defining Dysbiosis and Its Influence on Host Immunity and Disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Tanca, A.; Abbondio, M.; Palomba, A.; Fraumene, C.; Manghina, V.; Cucca, F.; Fiorillo, E.; Uzzau, S. Potential and Active Functions in the Gut Microbiota of a Healthy Human Cohort. Microbiome 2017, 5, 79. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.-H.; Huang, T.; Cai, Y.-D. Gene Expression Profiling Gut Microbiota in Different Races of Humans. Sci. Rep. 2016, 6, 23075. [Google Scholar] [CrossRef] [PubMed]
- De Roy, K.; Marzorati, M.; Van Den Abbeele, P.; Van De Wiele, T.; Boon, N. Minireview Synthetic Microbial Ecosystems: An Exciting Tool to Understand and Apply Microbial Communities. Environ. Microbiol. 2014, 16, 1472–1481. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Bortfeld-Miller, M.; Ackermann, M.; Vorholt, J.A. A Synthetic Community Approach Reveals Plant Genotypes Affecting the Phyllosphere Microbiota. PLoS Genet. 2014, 10, e1004283. [Google Scholar] [CrossRef]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A Unified Catalog of 204,938 Reference Genomes from the Human Gut Microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal Gene Transfer: Building the Web of Life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-D.; Liu, X.-M.; Huang, T.-L.; Xia, L.-C. The Statistical Power of K-Mer Based Aggregative Statistics for Alignment-Free Detection of Horizontal Gene Transfer. Synth. Syst. Biotechnol. 2019, 4, 150–156. [Google Scholar] [CrossRef]
- Mcgill, B.J.; Enquist, B.J.; Weiher, E.; Westoby, M. Rebuilding Community Ecology from Functional Traits. Trends Ecol. Evol. 2006, 21, 178–185. [Google Scholar] [CrossRef]
- Kiørboe, T.; Visser, A.; Andersen, K.H. A Trait-Based Approach to Ocean Ecology. ICES J. Mar. Sci. 2018, 75, 1849–1863. [Google Scholar] [CrossRef]
- Ho, A.; Kerckhof, F.-M.; Luke, C.; Reim, A.; Krause, S.; Boon, N.; Bodelier, P.L.E. Conceptualizing Functional Traits and Ecological Characteristics of Methane-Oxidizing Bacteria as Life Strategies. Environ. Microbiol. Rep. 2013, 5, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.; Le Roux, X.; Niklaus, P.A.; Van Bodegom, P.M.; Lennon, J.T.; Bertilsson, S.; Grossart, H.; Philippot, L.; Bodelier, P.L.E. Trait-Based Approaches for Understanding Microbial Biodiversity and Ecosystem Functioning. Front. Microbiol. 2014, 5, 251. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with BioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Nazeen, S.; Yu, Y.W.; Berger, B. Carnelian Uncovers Hidden Functional Patterns across Diverse Study Populations from Whole Metagenome Sequencing Reads. Genome Biol. 2020, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Ramesh, V.; Locasale, J.W. Acetate Metabolism in Physiology, Cancer, and Beyond. Trends Cell Biol. 2019, 29, 695–703. [Google Scholar] [CrossRef]
- Corfield, A.P. Mucins: A Biologically Relevant Glycan Barrier in Mucosal Protection. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Corfield, A.P.; Wagner, S.A.; O’Donnell, L.J.D.; Durdey, P.; Mountford, R.A.; Clamp, J.R. The Roles of Enteric Bacterial Sialidase, SialateO-Acetyl Esterase and Glycosulfatase in the Degradation of Human Colonic Mucin. Glycoconj. J. 1993, 10, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of Acetate to Butyrate Formation by Human Faecal Bacteria. Br. J. Nutr. 2004, 91, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial Degradation of Complex Carbohydrates in the Gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.-J. The Role of Butyrate on Colonic Function. Aliment. Pharmacol. Ther. 2007, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Sjövall, H.; Hansson, G.C. The Gastrointestinal Mucus System in Health and Disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361. [Google Scholar] [CrossRef]
- Lan, W.; Yang, C. Ruminal Methane Production: Associated Microorganisms and the Potential of Applying Hydrogen-Utilizing Bacteria for Mitigation. Sci. Total Environ. 2019, 654, 1270–1283. [Google Scholar] [CrossRef]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of Human Colonic Butyrate-Producing Bacteria Revealed by Analysis of the Butyryl-CoA: Acetate CoA-Transferase Gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef]
- Praharaj, A.B.; Dehury, B.; Mahapatra, N.; Kar, S.K.; Behera, S.K. Molecular Dynamics Insights into the Structure, Function, and Substrate Binding Mechanism of Mucin Desulfating Sulfatase of Gut Microbe Bacteroides Fragilis. J. Cell. Biochem. 2018, 119, 3618–3631. [Google Scholar] [CrossRef]
- Muyzer, G.; Stams, A.J.M. The Ecology and Biotechnology of Sulphate-Reducing Bacteria. Nat. Rev. Microbiol. 2008, 6, 441–454. [Google Scholar] [CrossRef]
- Pierce, E.; Xie, G.; Barabote, R.D.; Saunders, E.; Han, C.S.; Detter, J.C.; Richardson, P.; Brettin, T.S.; Das, A.; Ljungdahl, L.G.; et al. The Complete Genome Sequence of Moorella Thermoacetica (f. Clostridium Thermoaceticum). Environ. Microbiol. 2008, 10, 2550–2573. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin Dynamics and Enteric Pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. Enzymology of the Wood-Ljungdahl Pathway of Acetogenesis. Ann. N. Y. Acad. Sci. 2008, 1125, 129–136. [Google Scholar] [CrossRef]
- Rakoff-nahoum, S.; Coyne, M.J. An Ecological Network of Polysaccharide Utilization among Human Intestinal Symbionts. Curr. Biol. 2014, 24, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the in Vivo Metabolic Potential of Two Human Gut Acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef]
- Rey, F.E.; Gonzalez, M.D.; Cheng, J.; Wu, M.; Ahern, P.P.; Gordon, J.I. Metabolic Niche of a Prominent Sulfate-Reducing Human Gut Bacterium. Proc. Natl. Acad. Sci. USA 2013, 110, 13582–13587. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Muir, J.G.; Gibson, P.R. Does Butyrate Protect from Colorectal Cancer? J. Gastroenterol. Hepatol. 2006, 21, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the Bacterial Butyrate Synthesis Pathways by Analyzing (Meta)Genomic Data. MBio 2014, 5, e00889-14. [Google Scholar] [CrossRef]
- Wolin, M.J.; Miller, T.L. Interactions of Microbial Populations in Cellulose Fermentation. Fed. Proc. 1983, 42, 109–113. [Google Scholar]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of MRNA Abundance Using RNA-Seq Data: RPKM Measure Is Inconsistent among Samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic Microbial Community Profiling Using Unique Clade-Specific Marker Genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H. UniRef Clusters: A Comprehensive and Scalable Alternative for Improving Sequence Similarity Searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Ackerly, D.D.; Cornwell, W.K. A Trait-Based Approach to Community Assembly: Partitioning of Species Trait Values into within- and among-Community Components. Ecol. Lett. 2007, 10, 135–145. [Google Scholar] [CrossRef]
- Lajoie, G.; Kembel, S.W. Making the Most of Trait-Based Approaches for Microbial Ecology. Trends Microbiol. 2019, 27, 814–823. [Google Scholar] [CrossRef]
- Inkpen, S.A.; Douglas, G.M.; Brunet, T.D.P.; Leuschen, K.; Doolittle, W.F.; Langille, M.G.I. The Coupling of Taxonomy and Function in Microbiomes. Biol. Philos. 2017, 32, 1225–1243. [Google Scholar] [CrossRef]
- Omelchenko, M.V.; Galperin, M.Y.; Wolf, Y.I.; Koonin, E.V. Non-Homologous Isofunctional Enzymes: A Systematic Analysis of Alternative Solutions in Enzyme Evolution. Biol. Direct 2010, 5, 31. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantification Method | |||
| Abundance of the functional group based on TDGF identification | Abundance of the functional group based on taxonomic identification (16S rRNA) | ||
| Normalization method | Its proportion to the whole community | ||
| Its proportion to the total abundance of the distinguished functional groups | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kropochev, A.I.; Lashin, S.A.; Matushkin, Y.G.; Klimenko, A.I. Trait-Based Method of Quantitative Assessment of Ecological Functional Groups in the Human Intestinal Microbiome. Biology 2023, 12, 115. https://doi.org/10.3390/biology12010115
Kropochev AI, Lashin SA, Matushkin YG, Klimenko AI. Trait-Based Method of Quantitative Assessment of Ecological Functional Groups in the Human Intestinal Microbiome. Biology. 2023; 12(1):115. https://doi.org/10.3390/biology12010115
Chicago/Turabian StyleKropochev, Andrew I., Sergey A. Lashin, Yury G. Matushkin, and Alexandra I. Klimenko. 2023. "Trait-Based Method of Quantitative Assessment of Ecological Functional Groups in the Human Intestinal Microbiome" Biology 12, no. 1: 115. https://doi.org/10.3390/biology12010115
APA StyleKropochev, A. I., Lashin, S. A., Matushkin, Y. G., & Klimenko, A. I. (2023). Trait-Based Method of Quantitative Assessment of Ecological Functional Groups in the Human Intestinal Microbiome. Biology, 12(1), 115. https://doi.org/10.3390/biology12010115