
Comprehensive Analysis of Whole-Transcriptome Profiles in Response to Acute Hypersaline Challenge in Chinese Razor Clam Sinonovacula constricta
 ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Samples
2.2. Salinity Challenge and Tissue Sampling
2.3. RNA Sequencing
2.4. Identification of RNAs
2.5. Target Gene Prediction
2.6. GO and KEGG Analysis of Differentially Expressed RNAs
2.7. Construction of the lncRNA/circRNA-miRNA-mRNA Network
2.8. Quantitative Real-Time PCR Validation
3. Results
3.1. Sequencing Data
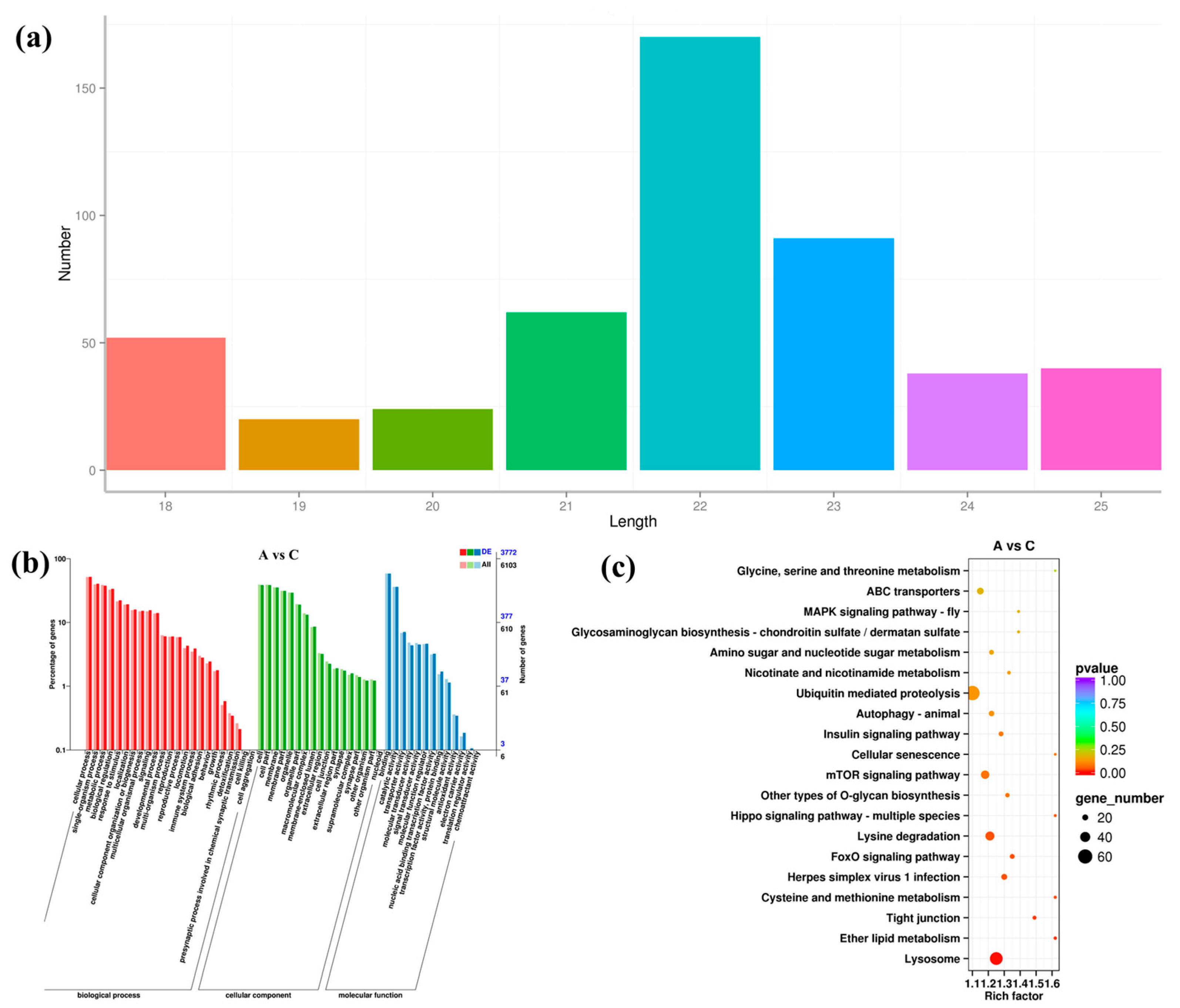
3.2. Expression Pattern of mRNA under High Salinity Stress
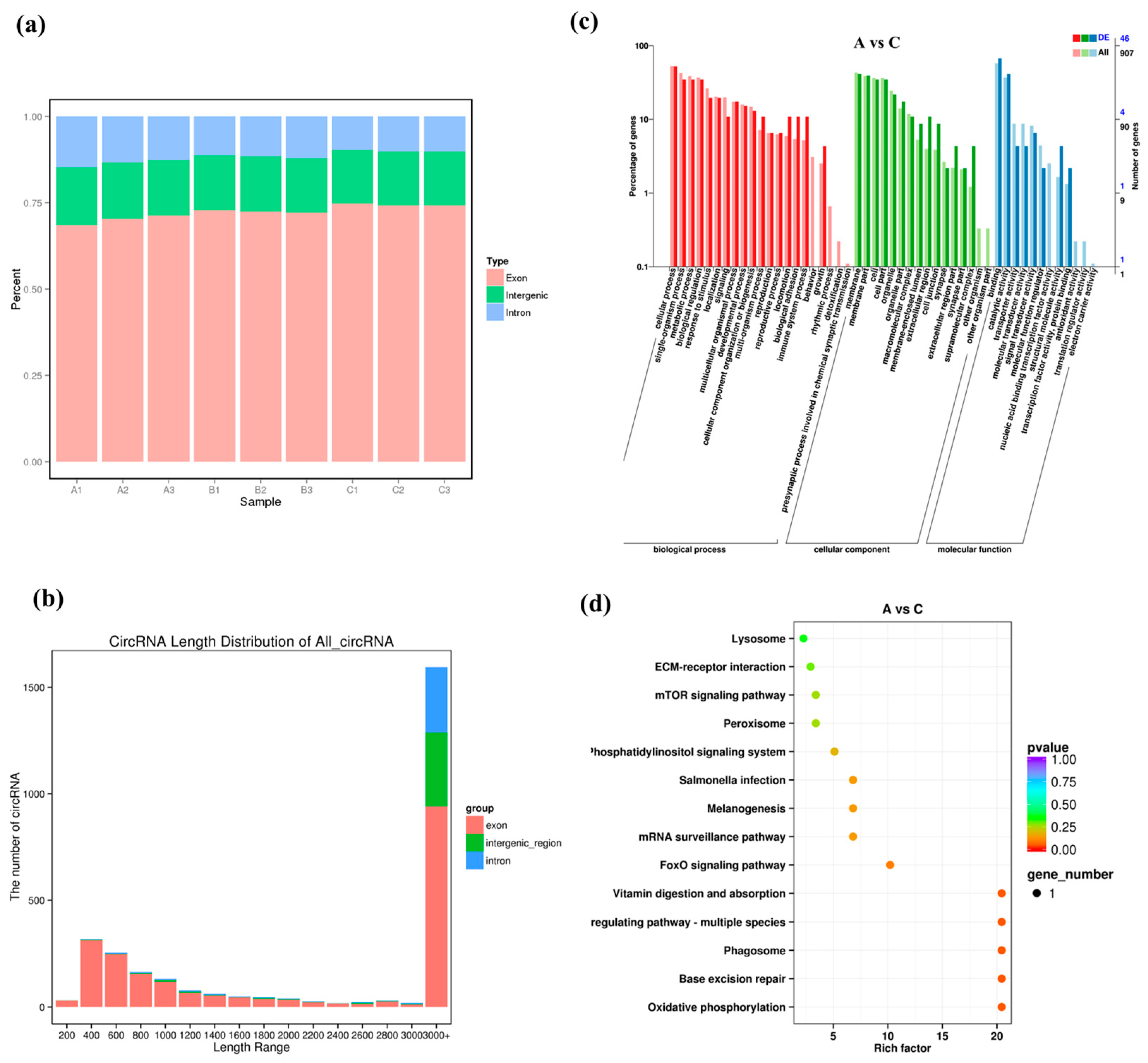
3.3. Expression Pattern of lncRNA under High Salinity Stress
3.4. Expression Patterns of circRNAs under High Salinity Stress
3.5. Expression Pattern of miRNA under High Salinity Stress
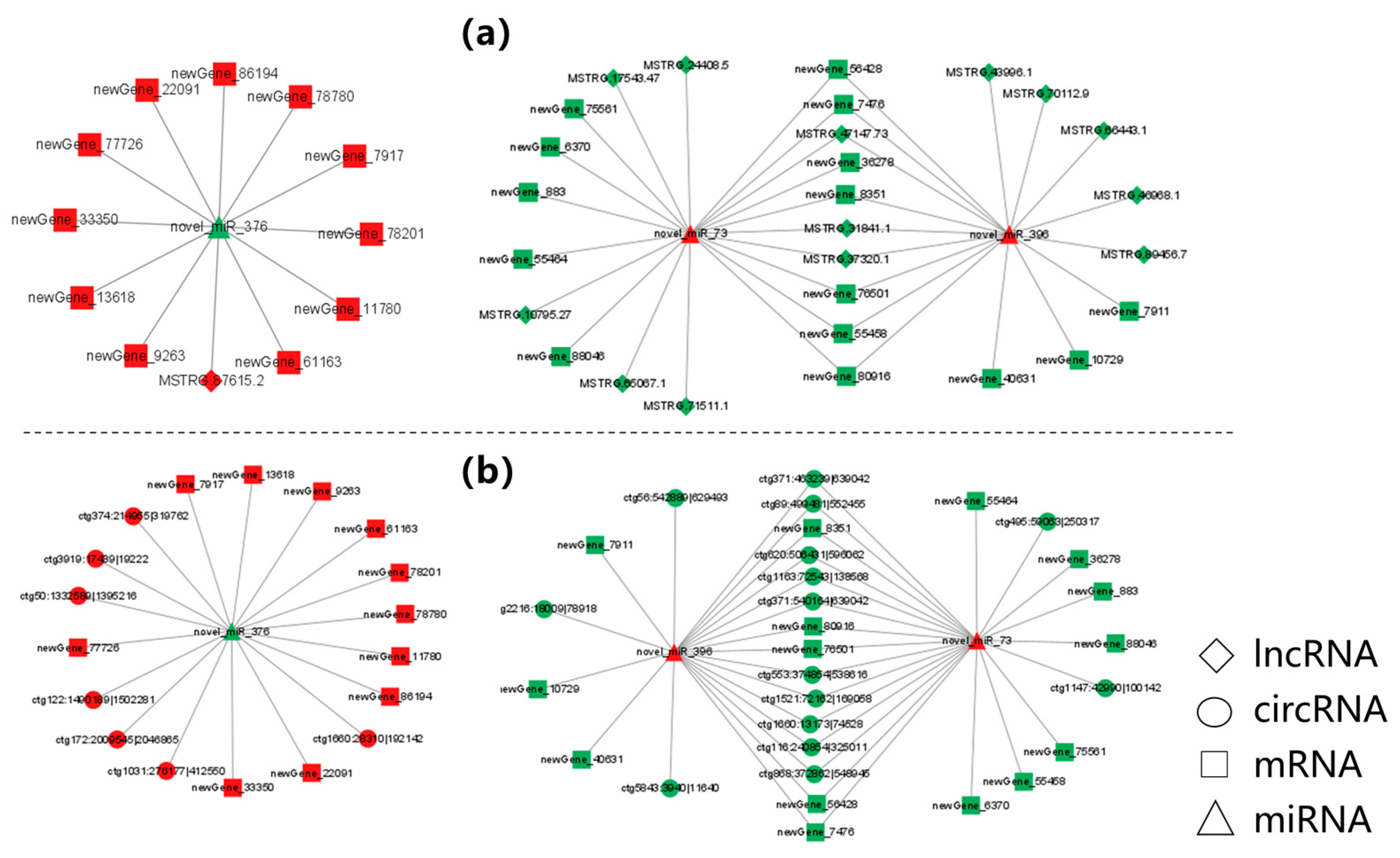
3.6. Construction of Potential lncRNA/circRNA-miRNA-mRNA Regulatory Networks
3.7. Quantitative Real-Time PCR Validation
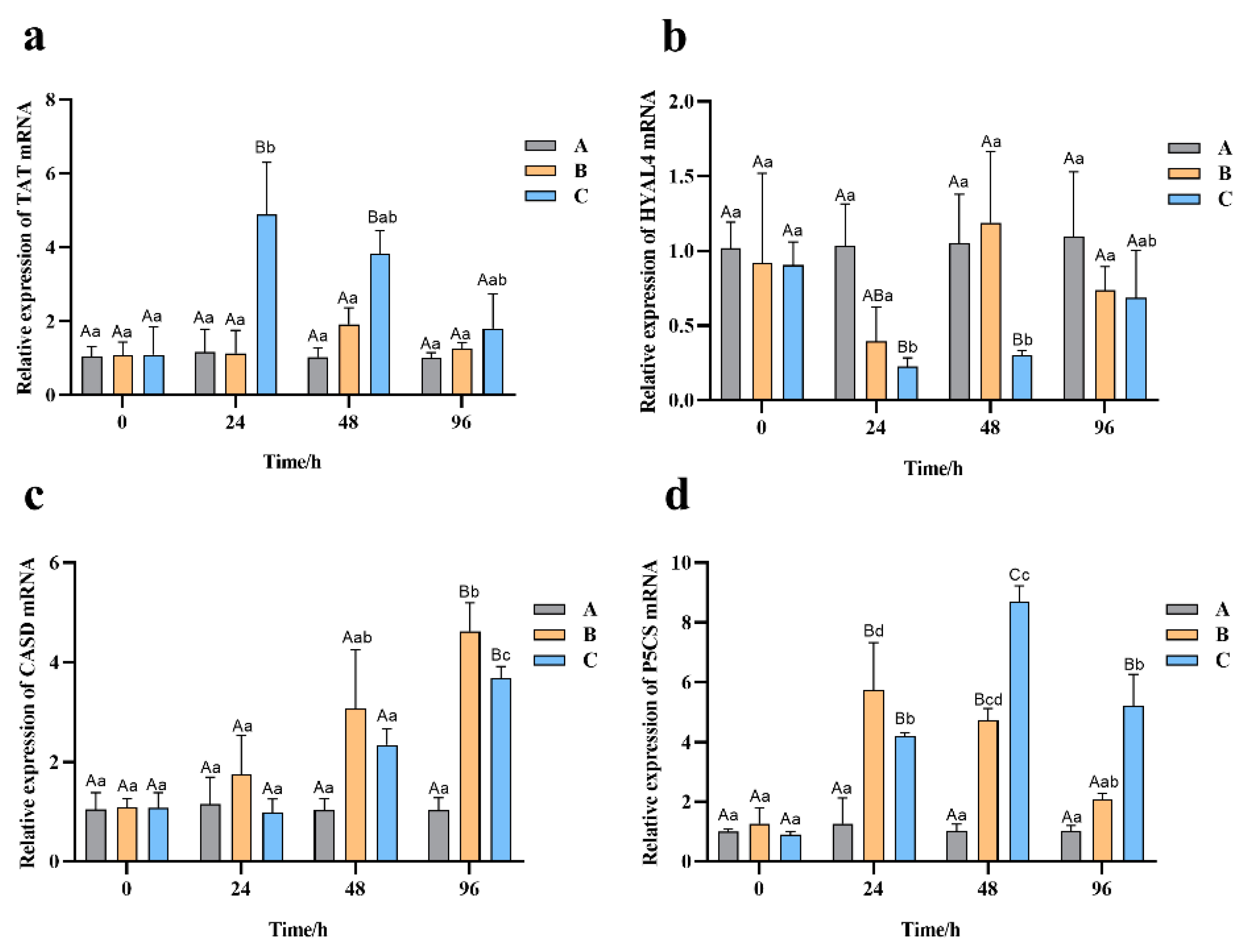
3.8. Effect of Salinity Stress on the Expression of Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, J.; Shu, M.; Xu, B.; Liu, G.; Ma, Y.; Guo, X.; Liu, Y. Mud crab Scylla paramamosain glutamate dehydrogenase: Molecular cloning, tissue expression and response to hyposmotic stress. Fisheries Sci. 2015, 81, 175–186. [Google Scholar] [CrossRef]
- Romano, N.; Zeng, C. Osmoregulation in decapod crustaceans: Implications to aquaculture productivity, methods for potential improvement and interactions with elevated ammonia exposure. Aquaculture 2012, 334–337, 12–23. [Google Scholar] [CrossRef]
- Berger, V.J.; Kharazova, A.D. Mechanisms of salinity adaptations in marine molluscs. Hydrobiologia 1997, 355, 115–126. [Google Scholar] [CrossRef]
- Carregosa, V.; Figueira, E.; Gil, A.M.; Pereira, S.; Pinto, J.; Soares, A.M.V.M.; Freitas, R. Tolerance of Venerupis philippinarum to salinity: Osmotic and metabolic aspects. Comp. Biochem. Physiol. A-Mol. Integr. Physiol. 2014, 171, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Munari, M.; Matozzo, V.; Marin, M.G. Combined effects of temperature and salinity on functional responses of haemocytes and survival in air of the clam Ruditapes philippinarum. Fish Shellfish Immun. 2011, 30, 1024–1030. [Google Scholar] [CrossRef]
- Agostini, M.; Ganini, C.; Candi, E.; Melino, G. The role of noncoding RNAs in epithelial cancer. Cell Death Discov. 2020, 6, 13. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef]
- Zhu, K.; Zhang, C.; Ma, X.; Hu, J.; Cai, T.; Zhang, L. Analyzing the Interactions of mRNAs and ncRNAs to Predict Competing Endogenous RNA Networks in Osteosarcoma Chemo-Resistance. Mol. Ther. 2019, 27, 518–530. [Google Scholar] [CrossRef]
- Chen, X.; Chen, J.; Shen, Y.; Bi, Y.; Hou, W.; Pan, G.; Wu, X. Transcriptional responses to low-salinity stress in the gills of adult female Portunus trituberculatus. Comp. Biochem. Physiol. D-Genom. Proteom. 2019, 29, 86–94. [Google Scholar] [CrossRef]
- Huang, B.; Liu, F.; Cao, D.; Li, G.; Lin, K.; Li, J.; Shi, G.; Zhang, J.; Wang, B.; Huang, J.; et al. Identification and characterization of miRNAs and target genes response to hypoosmotic adaption in the cobia, Rachycentron canadum. Aquac. Res. 2021, 52, 3322–3335. [Google Scholar] [CrossRef]
- Jin, F.; Sun, Y. Identification and characterization of salt-tolerance relative miRNAs in Procambarus clarkii by high-throughput sequencing. ExRNA 2019, 1, 18. [Google Scholar] [CrossRef]
- Shang, Y.; Tian, Y.; Wang, Y.; Guo, R. Novel lncRNA lncRNA001074 participates in the low salinity–induced response in the sea cucumber Apostichopus japonicus by targeting the let-7/NKAα axis. Cell Stress Chaperones 2021, 26, 785–798. [Google Scholar] [CrossRef]
- Tian, Y.; Shang, Y.; Guo, R.; Chang, Y.; Jiang, Y. Salinity stress-induced differentially expressed miRNAs and target genes in sea cucumbers Apostichopus japonicus. Cell Stress Chaperones 2019, 24, 719–733. [Google Scholar] [CrossRef]
- Wang, H.; Wei, H.; Tang, L.; Lu, J.; Mu, C.; Wang, C. Identification and characterization of miRNAs in the gills of the mud crab (Scylla paramamosain) in response to a sudden drop in salinity. BMC Genom. 2018, 19, 609. [Google Scholar] [CrossRef]
- Wang, X.; Yin, D.; Li, P.; Yin, S.; Wang, L.; Jia, Y.; Shu, X. MicroRNA-Sequence Profiling Reveals Novel Osmoregulatory MicroRNA Expression Patterns in Catadromous Eel Anguilla marmorata. PLoS ONE 2015, 10, e136383. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, P.; Gao, B.; Li, J. The identification and characteristics of salinity-related microRNAs in gills of Portunus trituberculatus. Cell Stress Chaperones 2016, 21, 63–74. [Google Scholar] [CrossRef]
- Tian, Y.; Shang, Y.; Guo, R.; Ding, J.; Li, X.; Chang, Y. miR-10 involved in salinity-induced stress responses and targets TBC1D5 in the sea cucumber, Apostichopus japonicas. Comp. Biochem. Physiol. B-Biochem. Mol. Biol. 2020, 242, 110406. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Liu, X.; Niu, D.; Yo, B.; Lan, T.; Dong, Z.; Li, J. Survival, growth and physiology of marine bivalve (Sinonovacula constricta) in long-term low-salt culture. Sci. Rep. 2019, 9, 2819. [Google Scholar] [CrossRef]
- Li, Y.; Niu, D.; Wu, Y.; Dong, Z.; Li, J. Integrated analysis of transcriptomic and metabolomic data to evaluate responses to hypersalinity stress in the gill of the razor clam (Sinonovacula constricta). Comp. Biochem. Physiol. D-Genom. Proteom. 2021, 38, 100793. [Google Scholar] [CrossRef]
- Cao, W.; Bi, S.; Chi, C.; Dong, Y.; Xia, S.; Liu, Z.; Zhou, L.; Sun, X.; Geng, Y.; Wu, B. Effects of High Salinity Stress on the Survival, Gill Tissue, Enzyme Activity and Free Amino Acid Content in Razor Clam Sinonovacula constricta. Front. Mar. Sci. 2022, 9, 839614. [Google Scholar] [CrossRef]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.; Liu, X.; Zhao, S.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2007, 36, D281–D288. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Heng, L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2007, 36, D149–D153. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Song, Y.; Han, H.; Xu, H.; Wei, D.; Smagghe, G.; Wang, J. Genome-wide analysis of long non-coding RNAs in adult tissues of the melon fly, Zeugodacus cucurbitae (Coquillett). BMC Genom. 2020, 21, 600. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, G.; Lin, Z.; Yao, H.; Dong, Y. The razor clam Sinonovacula constricta uses the strategy of conversion of toxic ammonia to glutamine in response to high environmental ammonia exposure. Mol. Biol. Rep. 2020, 47, 9579–9593. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Dong, Y.; Yao, H.; Lin, Z.; Sun, C. Identification and characteristic expression analysis of miR-8245a-5p target gene related to ammonia nitrogen stress in Sinonovacula constricta. Oceanol. Limnol. Sin. 2020, 51, 388–394. [Google Scholar] [CrossRef]
- Ma, B.; Ran, Z.; Xu, X.; Xu, J.; Liao, K.; Cao, J.; Yan, X. Comparative transcriptome analyses provide insights into the adaptation mechanisms to acute salt stresses in juvenile Sinonovacula constricta. Genes Genom. 2019, 41, 599–612. [Google Scholar] [CrossRef]
- Hosoi, M.; Shinzato, C.; Takagi, M.; Hosoi-Tanabe, S.; Sawada, H.; Terasawa, E.; Toyohara, H. Taurine transporter from the giant Pacific oyster Crassostrea gigas: Function and expression in response to hyper- and hypo-osmotic stress. Fisheries Sci. 2007, 73, 385–394. [Google Scholar] [CrossRef]
- Schomburg, I.; Chang, A.; Placzek, S.; Söhngen, C.; Rother, M.; Lang, M.; Munaretto, C.; Ulas, S.; Stelzer, M.; Grote, A.; et al. BRENDA in 2013: Integrated reactions, kinetic data, enzyme function data, improved disease classification: New options and contents in BRENDA. Nucleic Acids Res. 2012, 41, D764–D772. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Sun, Y.; Qimuge, N.; Wang, G.; Wang, Y.; Chu, G.; Yu, T.; Yang, G.; Pang, W. Adiponectin AS lncRNA inhibits adipogenesis by transferring from nucleus to cytoplasm and attenuating Adiponectin mRNA translation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 420–432. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Li, S.; Wang, G.X.; Yu, Q.; Lin, J.D. A long noncoding RNA transcriptional regulatory circuit drives thermogenic adipocyte differentiation. Mol. Cell 2014, 55, 372–382. [Google Scholar] [CrossRef]
- Zhu, E.; Zhang, J.; Li, Y.; Yuan, H.; Zhou, J.; Wang, B. Long noncoding RNA Plnc1 controls adipocyte differentiation by regulating peroxisome proliferator-activated receptor γ. FASEB J. 2019, 33, 2396–2408. [Google Scholar] [CrossRef] [PubMed]
- Dreher, K.; Callis, J. Ubiquitin, Hormones and Biotic Stress in Plants. Ann. Bot.-Lond. 2007, 99, 787–822. [Google Scholar] [CrossRef] [PubMed]
- Kornitzer, D.; Ciechanover, A. Modes of regulation of ubiquitin-mediated protein degradation. J. Cell. Physiol. 2000, 182, 1–11. [Google Scholar] [CrossRef]
- Huo, D.; Su, F.; Cui, W.; Liu, S.; Zhang, L.; Yang, H.; Sun, L. Heat stress and evisceration caused lipid metabolism and neural transduction changes in sea cucumber: Evidence from metabolomics. Mar. Pollut. Bull. 2022, 182, 113993. [Google Scholar] [CrossRef]
- Yao, H.; Li, X.; Chen, Y.; Liang, G.; Gao, G.; Wang, H.; Wang, C.; Mu, C. Metabolic Changes in Scylla paramamosain During Adaptation to an Acute Decrease in Salinity. Front. Mar. Sci. 2021, 8, 734519. [Google Scholar] [CrossRef]
- Rong, D.; Sun, H.; Li, Z.; Liu, S.; Dong, C.; Fu, K.; Tang, W.; Cao, H. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget 2017, 8, 73271–73281. [Google Scholar] [CrossRef]
- Chen, L. Non-canonical Hippo signaling regulates immune responses. Adv. Immunol. 2019, 144, 87–119. [Google Scholar] [CrossRef]
- Wang, H.; Dong, Q.; Duan, D.; Zhao, S.; Li, M.; van Nocker, S.; Ma, F.; Mao, K. Comprehensive genomic analysis of the TYROSINE AMINOTRANSFERASE (TAT) genes in apple (Malus domestica) allows the identification of MdTAT2 conferring tolerance to drought and osmotic stresses in plants. Plant Physiol. Bioch. 2018, 133, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Xiao, J.; Chen, X.; Hu, X.; Li, R.; Chen, S.; Feng, X.; Shen, S.; Ma, H.; Zhuang, J.; et al. G protein pathway suppressor 2 enhanced the renal large-conductance Ca2+-activated potassium channel expression via inhibiting ERK1/2 signaling pathway. Am. J. Physiol.-Renal. 2018, 315, F503–F511. [Google Scholar] [CrossRef]
- Dzgoev, S.G. Selective V2-Agonist of Vasopressin Desmopressin Stimulates Activity of Serum Hyaluronidase. Bull. Exp. Biol. Med. 2015, 159, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Stridh, S.; Palm, F.; Hansell, P. Renal interstitial hyaluronan: Functional aspects during normal and pathological conditions. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2012, 302, R1235–R1249. [Google Scholar] [CrossRef]
- Meng, J.; Zhu, Q.; Zhang, L.; Li, C.; Li, L.; She, Z.; Huang, B.; Zhang, G. Genome and transcriptome analyses provide insight into the euryhaline adaptation mechanism of Crassostrea gigas. PLoS ONE 2013, 8, e58563. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Gene Name | Sequences (5′→3′) |
|---|---|---|
| mRNA | E3 ubiquitin-protein ligase UBR4 (UBR4) | F: TCTGGAGGTACTGGATGGAGCATTC R: GGAGAGGATGGCAGAGTGAGGAG |
| tyrosine aminotransferase (TAT) | F: TAGCGGAGTCTCTGGGCATACAC R: TGGACGGACTGTTGACAATAATGGC | |
| galectin-9 (Gal9) | F: GGGACTGAGGAGACTGCCATACC R: GTGTTTGTCGTTCACATCCACCTTG | |
| serine racemase (SR) | F: TGTGGAGGTGGAGGACTGGTTG R: GGTTGGTTCCACTGCATACACTCTG | |
| cysteine sulfinic acid decarboxylase (CASD) | F: CCTGACGAGTTGACTGCCATTCTG R: TGACGCTGTACTTGACGATCTTGTG | |
| ∆1-pyrroline-5-carboxylate synthase (P5CS) | F: CAAGCAAAAATGAACGG R: CCAAGGGAGAGACCACA | |
| hyaluronidase 4 (HYAL4) | F: ATCAGCAAACAAGCACCGTTCAATC R: CGTATCCAGCAAGGAGAGGTTCAAG | |
| lncRNA | MSTRG.38153.5 | F: CAGGGTGGGTGTCCATGATTTAAGG R: CCGTGAAGTGGTTTGCGTGAAATG |
| MSTRG.20715.2 | F: AAGGGAGGTGCAATGTCGATGTG R: GGACCTCTGCCTTATGTGTTACTGG | |
| MSTRG.87615.2 | F: GCAGAGTCTCTAGCACTGTGTCTTG R: GTCATGGTCAGGCTCGATCACAC | |
| MSTRG.4211.1 | F: GGTGCCGAAGATGCCATCAGTC R: CCTGGTGTGTTAATCGCCTCCTTC | |
| MSTRG.78885.7 | F: AGGACGCTGATTACTCGATTAACGG R: GAGGAGAGTGTGAACTGTGCAAGAC | |
| MSTRG.52430.2 | F: ACCCAGCCCTATGTTGCCATTTG R: TTCTGTCTGTGTGCAGTGATTCTCC | |
| MSTRG.14539.4 | F: AGTCCTACTGGCTGCTGATCCG R: CCTGCTGTGTTTAGACAACCTGGAG | |
| circRNA | ctg122:1490189|1502281 | F: TTCAATCCAGCGAACTGCGA R: GTTCGAGAGTTTCGCACGC |
| ctg2216:18009|78918 | F: GCTGATGTAGTCAGGAAGACGG R: GACGTAGGTCGGGTCATGGA | |
| miRNA | novel_miR_376 | F: AATTGTTTGACCGAGGATGGTCA |
| novel_miR_73 | F: AATCCAGTGACTGGGTGTGGTA | |
| novel_miR_396 | F: AAGCGACCGGTGTCAGGATAA | |
| qRT-PCR of control | Ribosomal protein S9 (RS9) | F: TGAAGTCTGGCGTGTCAAGT R: CGTCTCAAAAGGGCATTACC |
| U6 snRNA (U6) | F: CTCGCTTCGGCAGCACA R: AACGCTTCACGAATTTGCGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, W.; Dong, Y.; Geng, Y.; Bi, S.; Liu, Z.; Zhou, L.; Sun, X.; Xia, S.; Chi, C.; Wu, B. Comprehensive Analysis of Whole-Transcriptome Profiles in Response to Acute Hypersaline Challenge in Chinese Razor Clam Sinonovacula constricta. Biology 2023, 12, 106. https://doi.org/10.3390/biology12010106
Cao W, Dong Y, Geng Y, Bi S, Liu Z, Zhou L, Sun X, Xia S, Chi C, Wu B. Comprehensive Analysis of Whole-Transcriptome Profiles in Response to Acute Hypersaline Challenge in Chinese Razor Clam Sinonovacula constricta. Biology. 2023; 12(1):106. https://doi.org/10.3390/biology12010106
Chicago/Turabian StyleCao, Wei, Yinghui Dong, Yusong Geng, Siqi Bi, Zhihong Liu, Liqing Zhou, Xiujun Sun, Sudong Xia, Changfeng Chi, and Biao Wu. 2023. "Comprehensive Analysis of Whole-Transcriptome Profiles in Response to Acute Hypersaline Challenge in Chinese Razor Clam Sinonovacula constricta" Biology 12, no. 1: 106. https://doi.org/10.3390/biology12010106
APA StyleCao, W., Dong, Y., Geng, Y., Bi, S., Liu, Z., Zhou, L., Sun, X., Xia, S., Chi, C., & Wu, B. (2023). Comprehensive Analysis of Whole-Transcriptome Profiles in Response to Acute Hypersaline Challenge in Chinese Razor Clam Sinonovacula constricta. Biology, 12(1), 106. https://doi.org/10.3390/biology12010106