
Drug Discovery Strategies for Inherited Retinal Degenerations



{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Targets and Drug Discoveries for Modulating Pathways in Photoreceptors and Retinal Pigment Epithelial Cells
3. Targets and Drug Discoveries for RGC Degeneration
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodieck, R.W.; Rodieck, R.W. The First steps in Seeing; Sinauer Associates: Sunderland, MA, USA, 1998; Volume 1. [Google Scholar]
- Lamba, D.; Karl, M.; Reh, T. Neural Regeneration and Cell Replacement: A View from the Eye. Cell Stem Cell 2008, 2, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A. Understanding Glaucomatous Optic Neuropathy: The Synergy Between Clinical Observation and Investigation. Annu. Rev. Vis. Sci. 2016, 2, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Hejtmancik, J.F.; Daiger, S.P. Understanding the genetic architecture of human retinal degenerations. Proc. Natl. Acad. Sci. USA 2020, 117, 3904–3906. [Google Scholar] [CrossRef]
- Wright, A.F.; Chakarova, C.F.; El-Aziz, M.M.A.; Bhattacharya, S.S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010, 11, 273–284. [Google Scholar] [CrossRef]
- Okawa, H.; Sampath, A.P.; Laughlin, S.B.; Fain, G.L. ATP Consumption by Mammalian Rod Photoreceptors in Darkness and in Light. Curr. Biol. 2008, 18, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.B. Warburg’s vision. eLife 2017, 6, e29217. [Google Scholar] [CrossRef]
- Clarke, G.; Collins, R.A.; Leavitt, B.R.; Andrews, D.F.; Hayden, M.; Lumsden, C.J.; McInnes, R.R. A one-hit model of cell death in inherited neuronal degenerations. Nature 2000, 406, 195–199. [Google Scholar] [CrossRef]
- Yao, J.; Jia, L.; Feathers, K.; Lin, C.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Zacks, D.N. Autophagy-mediated catabolism of visual transduction proteins prevents retinal degeneration. Autophagy 2016, 12, 2439–2450. [Google Scholar] [CrossRef]
- Wang, W.; Kini, A.; Wang, Y.; Liu, T.; Chen, Y.; Vukmanic, E.; Emery, D.; Liu, Y.; Lu, X.; Jin, L.; et al. Metabolic Deregulation of the Blood-Outer Retinal Barrier in Retinitis Pigmentosa. Cell Rep. 2019, 28, 1323–1334.e4. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.D.; de Guimaraes, T.A.C.; Georgiou, M.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Current management and clinical trials. Br. J. Ophthalmol. 2022, 106, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Berson, D.M. Strange vision: Ganglion cells as circadian photoreceptors. Trends Neurosci. 2003, 26, 314–320. [Google Scholar] [CrossRef]
- Wong, K.Y.; Dunn, F.A.; Berson, D.M. Photoreceptor Adaptation in Intrinsically Photosensitive Retinal Ganglion Cells. Neuron 2005, 48, 1001–1010. [Google Scholar] [CrossRef]
- Kirches, E. LHON: Mitochondrial Mutations and More. Curr. Genom. 2011, 12, 44–54. [Google Scholar] [CrossRef]
- Lenaers, G.; Hamel, C.P.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef]
- Wolf, C.; Gramer, E.; Müller-Myhsok, B.; Pasutto, F.; Reinthal, E.; Wissinger, B.; Weisschuh, N. Evaluation of nine candidate genes in patients with normal tension glaucoma: A case control study. BMC Med Genet. 2009, 10, 91. [Google Scholar] [CrossRef]
- Yuwaiman, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited mitochondrial optic neuropathies. J. Med Genet. 2008, 46, 145–158. [Google Scholar] [CrossRef]
- Kleesattel, D.; Crish, S.D.; Inman, D.M. Decreased Energy Capacity and Increased Autophagic Activity in Optic Nerve Axons With Defective Anterograde Transport. Investig. Opthalmology Vis. Sci. 2015, 56, 8215–8227. [Google Scholar] [CrossRef]
- Baltan, S.; Inman, D.; Danilov, C.A.; Morrison, R.S.; Calkins, D.J.; Horner, P.J. Metabolic Vulnerability Disposes Retinal Ganglion Cell Axons to Dysfunction in a Model of Glaucomatous Degeneration. J. Neurosci. 2010, 30, 5644–5652. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Palmer, P.G.; Smith, M.A.; Gevorgyan, V.; Wilson, G.N.; Crish, S.D.; Inman, D.M. Structural and Functional Rescue of Chronic Metabolically Stressed Optic Nerves through Respiration. J. Neurosci. 2018, 38, 5122–5139. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, V.; Rezania, F.; A Trounce, I.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.C.; Campanelli, J.; Sande, P.; Sáenz, D.A.; Sarmiento, M.I.K.; Rosenstein, R.E. Retinal Oxidative Stress Induced by High Intraocular Pressure. Free Radic. Biol. Med. 2004, 37, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, L.; Morrison, R.S.; Horner, P.J.; Inman, D.M. Mitochondrial Morphology Differences and Mitophagy Deficit in Murine Glaucomatous Optic Nerve. Investig. Opthalmology Vis. Sci. 2015, 56, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Kim, K.; Noh, Y.H.; Hoshijima, M.; Lukas, T.J.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A. Increased mitochondrial fission and volume density by blocking glutamate excitotoxicity protect glaucomatous optic nerve head astrocytes. Glia 2015, 63, 736–753. [Google Scholar] [CrossRef]
- Fernandes, K.A.; Harder, J.M.; Fornarola, L.B.; Freeman, R.S.; Clark, A.F.; Pang, I.-H.; John, S.W.; Libby, R.T. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol. Dis. 2012, 46, 393–401. [Google Scholar] [CrossRef]
- Dreyer, E.B. A Proposed Role for Excitotoxicity in Glaucoma. J. Glaucoma 1998, 7, 62–67. [Google Scholar] [CrossRef]
- Lotery, A.J. Glutamate excitotoxicity in glaucoma: Truth or fiction? Eye 2005, 19, 369–370. [Google Scholar] [CrossRef]
- Salt, T.E.; Cordeiro, M.F. Glutamate excitotoxicity in glaucoma: Throwing the baby out with the bathwater? Eye 2006, 20, 730–731. [Google Scholar] [CrossRef]
- Osborne, N.N.; Chidlow, G.; Wood, J.P.M. Glutamate excitotoxicity in glaucoma: Truth or fiction? By AJ Lotery. Eye 2006, 20, 1392–1394. [Google Scholar] [CrossRef]
- Gharahkhani, P.; Jorgenson, E.; Hysi, P.; Khawaja, A.P.; Pendergrass, S.; Han, X.; Ong, J.S.; Hewitt, A.W.; Segrè, A.V.; Rouhana, J.M.; et al. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 2021, 12, 1258. [Google Scholar] [CrossRef] [PubMed]
- Storgaard, L.; Tran, T.L.; Freiberg, J.C.; Hauser, A.S.; Kolko, M. Glaucoma Clinical Research: Trends in Treatment Strategies and Drug Development. Front. Med. 2021, 8, 733080. [Google Scholar] [CrossRef] [PubMed]
- Faiq, M.A.; Wollstein, G.; Schuman, J.S.; Chan, K.C. Cholinergic nervous system and glaucoma: From basic science to clinical applications. Prog. Retin. Eye Res. 2019, 72, 100767. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Bojanowski, C.M.; Chan, C.-C. Genetic factors of age-related macular degeneration. Prog. Retin. Eye Res. 2004, 23, 229–249. [Google Scholar] [CrossRef]
- Liu, H.; Tang, J.; Du, Y.; Lee, C.A.; Golczak, M.; Muthusamy, A.; Antonetti, D.A.; Veenstra, A.A.; Amengual, J.; von Lintig, J.; et al. Retinylamine Benefits Early Diabetic Retinopathy in Mice. J. Biol. Chem. 2015, 290, 21568–21579. [Google Scholar] [CrossRef]
- Yokomizo, H.; Maeda, Y.; Park, K.; Clermont, A.C.; Hernandez, S.L.; Fickweiler, W.; Li, Q.; Wang, C.-H.; Paniagua, S.M.; Simao, F.; et al. Retinol binding protein 3 is increased in the retina of patients with diabetes resistant to diabetic retinopathy. Sci. Transl. Med. 2019, 11, eaau6627. [Google Scholar] [CrossRef]
- Collin, R.W.; Garanto, A. Applications of antisense oligonucleotides for the treatment of inherited retinal diseases. Curr. Opin. Ophthalmol. 2017, 28, 260–266. [Google Scholar] [CrossRef]
- Bull, N.D.; Martin, K.R. Concise Review: Toward Stem Cell-Based Therapies for Retinal Neurodegenerative Diseases. Stem Cells 2011, 29, 1170–1175. [Google Scholar] [CrossRef]
- Garafalo, A.V.; Cideciyan, A.V.; Héon, E.; Sheplock, R.; Pearson, A.; Yu, C.W.; Sumaroka, A.; Aguirre, G.D.; Jacobson, S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020, 77, 100827. [Google Scholar] [CrossRef]
- Shen, Y. Stem cell therapies for retinal diseases: From bench to bedside. J. Mol. Med. 2020, 98, 1347–1368. [Google Scholar] [CrossRef]
- Uyama, H.; Mandai, M.; Takahashi, M. Stem---cell---based therapies for retinal degenerative diseases: Current challenges in the establishment of new treatment strategies. Dev. Growth Differ. 2021, 63, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, Q.; Temple, S. Stem cell therapies for retinal diseases: Recapitulating development to replace degenerated cells. Development 2017, 144, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Arshavsky, V.Y.; Lamb, T.D.; Pugh, E.N. G Proteins and Phototransduction. Annu. Rev. Physiol. 2002, 64, 153–187. [Google Scholar] [CrossRef] [PubMed]
- Ridge, K.D.; Abdulaev, N.G.; Sousa, M.; Palczewski, K. Phototransduction: Crystal clear. Trends Biochem. Sci. 2003, 28, 479–487. [Google Scholar] [CrossRef]
- Chang, B.; Hawes, N.; Hurd, R.; Davisson, M.; Nusinowitz, S.; Heckenlively, J. Retinal degeneration mutants in the mouse. Vis. Res. 2002, 42, 517–525. [Google Scholar] [CrossRef]
- Dryja, T.P.; Rucinski, D.E.; Chen, S.H.; Berson, E.L. Frequency of mutations in the gene encoding the alpha subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1859–1865. [Google Scholar]
- Paquet-Durand, F.; Hauck, S.; Van Veen, T.; Ueffing, M.; Ekström, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Niemi, G.A.; Reh, T.A.; Hurley, J.B. Leber congenital amaurosis linked to AIPL1: A mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13897–13902. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S.; Doan, T.; Rieke, F.; Detwiler, P.B.; Frederick, J.M.; Baehr, W. Deletion of PrBP/δ impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl. Acad. Sci. USA 2007, 104, 8857–8862. [Google Scholar] [CrossRef]
- Xu, J.; Morris, L.; Thapa, A.; Ma, H.; Michalakis, S.; Biel, M.; Baehr, W.; Peshenko, I.V.; Dizhoor, A.; Ding, X.-Q. cGMP accumulation causes photoreceptor degeneration in CNG channel deficiency: Evidence of cGMP cytotoxicity independently of enhanced CNG channel function. J. Neurosci. 2013, 33, 14939–14948. [Google Scholar] [CrossRef]
- Paquet-Durand, F.; Marigo, V.; Ekström, P. RD Genes Associated with High Photoreceptor cGMP-Levels (Mini-Review). Adv. Exp. Med. Biol. 2019, 1185, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tsang, S.H.; Chen, J. Two pathways of rod photoreceptor cell death induced by elevated cGMP. Hum. Mol. Genet. 2017, 26, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Strassmaier, T.; Brady, J.; Karpen, J. The Pharmacology of Cyclic Nucleotide-Gated Channels: Emerging from the Darkness. Curr. Pharm. Des. 2006, 12, 3597–3613. [Google Scholar] [CrossRef] [PubMed]
- Frasson, M.; Sahel, J.A.; Fabre, M.; Simonutti, M.; Dreyfus, H.; Picaud, S. Retinitis pigmentosa: Rod photoreceptor rescue by a calcium-channel blocker in the rd mouse. Nat. Med. 1999, 5, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Vighi, E.; Trifunovic, D.; Veiga-Crespo, P.; Rentsch, A.; Hoffmann, D.; Sahaboglu, A.; Strasser, T.; Kulkarni, M.; Bertolotti, E.; van den Heuvel, A.; et al. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2997–E3006. [Google Scholar] [CrossRef]
- Tolone, A.; Haq, W.; Fachinger, A.; Rentsch, A.; Herberg, F.W.; Schwede, F.; Paquet-Durand, F. Retinal degeneration: Multilevel protection of photoreceptor and ganglion cell viability and function with the novel PKG inhibitor CN238. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kiser, P.D.; Golczak, M.; Maeda, A.; Palczewski, K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2012, 1821, 137–151. [Google Scholar] [CrossRef]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the Function of Rim Protein in Photoreceptors and Etiology of Stargardt’s Disease from the Phenotype in abcr Knockout Mice. Cell 1999, 98, 13–23. [Google Scholar] [CrossRef]
- Lenis, T.L.; Hu, J.; Ng, S.Y.; Jiang, Z.; Sarfare, S.; Lloyd, M.B.; Esposito, N.J.; Samuel, W.; Jaworski, C.; Bok, D.; et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E11120–E11127. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Fishkin, N.; Zhou, J.; Cai, B.; Jang, Y.P.; Krane, S.; Itagaki, Y.; Nakanishi, K. A2E, a byproduct of the visual cycle. Vis. Res. 2003, 43, 2983–2990. [Google Scholar] [CrossRef]
- Chen, Y.; Okano, K.; Maeda, T.; Chauhan, V.; Golczak, M.; Maeda, A.; Palczewski, K. Mechanism of All-trans-retinal Toxicity with Implications for Stargardt Disease and Age-related Macular Degeneration*. J. Biol. Chem. 2012, 287, 5059–5069. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A Membrane Receptor for Retinol Binding Protein Mediates Cellular Uptake of Vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 Disrupts Vitamin A Uptake Homeostasis in a STRA6-Deficient Animal Model for Matthew-Wood Syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Clarke, O.B.; Kim, J.; Stowe, S.; Kim, Y.-K.; Assur, Z.; Cavalier, M.; Godoy-Ruiz, R.; von Alpen, D.C.; Manzini, C.; et al. Structure of the STRA6 receptor for retinol uptake. Science 2016, 353, aad8266. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S. Retinol-Binding Protein: The Serum Transport Protein for Vitamin A*. Endocr. Rev. 1989, 10, 308–316. [Google Scholar] [CrossRef]
- Katz, M.L.; Redmond, T.M. Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3023–3030. [Google Scholar]
- Bui, T.V.; Han, Y.; Reid, S.N.M.; Phan, K.B.; Mata, N.L. Reduction of Serum Retinol Binding Protein Arrests the Accumulation of Toxic Retinal Fluorophores: A Nonpharmacological Approach. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3244. [Google Scholar]
- Hussain, R.M.; Gregori, N.Z.; Ciulla, T.A.; Lam, B.L. Pharmacotherapy of retinal disease with visual cycle modulators. Expert Opin. Pharmacother. 2018, 19, 471–481. [Google Scholar] [CrossRef]
- Radu, R.A.; Han, Y.; Bui, T.V.; Nusinowitz, S.; Bok, D.; Lichter, J.; Widder, K.; Travis, G.H.; Mata, N.L. Reductions in Serum Vitamin A Arrest Accumulation of Toxic Retinal Fluorophores: A Potential Therapy for Treatment of Lipofuscin-Based Retinal Diseases. Investig. Opthalmology Vis. Sci. 2005, 46, 4393–4401. [Google Scholar] [CrossRef]
- Golczak, M.; Kuksa, V.; Maeda, T.; Moise, A.R.; Palczewski, K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc. Natl. Acad. Sci. USA 2005, 102, 8162–8167. [Google Scholar] [CrossRef]
- Kubota, R.; Boman, N.L.; David, R.; Mallikaarjun, S.; Patil, S.; Birch, D. Safety and effect on rod function of acu-4429, a novel small-molecule visual cycle modulator. Retina 2012, 32, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Priefer, R. Retinol binding protein 4 antagonists and protein synthesis inhibitors: Potential for therapeutic development. Eur. J. Med. Chem. 2021, 226, 113856. [Google Scholar] [CrossRef] [PubMed]
- Berni, R.; Formelli, F. In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett. 1992, 308, 43–45. [Google Scholar] [CrossRef]
- Dobri, N.; Qin, Q.; Kong, J.; Yamamoto, K.; Liu, Z.; Moiseyev, G.; Ma, J.-X.; Allikmets, R.; Sparrow, J.R.; Petrukhin, K. A1120, a Nonretinoid RBP4 Antagonist, Inhibits Formation of Cytotoxic Bisretinoids in the Animal Model of Enhanced Retinal Lipofuscinogenesis. Investig. Opthalmology Vis. Sci. 2013, 54, 85–95. [Google Scholar] [CrossRef]
- Issa, P.C.; Barnard, A.R.; Herrmann, P.; Washington, I.; MacLaren, R.E. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc. Natl. Acad. Sci. USA 2015, 112, 8415–8420. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.; DeBartolomeo, G.; Washington, I.; Saad, L. ALK-001 (C20-D3-Vitamin A) slows the growth of atrophic lesions in ABCA4-related Stargardt Disease: Results of a Phase 2 placebo-controlled clinical trial (TEASE study). Investig. Ophthalmol. Vis. Sci. 2022, 63, 38. [Google Scholar]
- Bavik, C.; Henry, S.H.; Zhang, Y.; Mitts, K.; McGinn, T.; Budzynski, E.; Pashko, A.; Lieu, K.L.; Zhong, S.; Blumberg, B.; et al. Visual Cycle Modulation as an Approach toward Preservation of Retinal Integrity. PLoS ONE 2015, 10, e0124940. [Google Scholar] [CrossRef]
- Maeda, A.; Golczak, M.; Chen, Y.; Okano, K.; Kohno, H.; Shiose, S.; Ishikawa, K.; Harte, W.; Palczewska, G.; Maeda, T.; et al. Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol. 2011, 8, 170–178. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in Mice Induced by Disrupted All-trans-retinal Clearance. J. Biol. Chem. 2008, 283, 26684–26693. [Google Scholar] [CrossRef]
- Chen, Y.; Palczewska, G.; Mustafi, D.; Golczak, M.; Dong, Z.; Sawada, O.; Maeda, T.; Maeda, A.; Palczewski, K. Systems pharmacology identifies drug targets for Stargardt disease–associated retinal degeneration. J. Clin. Investig. 2013, 123, 5119–5134. [Google Scholar] [CrossRef]
- Li, Z.Y.; Kljavin, I.J.; Milam, A.H. Rod photoreceptor neurite sprouting in retinitis pigmentosa. J. Neurosci. 1995, 15, 5429–5438. [Google Scholar] [CrossRef] [PubMed]
- Alfinito, P.D.; Townes-Anderson, E. Activation of mislocalized opsin kills rod cells: A novel mechanism for rod cell death in retinal disease. Proc. Natl. Acad. Sci. USA 2002, 99, 5655–5660. [Google Scholar] [CrossRef] [PubMed]
- Nakao, T.; Tsujikawa, M.; Notomi, S.; Ikeda, Y.; Nishida, K. The Role of Mislocalized Phototransduction in Photoreceptor Cell Death of Retinitis Pigmentosa. PLoS ONE 2012, 7, e32472. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Ghosh, E.; Shukla, A.K. Emerging Approaches to GPCR Ligand Screening for Drug Discovery. Trends Mol. Med. 2015, 21, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Irwin, J.J.; Shoichet, B.K. Discovery of new GPCR ligands to illuminate new biology. Nat. Chem. Biol. 2017, 13, 1143–1151. [Google Scholar] [CrossRef]
- Milam, A.H.; Li, Z.Y.; Fariss, R.N. Histopathology of the human retina in retinitis pigmentosa. Prog. Retin. Eye Res. 1998, 17, 175–205. [Google Scholar] [CrossRef]
- Wang, W.; Lee, S.J.; Scott, P.A.; Lu, X.; Emery, D.; Liu, Y.; Ezashi, T.; Roberts, M.R.; Ross, J.W.; Kaplan, H.J.; et al. Two-Step Reactivation of Dormant Cones in Retinitis Pigmentosa. Cell Rep. 2016, 15, 372–385. [Google Scholar] [CrossRef]
- Ruggiero, L.; Connor, M.P.; Chen, J.; Langen, R.; Finnemann, S.C. Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5 −/− or Mfge8 −/− mouse retina. Proc. Natl. Acad. Sci. USA 2012, 109, 8145–8148. [Google Scholar] [CrossRef]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-Dependent Degradation of TXNIP upon Energy Stress Leads to Enhanced Glucose Uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef]
- Xu, L.; Ash, J.D. The Role of AMPK Pathway in Neuroprotection. Adv. Exp. Med. Biol. 2016, 854, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kong, L.; Wang, J.; Ash, J.D. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2018, 115, 10475–10480. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Bao, S.; Zhang, C.; Zhang, J.; Lv, J.; Li, X.; Chudhary, M.; Ren, X.; Kong, L. Stimulation of AMPK Prevents Diabetes-Induced Photoreceptor Cell Degeneration. Oxidative Med. Cell. Longev. 2021, 2021, 5587340. [Google Scholar] [CrossRef]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Zhang, L.; Du, J.; Justus, S.; Hsu, C.-W.; Bonet-Ponce, L.; Wu, W.-H.; Tsai, Y.-T.; Wu, W.-P.; Jia, Y.; Duong, J.K.; et al. Reprogramming metabolism by targeting sirtuin 6 attenuates retinal degeneration. J. Clin. Investig. 2016, 126, 4659–4673. [Google Scholar] [CrossRef]
- Fiorentino, F.; Mai, A.; Rotili, D. Emerging Therapeutic Potential of SIRT6 Modulators. J. Med. Chem. 2021, 64, 9732–9758. [Google Scholar] [CrossRef]
- Zode, G.S.; Bugge, K.E.; Mohan, K.; Grozdanic, S.D.; Peters, J.C.; Koehn, D.R.; Anderson, M.; Kardon, R.; Stone, E.M.; Sheffield, V.C. Topical Ocular Sodium 4-Phenylbutyrate Rescues Glaucoma in a Myocilin Mouse Model of Primary Open-Angle Glaucoma. Investig. Opthalmology Vis. Sci. 2012, 53, 1557–1565. [Google Scholar] [CrossRef]
- Hanrahan, J.W.; Sampson, H.M.; Thomas, D.Y. Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol. Sci. 2013, 34, 119–125. [Google Scholar] [CrossRef]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef]
- Noorwez, S.M.; Kuksa, V.; Imanishi, Y.; Zhu, L.; Filipek, S.; Palczewski, K.; Kaushal, S. Pharmacological Chaperone-mediated in Vivo Folding and Stabilization of the P23H-opsin Mutant Associated with Autosomal Dominant Retinitis Pigmentosa. J. Biol. Chem. 2003, 278, 14442–14450. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Zhou, Y.; Hajkova, D.; Miyagi, M.; Dinculescu, A.; Hauswirth, W.W.; Palczewski, K.; Geng, R.; Alagramam, K.N.; Isosomppi, J.; et al. Clarin-1, Encoded by the Usher Syndrome III Causative Gene, Forms a Membranous Microdomain: Possible role of clarin-1 in organizing the actin cytoskeleton. J. Biol. Chem. 2009, 284, 18980–18993. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Samardzija, M.; Yang, Z.; Grimm, C.; Jin, M. Pharmacological Amelioration of Cone Survival and Vision in a Mouse Model for Leber Congenital Amaurosis. J. Neurosci. 2016, 36, 5808–5819. [Google Scholar] [CrossRef]
- Wang, T.; Reingruber, J.; Woodruff, M.L.; Majumder, A.; Camarena, A.; Artemyev, N.; Fain, G.; Chen, J. The PDE6 mutation in the rd10 retinal degeneration mouse model causes protein mislocalization and instability and promotes cell death through increased ion influx. J. Biol. Chem. 2018, 293, 15332–15346. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.H.; Davenport, C.M.; Hennessey, J.C.; Maumenee, I.H.; Jacobson, S.G.; Heckenlively, J.R.; Nowakowski, R.; Fishman, G.; Gouras, P.; Nathans, J. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1991, 88, 6481–6485. [Google Scholar] [CrossRef]
- Sakami, S.; Maeda, T.; Bereta, G.; Okano, K.; Golczak, M.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G.; Palczewski, K. Probing Mechanisms of Photoreceptor Degeneration in a New Mouse Model of the Common Form of Autosomal Dominant Retinitis Pigmentosa due to P23H Opsin Mutations. J. Biol. Chem. 2011, 286, 10551–10567. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.E.; Gordon, J.W.; Pawlyk, B.S.; Roof, D.; Hayes, A.; Molday, R.S.; Mukai, S.; Cowley, G.S.; Berson, E.L.; Dryja, T.P. Transgenic mice with a rhodopsin mutation (Pro23His): A mouse model of autosomal dominant retinitis pigmentosa. Neuron 1992, 9, 815–830. [Google Scholar] [CrossRef]
- Chiang, W.-C.; Kroeger, H.; Sakami, S.; Messah, C.; Yasumura, D.; Matthes, M.T.; Coppinger, J.A.; Palczewski, K.; LaVail, M.M.; Lin, J.H. Robust Endoplasmic Reticulum-Associated Degradation of Rhodopsin Precedes Retinal Degeneration. Mol. Neurobiol. 2015, 52, 679–695. [Google Scholar] [CrossRef]
- Li, S.; Izumi, T.; Hu, J.; Jin, H.H.; Siddiqui, A.-A.A.; Jacobson, S.; Bok, D.; Jin, M. Rescue of Enzymatic Function for Disease-associated RPE65 Proteins Containing Various Missense Mutations in Non-active Sites. J. Biol. Chem. 2014, 289, 18943–18956. [Google Scholar] [CrossRef]
- Adato, A.; Vreugde, S.; Joensuu, T.; Avidan, N.; Hamalainen, R.H.; Belenkiy, O.; Olender, T.; Bonne-Tamir, B.; Ben-Asher, E.; Espinós, C.; et al. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur. J. Hum. Genet. 2002, 10, 339–350. [Google Scholar] [CrossRef]
- Geng, R.; Geller, S.F.; Hayashi, T.; Ray, C.A.; Reh, T.A.; Bermingham-McDonogh, O.; Jones, S.M.; Wright, C.G.; Melki, S.; Imanishi, Y.; et al. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum. Mol. Genet. 2009, 18, 2748–2760. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Melki, S.; Chen, D.H.-C.; Tian, G.; Furness, D.N.; Oshima-Takago, T.; Neef, J.; Moser, T.; Askew, C.; Horwitz, G.; et al. The Mechanosensory Structure of the Hair Cell Requires Clarin-1, a Protein Encoded by Usher Syndrome III Causative Gene. J. Neurosci. 2012, 32, 9485–9498. [Google Scholar] [CrossRef]
- Tian, G.; Lee, R.; Ropelewski, P.; Imanishi, Y. Impairment of Vision in a Mouse Model of Usher Syndrome Type III. Investig. Opthalmology Vis. Sci. 2016, 57, 866. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.R.; Zhou, G.; Huang, D.; Davis, J.R.; Möller, C.; Jacobson, S.G.; Kimberling, W.J.; Sumegi, J. Usher Syndrome Type III: Revised Genomic Structure of the USH3 Gene and Identification of Novel Mutations. Am. J. Hum. Genet. 2002, 71, 607–617. [Google Scholar] [CrossRef][Green Version]
- Isosomppi, J.; Västinsalo, H.; Geller, S.F.; Heon, E.; Flannery, J.G.; Sankila, E.-M. Disease-causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane. Mol. Vis. 2009, 15, 1806–1818. [Google Scholar] [PubMed]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos---Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef]
- Samardzija, M.; Von Lintig, J.; Tanimoto, N.; Oberhauser, V.; Thiersch, M.; Remé, C.E.; Seeliger, M.; Grimm, C.; Wenzel, A. R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum. Mol. Genet. 2007, 17, 281–292. [Google Scholar] [CrossRef]
- Burns, J.N.; Turnage, K.C.; Walker, C.A.; Lieberman, R.L. The Stability of Myocilin Olfactomedin Domain Variants Provides New Insight into Glaucoma as a Protein Misfolding Disorder. Biochemistry 2011, 50, 5824–5833. [Google Scholar] [CrossRef]
- Kusaczuk, M. Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef]
- Boatright, J.H.; Moring, A.G.; McElroy, C.; Phillips, M.J.; Do, V.T.; Chang, B.; Hawes, N.L.; Boyd, A.P.; Sidney, S.S.; Stewart, R.E.; et al. Tool from ancient pharmacopoeia prevents vision loss. Mol. Vis. 2006, 12, 1706–1714. [Google Scholar]
- Behnen, P.; Felline, A.; Comitato, A.; Di Salvo, M.T.; Raimondi, F.; Gulati, S.; Kahremany, S.; Palczewski, K.; Marigo, V.; Fanelli, F. A Small Chaperone Improves Folding and Routing of Rhodopsin Mutants Linked to Inherited Blindness. iScience 2018, 4, 1–19. [Google Scholar] [CrossRef]
- Van Hooser, J.P.; Aleman, T.S.; He, Y.-G.; Cideciyan, A.V.; Kuksa, V.; Pittler, S.J.; Stone, E.M.; Jacobson, S.G.; Palczewski, K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc. Natl. Acad. Sci. USA 2000, 97, 8623–8628. [Google Scholar] [CrossRef] [PubMed]
- Van Hooser, J.P.; Liang, Y.; Maeda, T.; Kuksa, V.; Jang, G.-F.; He, Y.-G.; Rieke, F.; Fong, H.K.W.; Detwiler, P.B.; Palczewski, K. Recovery of Visual Functions in a Mouse Model of Leber Congenital Amaurosis. J. Biol. Chem. 2002, 277, 19173–19182. [Google Scholar] [CrossRef] [PubMed]
- Koenekoop, R.K.; Sui, R.; Sallum, J.; Born, L.I.V.D.; Ajlan, R.; Khan, A.; Hollander, A.I.D.; Cremers, F.P.M.; Mendola, J.D.; Bittner, A.K.; et al. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: An open-label phase 1b trial. Lancet 2014, 384, 1513–1520. [Google Scholar] [CrossRef]
- Vats, A.; Xi, Y.; Feng, B.; Clinger, O.D.; Leger, A.J.S.; Liu, X.; Ghosh, A.; Dermond, C.D.; Lathrop, K.L.; Tochtrop, G.P.; et al. Nonretinoid chaperones improve rhodopsin homeostasis in a mouse model of retinitis pigmentosa. JCI Insight 2022, 7, e153717. [Google Scholar] [CrossRef] [PubMed]
- Alagramam, K.N.; Gopal, S.R.; Geng, R.; Chen, D.H.-C.; Nemet, I.; Lee, R.; Tian, G.; Miyagi, M.; Malagu, K.F.; Lock, C.J.; et al. A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat. Chem. Biol. 2016, 12, 444–451. [Google Scholar] [CrossRef]
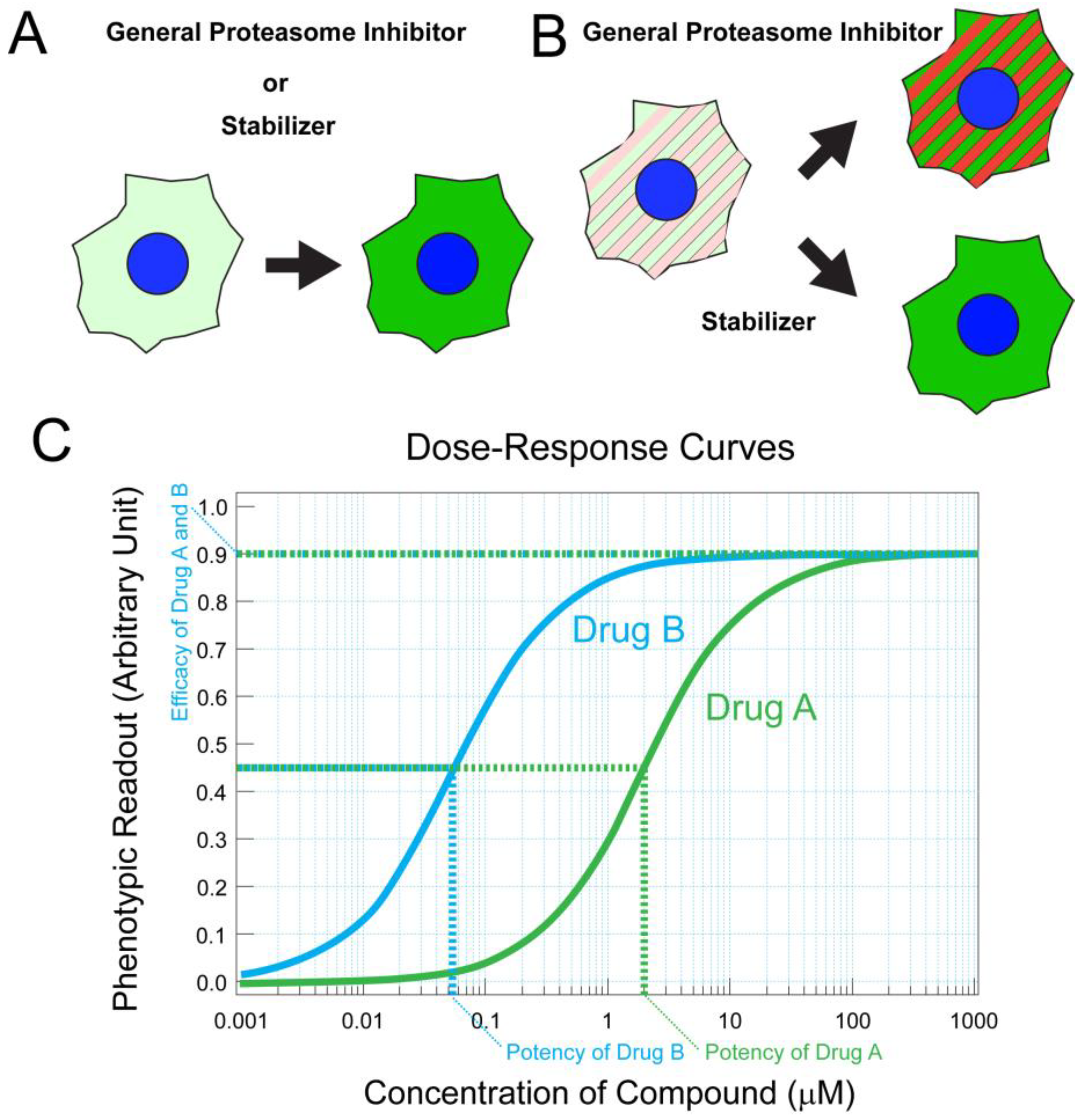
- Lobanova, E.S.; Finkelstein, S.; Skiba, N.P.; Arshavsky, V.Y. Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9986–9991. [Google Scholar] [CrossRef]
- Lee, J.N.; Kim, S.-G.; Lim, J.-Y.; Kim, S.-J.; Choe, S.-K.; Park, R. Proteasome inhibitors induce auditory hair cell death through peroxisome dysfunction. Biochem. Biophys. Res. Commun. 2015, 456, 269–274. [Google Scholar] [CrossRef]
- Meissner, F.; Geddes-McAlister, J.; Mann, M.; Bantscheff, M. The emerging role of mass spectrometry-based proteomics in drug discovery. Nat. Rev. Drug Discov. 2022, 21, 637–654. [Google Scholar] [CrossRef]
- Arango-Gonzalez, B.; Sen, M.; Guarascio, R.; Ziaka, K.; del Amo, E.M.; Hau, K.; Poultney, H.; Asfahani, R.; Urtti, A.; Chou, T.-F.; et al. Inhibition of VCP preserves retinal structure and function in autosomal dominant retinal degeneration. bioRxiv 2020. [Google Scholar] [CrossRef]
- Newman, N.J. Hereditary Optic Neuropathies: From the Mitochondria to the Optic Nerve. Am. J. Ophthalmol. 2005, 140, 517–523. [Google Scholar] [CrossRef] [PubMed]
- E Ogden, T.; Miller, R.F. Studies of the optic nerve of the rhesus monkey: Nerve fiber spectrum and physiological properties. Vis. Res. 1966, 6, 485–506. [Google Scholar] [CrossRef]
- Hollander, H.; Makarov, F.; Stefani, F.; Stone, J. Evidence of Constriction of Optic Nerve Axons at the Lamina cribrosa in the Normotensive Eye in Humans and Other Mammals. Ophthalmic Res. 1995, 27, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Bristow, E.A.; Griffiths, P.G.; Andrews, R.M.; Johnson, M.A.; Turnbull, D.M. The Distribution of Mitochondrial Activity in Relation to Optic Nerve Structure. Arch. Ophthalmol. 2002, 120, 791–796. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Das, A.; Bell, C.M.; Berlinicke, C.A.; Marsh-Armstrong, N.; Zack, D.J. Programmed switch in the mitochondrial degradation pathways during human retinal ganglion cell differentiation from stem cells is critical for RGC survival. Redox Biol. 2020, 34, 101465. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.-E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome–lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Esteban-Martínez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Héon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-Onset Primary Open-Angle Glaucoma Caused by Mutations in Optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Varma, R.; Tielsch, J.; Katz, J.; Sommer, A.; Gilbert, D.L. The relationship between optic disc area and open-angle glaucoma: The Baltimore Eye Survey. J. Glaucoma 1999, 8, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Ritch, R.; Darbro, B.; Menon, G.; Khanna, C.L.; Solivan-Timpe, F.; Roos, B.R.; Sarfarzi, M.; Kawase, K.; Yamamoto, T.; Robin, A.L.; et al. TBK1Gene Duplication and Normal-Tension Glaucoma. JAMA Ophthalmol. 2014, 132, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xie, X.; Wang, Y.; Liu, J.; Cheng, X.; Guo, Y.; Gong, Y.; Hu, S.; Pan, L. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat. Commun. 2016, 7, 12708. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, Y.; Iejima, D.; Kobayashi, H.; Chi, Z.-L.; Kawase, K.; Yamamoto, T.; Seki, T.; Yuasa, S.; Fukuda, K.; Iwata, T. Enhanced optineurin E50K–TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum. Mol. Genet. 2013, 22, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.-L.; Akahori, M.; Obazawa, M.; Minami, M.; Noda, T.; Nakaya, N.; Tomarev, S.; Kawase, K.; Yamamoto, T.; Noda, S.; et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum. Mol. Genet. 2010, 19, 2606–2615. [Google Scholar] [CrossRef] [PubMed]
- Bell, J. Amlexanox for the Treatment of Recurrent Aphthous Ulcers. Clin. Drug Investig. 2005, 25, 555–566. [Google Scholar] [CrossRef]
- Reilly, S.M.; Chiang, S.-H.; Decker, S.J.; Chang, L.; Uhm, M.; Larsen, M.J.; Rubin, J.R.; Mowers, J.; White, N.M.; Hochberg, I.; et al. An inhibitor of the protein kinases TBK1 and IKK-ɛ improves obesity-related metabolic dysfunctions in mice. Nat. Med. 2013, 19, 313–321. [Google Scholar] [CrossRef]
- Minegishi, Y.; Nakayama, M.; Iejima, D.; Kawase, K.; Iwata, T. Significance of optineurin mutations in glaucoma and other diseases. Prog. Retin. Eye Res. 2016, 55, 149–181. [Google Scholar] [CrossRef]
- Clark, K.; Plater, L.; Peggie, M.; Cohen, P. Use of the Pharmacological Inhibitor BX795 to Study the Regulation and Physiological Roles of TBK1 and IκB Kinase ϵ: A distinct upstream kinase mediates Ser-172 phosphorylation and activation. J. Biol. Chem. 2009, 284, 14136–14146. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Larrea, D.; Pera, M.; Gonnelli, A.; Quintana-Cabrera, R.; Akman, H.O.; Guardia-Laguarta, C.; Velasco, K.R.; Area-Gomez, E.; Dal Bello, F.; De Stefani, D.; et al. MFN2 mutations in Charcot–Marie–Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum. Mol. Genet. 2019, 28, 1782–1800. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.-W.; Wang, J.-K.; Wang, L.-C.; Guo, Q.; Liu, T.-T.; Wang, F.-J.; Feng, N.; Zhang, X.-W.; Liao, L.-X.; Zhao, M.-M.; et al. Small molecule induces mitochondrial fusion for neuroprotection via targeting CK2 without affecting its conventional kinase activity. Signal Transduct. Target. Ther. 2021, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Malty, R.H.; Jessulat, M.; Jin, K.; Musso, G.; Vlasblom, J.; Phanse, S.; Zhang, Z.; Babu, M. Mitochondrial Targets for Pharmacological Intervention in Human Disease. J. Proteome Res. 2015, 14, 5–21. [Google Scholar] [CrossRef]
- Geyman, L.S.; Suwan, Y.; Garg, R.; Field, M.G.; Krawitz, B.D.; Mo, S.; Pinhas, A.; Ritch, R.; Rosen, R.B. Noninvasive Detection of Mitochondrial Dysfunction in Ocular Hypertension and Primary Open-angle Glaucoma. J. Glaucoma 2018, 27, 592–599. [Google Scholar] [CrossRef]
- Izzotti, A.; Bagnis, A.; Saccὰ, S. The role of oxidative stress in glaucoma. Mutat. Res. Mutat. Res. 2006, 612, 105–114. [Google Scholar] [CrossRef]
- Gaskin, J.F.; Shah, M.; Chan, E. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants 2021, 10, 238. [Google Scholar] [CrossRef]
- Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.K.; Hamann, S.; Kolko, M. Oxidative Stress in Optic Neuropathies. Antioxidants 2021, 10, 1538. [Google Scholar] [CrossRef]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by Protein Kinase C Regulates Antioxidant Response Element-mediated Transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, S.; Yan, J.; Li, Y.; Xin, Z.; Lin, Y.; Qu, Y. An overview of the molecular mechanisms and novel roles of Nrf2 in neurodegenerative disorders. Cytokine Growth Factor Rev. 2015, 26, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Naguib, S.; Backstrom, J.R.; Gil, M.; Calkins, D.J.; Rex, T.S. Retinal oxidative stress activates the NRF2/ARE pathway: An early endogenous protective response to ocular hypertension. Redox Biol. 2021, 42, 101883. [Google Scholar] [CrossRef]
- Fujita, K.; Nishiguchi, K.M.; Shiga, Y.; Nakazawa, T. Spatially and Temporally Regulated NRF2 Gene Therapy Using Mcp-1 Promoter in Retinal Ganglion Cell Injury. Mol. Ther.-Methods Clin. Dev. 2017, 5, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Himori, N.; Yamamoto, K.; Maruyama, K.; Ryu, M.; Taguchi, K.; Yamamoto, M.; Nakazawa, T. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J. Neurochem. 2013, 127, 669–680. [Google Scholar] [CrossRef]
- Mori, S.; Kurimoto, T.; Maeda, H.; Nakamura, M. Dimethyl Fumarate Promotes the Survival of Retinal Ganglion Cells after Optic Nerve Injury, Possibly through the Nrf2/HO-1 Pathway. Int. J. Mol. Sci. 2020, 22, 297. [Google Scholar] [CrossRef]
- Chien, J.-Y.; Chou, Y.-Y.; Ciou, J.-W.; Liu, F.-Y.; Huang, S.-P. The Effects of Two Nrf2 Activators, Bardoxolone Methyl and Omaveloxolone, on Retinal Ganglion Cell Survival during Ischemic Optic Neuropathy. Antioxidants 2021, 10, 1466. [Google Scholar] [CrossRef]
- Pan, H.; He, M.; Liu, R.; Brecha, N.C.; Yu, A.C.H.; Pu, M. Sulforaphane Protects Rodent Retinas against Ischemia-Reperfusion Injury through the Activation of the Nrf2/HO-1 Antioxidant Pathway. PLoS ONE 2014, 9, e114186. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The Multifaceted Role of Nrf2 in Mitochondrial Function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A ΔΨm Independent Pharmacological Regulator of Mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-J.; Lin, S.-H.; Liu, Y.-T.; Lin, H.-C.; Li, T.-N.; Yao, C.-K. A circuit-dependent ROS feedback loop mediates glutamate excitotoxicity to sculpt the Drosophila motor system. eLife 2019, 8, e47372. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Cheung, W.; Guo, L.; Cordeiro, M.F. Neuroprotection in Glaucoma: Drug-Based Approaches. Optom. Vis. Sci. 2008, 85, E406–E416. [Google Scholar] [CrossRef]
- Hare, W.A.; WoldeMussie, E.; Weinreb, R.N.; Ton, H.; Ruiz, G.; Wijono, M.; Feldmann, B.; Zangwill, L.; Wheeler, L. Efficacy and Safety of Memantine Treatment for Reduction of Changes Associated with Experimental Glaucoma in Monkey, II: Structural Measures. Investig. Opthalmology Vis. Sci. 2004, 45, 2640–2651. [Google Scholar] [CrossRef]
- Guo, X.; Zhou, J.; Starr, C.; Mohns, E.J.; Li, Y.; Chen, E.P.; Yoon, Y.; Kellner, C.P.; Tanaka, K.; Wang, H.; et al. Preservation of vision after CaMKII-mediated protection of retinal ganglion cells. Cell 2021, 184, 4299–4314.e12. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Liebmann, J.M.; Cioffi, G.A.; Goldberg, I.; Brandt, J.D.; Johnson, C.A.; Zangwill, L.M.; Schneider, S.; Badger, H.; Bejanian, M. Oral Memantine for the Treatment of Glaucoma: Design and Results of 2 Randomized, Placebo-Controlled, Phase 3 Studies. Ophthalmology 2018, 125, 1874–1885. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Nicoll, R.A.; Roche, K.W. Diversity in NMDA Receptor Composition: Many regulators, many consequences. Neuroscientist 2013, 19, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Libby, R.; Jakobs, T.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Mahesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef]
- Davis, R.J. Signal Transduction by the JNK Group of MAP Kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Perrin, V.; Dufour, N.; Raoul, C.; Hassig, R.; Brouillet, E.; Aebischer, P.; Luthi-Carter, R.; Déglon, N. Implication of the JNK pathway in a rat model of Huntington’s disease. Exp. Neurol. 2009, 215, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.G.; Harding, T.; Weller, M.; Bieneman, A.; Uney, J.B.; Schulz, J.B. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 10433–10438. [Google Scholar] [CrossRef]
- Welsbie, D.S.; Yang, Z.; Ge, Y.; Mitchell, K.L.; Zhou, X.; Martin, S.E.; Berlinicke, C.A.; Hackler, L., Jr.; Fuller, J.; Fu, J.; et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc. Natl. Acad. Sci. USA 2013, 110, 4045–4050. [Google Scholar] [CrossRef]
- Welsbie, D.S.; Mitchell, K.L.; Jaskula-Ranga, V.; Sluch, V.; Yang, Z.; Kim, J.; Buehler, E.; Patel, A.; Martin, S.E.; Zhang, P.-W.; et al. Enhanced Functional Genomic Screening Identifies Novel Mediators of Dual Leucine Zipper Kinase-Dependent Injury Signaling in Neurons. Neuron 2017, 94, 1142–1154.e6. [Google Scholar] [CrossRef]
- Harrington, E.A.; Bebbington, D.; Moore, J.; Rasmussen, R.K.; Ajose-Adeogun, A.O.; Nakayama, T.; Graham, J.A.; Demur, C.; Hercend, T.; Diu-Hercend, A.; et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004, 10, 262–267. [Google Scholar] [CrossRef]
- Peng, Y.-R.; Shekhar, K.; Yan, W.; Herrmann, D.; Sappington, A.; Bryman, G.S.; van Zyl, T.; Do, M.T.H.; Regev, A.; Sanes, J.R. Molecular Classification and Comparative Taxonomics of Foveal and Peripheral Cells in Primate Retina. Cell 2019, 176, 1222–1237.e22. [Google Scholar] [CrossRef]
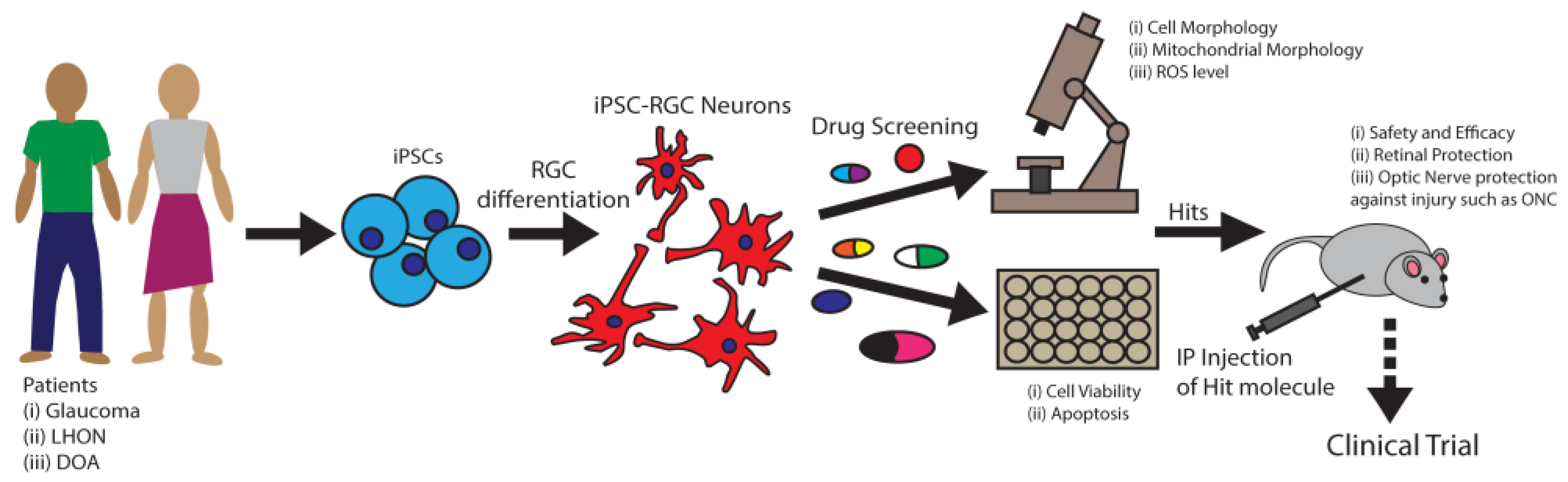
- Sluch, V.M.; Chamling, X.; Liu, M.M.; Berlinicke, C.A.; Cheng, J.; Mitchell, K.L.; Welsbie, D.S.; Zack, D.J. Enhanced Stem Cell Differentiation and Immunopurification of Genome Engineered Human Retinal Ganglion Cells. STEM CELLS Transl. Med. 2017, 6, 1972–1986. [Google Scholar] [CrossRef]
- Sluch, V.M.; Davis, C.-H.O.; Ranganathan, V.; Kerr, J.M.; Krick, K.; Martin, R.; Berlinicke, C.A.; Marsh-Armstrong, N.; Diamond, J.S.; Mao, H.-Q.; et al. Differentiation of human ESCs to retinal ganglion cells using a CRISPR engineered reporter cell line. Sci. Rep. 2015, 5, 16595. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.-C.; Sun, C.; Cameron, E.G.; Madaan, A.; Wu, S.; Xia, X.; Zhang, X.; Tenerelli, K.; Nahmou, M.; Knasel, C.M.; et al. Opposing Effects of Growth and Differentiation Factors in Cell-Fate Specification. Curr. Biol. 2019, 29, 1963–1975.e5. [Google Scholar] [CrossRef] [PubMed]
- Teotia, P.; Chopra, D.A.; Dravid, S.M.; Van Hook, M.J.; Qiu, F.; Morrison, J.; Rizzino, A.; Ahmad, I. Generation of Functional Human Retinal Ganglion Cells with Target Specificity from Pluripotent Stem Cells by Chemically Defined Recapitulation of Developmental Mechanism. Stem Cells 2017, 35, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Ohlemacher, S.K.; Sridhar, A.; Xiao, Y.; Hochstetler, A.E.; Sarfarazi, M.; Cummins, T.R.; Meyer, J.S. Stepwise Differentiation of Retinal Ganglion Cells from Human Pluripotent Stem Cells Enables Analysis of Glaucomatous Neurodegeneration. Stem Cells 2016, 34, 1553–1562. [Google Scholar] [CrossRef]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef]
- Badea, T.C.; Cahill, H.; Ecker, J.; Hattar, S.; Nathans, J. Distinct Roles of Transcription Factors Brn3a and Brn3b in Controlling the Development, Morphology, and Function of Retinal Ganglion Cells. Neuron 2009, 61, 852–864. [Google Scholar] [CrossRef]
- Badea, T.C.; Williams, J.; Smallwood, P.; Shi, M.; Motajo, O.; Nathans, J. Combinatorial Expression of Brn3 Transcription Factors in Somatosensory Neurons: Genetic and Morphologic Analysis. J. Neurosci. 2012, 32, 995–1007. [Google Scholar] [CrossRef]
- Gan, L.; Xiang, M.; Zhou, L.; Wagner, D.S.; Klein, W.H.; Nathans, J. POU domain factor Brn-3b is required for the development of a large set of retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1996, 93, 3920–3925. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.-R.; Li, L.-H.; Park, H.-J.; Park, J.-H.; Lee, K.Y.; Kim, M.-K.; Shin, B.A.; Choi, S.-Y. High Cleavage Efficiency of a 2A Peptide Derived from Porcine Teschovirus-1 in Human Cell Lines, Zebrafish and Mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Tuffy, C.; Mertz, J.; Quillen, S.; Wechsler, L.; Quigley, H.; Zack, D.J.; Johnson, T.V. Role of the Internal Limiting Membrane in Structural Engraftment and Topographic Spacing of Transplanted Human Stem Cell-Derived Retinal Ganglion Cells. Stem Cell Rep. 2021, 16, 149–167. [Google Scholar] [CrossRef]
- Zhou, T.; Tan, L.; Cederquist, G.Y.; Fan, Y.; Hartley, B.J.; Mukherjee, S.; Tomishima, M.; Brennand, K.J.; Zhang, Q.; Schwartz, R.E.; et al. High-Content Screening in hPSC-Neural Progenitors Identifies Drug Candidates that Inhibit Zika Virus Infection in Fetal-like Organoids and Adult Brain. Cell Stem Cell 2017, 21, 274–283.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Zhong, P.; Zheng, W.; Beekman, J.M. Pharmacological analysis of CFTR variants of cystic fibrosis using stem cell-derived organoids. Drug Discov. Today 2019, 24, 2126–2138. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A. The Flaws and Human Harms of Animal Experimentation. Camb. Q. Health Ethic 2015, 24, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e4. [Google Scholar] [CrossRef] [PubMed]
- Maruotti, J.; Wahlin, K.; Gorrell, D.; Bhutto, I.; Lutty, G.; Zack, D.J. A Simple and Scalable Process for the Differentiation of Retinal Pigment Epithelium From Human Pluripotent Stem Cells. STEM CELLS Transl. Med. 2013, 2, 341–354. [Google Scholar] [CrossRef]
- Kallman, A.; Capowski, E.E.; Wang, J.; Kaushik, A.M.; Jansen, A.D.; Edwards, K.L.; Chen, L.; Berlinicke, C.A.; Phillips, M.J.; Pierce, E.A.; et al. Investigating cone photoreceptor development using patient-derived NRL null retinal organoids. Commun. Biol. 2020, 3, 82. [Google Scholar] [CrossRef]
- Wahlin, K.J.; Maruotti, J.A.; Sripathi, S.R.; Ball, J.; Angueyra, J.M.; Kim, C.; Grebe, R.; Li, W.; Jones, B.W.; Zack, D.J. Photoreceptor Outer Segment-like Structures in Long-Term 3D Retinas from Human Pluripotent Stem Cells. Sci. Rep. 2017, 7, 766. [Google Scholar] [CrossRef]
- Ito, S.-I.; Onishi, A.; Takahashi, M. Chemically-induced photoreceptor degeneration and protection in mouse iPSC-derived three-dimensional retinal organoids. Stem Cell Res. 2017, 24, 94–101. [Google Scholar] [CrossRef]
- Grotz, S.; Schäfer, J.; A Wunderlich, K.; Ellederova, Z.; Auch, H.; Bähr, A.; Runa---Vochozkova, P.; Fadl, J.; Arnold, V.; Ardan, T.; et al. Early disruption of photoreceptor cell architecture and loss of vision in a humanized pig model of usher syndromes. EMBO Mol. Med. 2022, 14, e14817. [Google Scholar] [CrossRef]
- Harada, C.; Kimura, A.; Guo, X.; Namekata, K.; Harada, T. Recent advances in genetically modified animal models of glaucoma and their roles in drug repositioning. Br. J. Ophthalmol. 2019, 103, 161–166. [Google Scholar] [CrossRef]
- Telias, M.; Sit, K.K.; Frozenfar, D.; Smith, B.; Misra, A.; Goard, M.J.; Kramer, R.H. Retinoic acid inhibitors mitigate vision loss in a mouse model of retinal degeneration. Sci. Adv. 2022, 8, eabm4643. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, A.; Imanishi, Y. Drug Discovery Strategies for Inherited Retinal Degenerations. Biology 2022, 11, 1338. https://doi.org/10.3390/biology11091338
Das A, Imanishi Y. Drug Discovery Strategies for Inherited Retinal Degenerations. Biology. 2022; 11(9):1338. https://doi.org/10.3390/biology11091338
Chicago/Turabian StyleDas, Arupratan, and Yoshikazu Imanishi. 2022. "Drug Discovery Strategies for Inherited Retinal Degenerations" Biology 11, no. 9: 1338. https://doi.org/10.3390/biology11091338
APA StyleDas, A., & Imanishi, Y. (2022). Drug Discovery Strategies for Inherited Retinal Degenerations. Biology, 11(9), 1338. https://doi.org/10.3390/biology11091338