
Identification of 11-Hydroxytephrosin and Torosaflavone A as Potential Inhibitors of 3-Phosphoinositide-Dependent Protein Kinase 1 (PDPK1): Toward Anticancer Drug Discovery

 ,
,  , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Computer Environment and Web Resources
2.2. Receptor Preparation
2.3. Databases Used for Screening
2.4. Molecular Docking-Based Virtual Screening
2.5. ADMET Properties of Compounds
2.6. PASS Analysis
2.7. MD Simulations
2.8. Principal Component Analysis and Free Energy Landscapes
3. Results and Discussion
3.1. Molecular Docking-Based Virtual Screening
3.2. ADMET Properties of Compounds
3.3. PASS Analysis
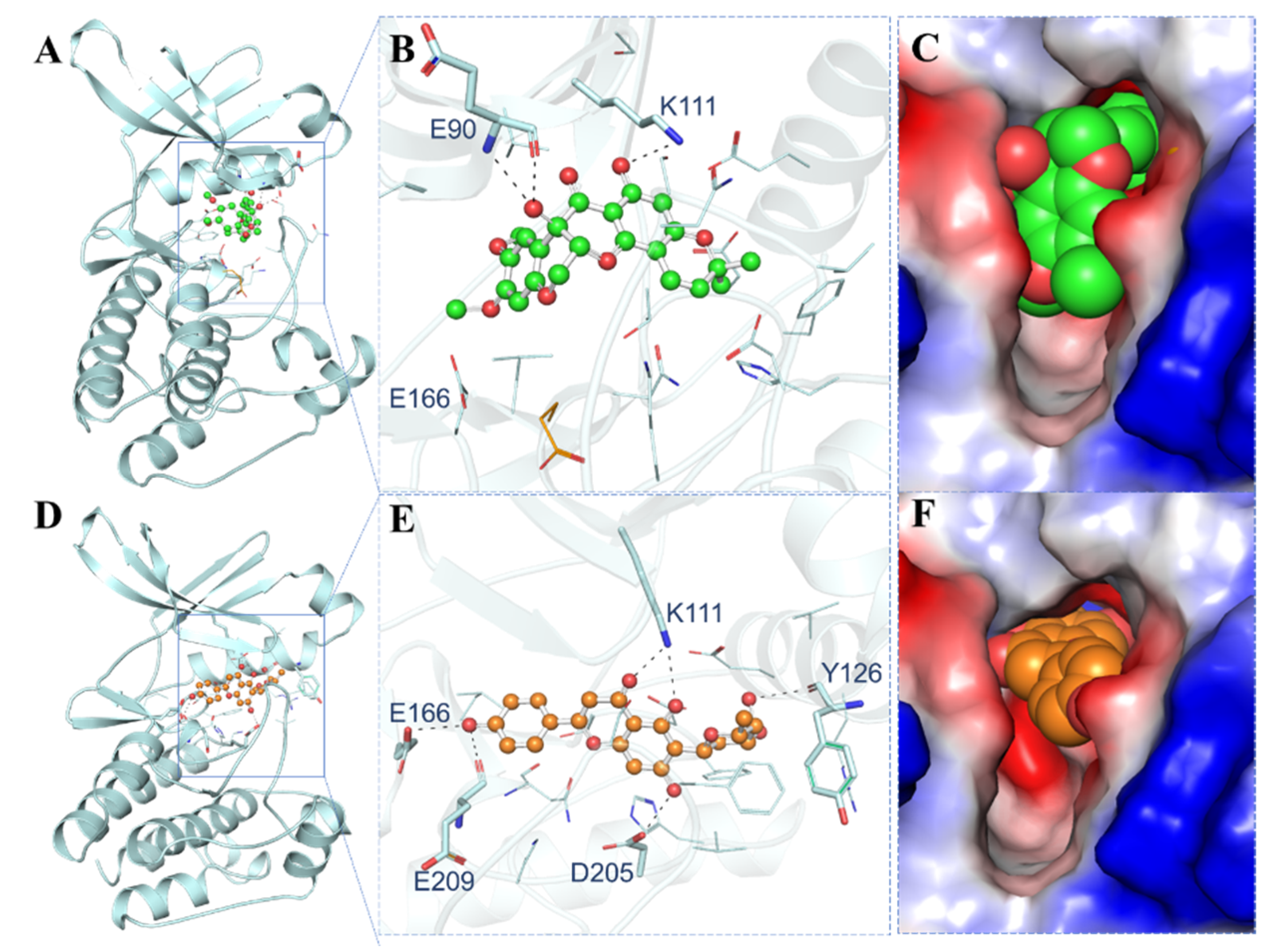
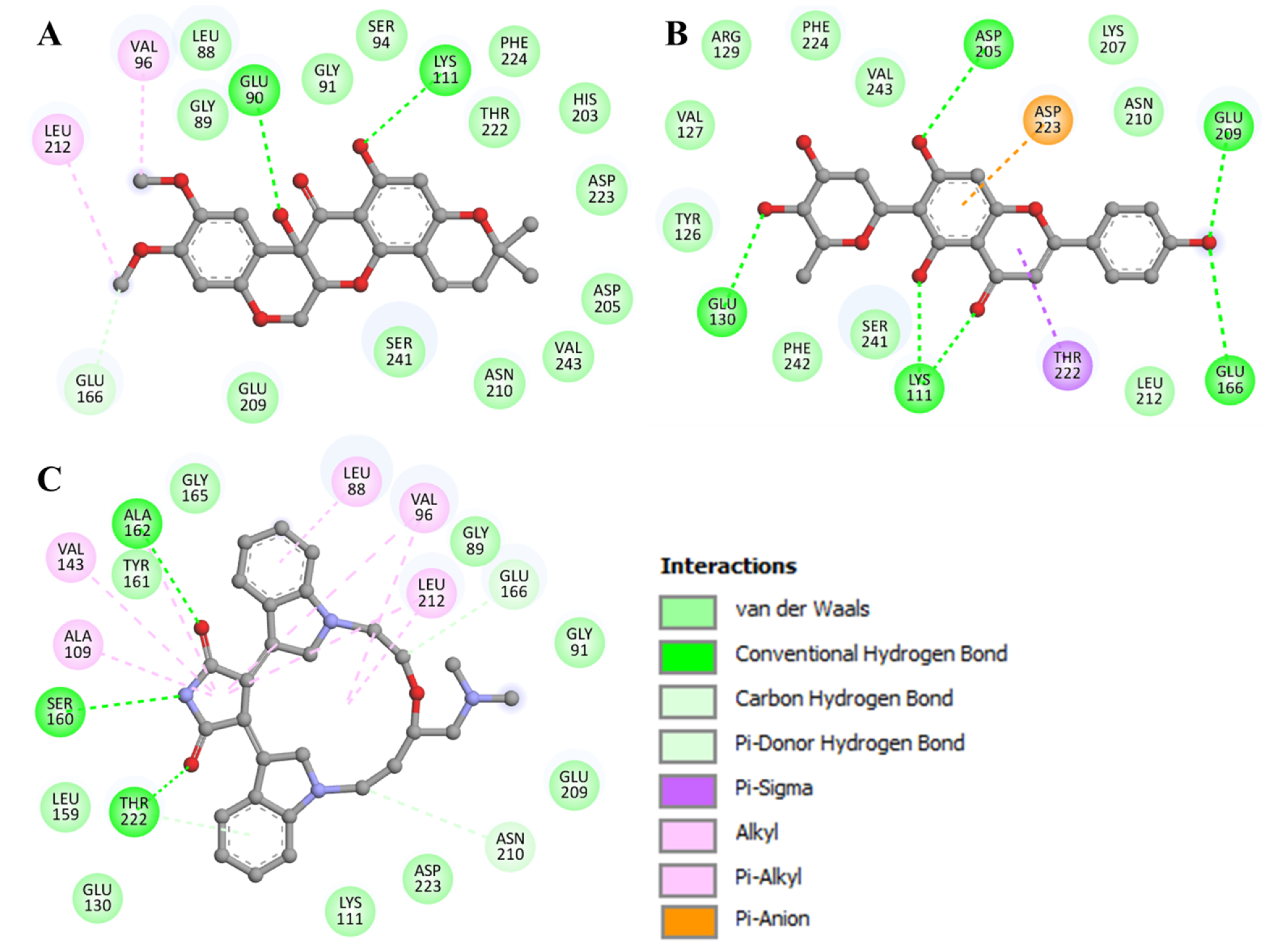
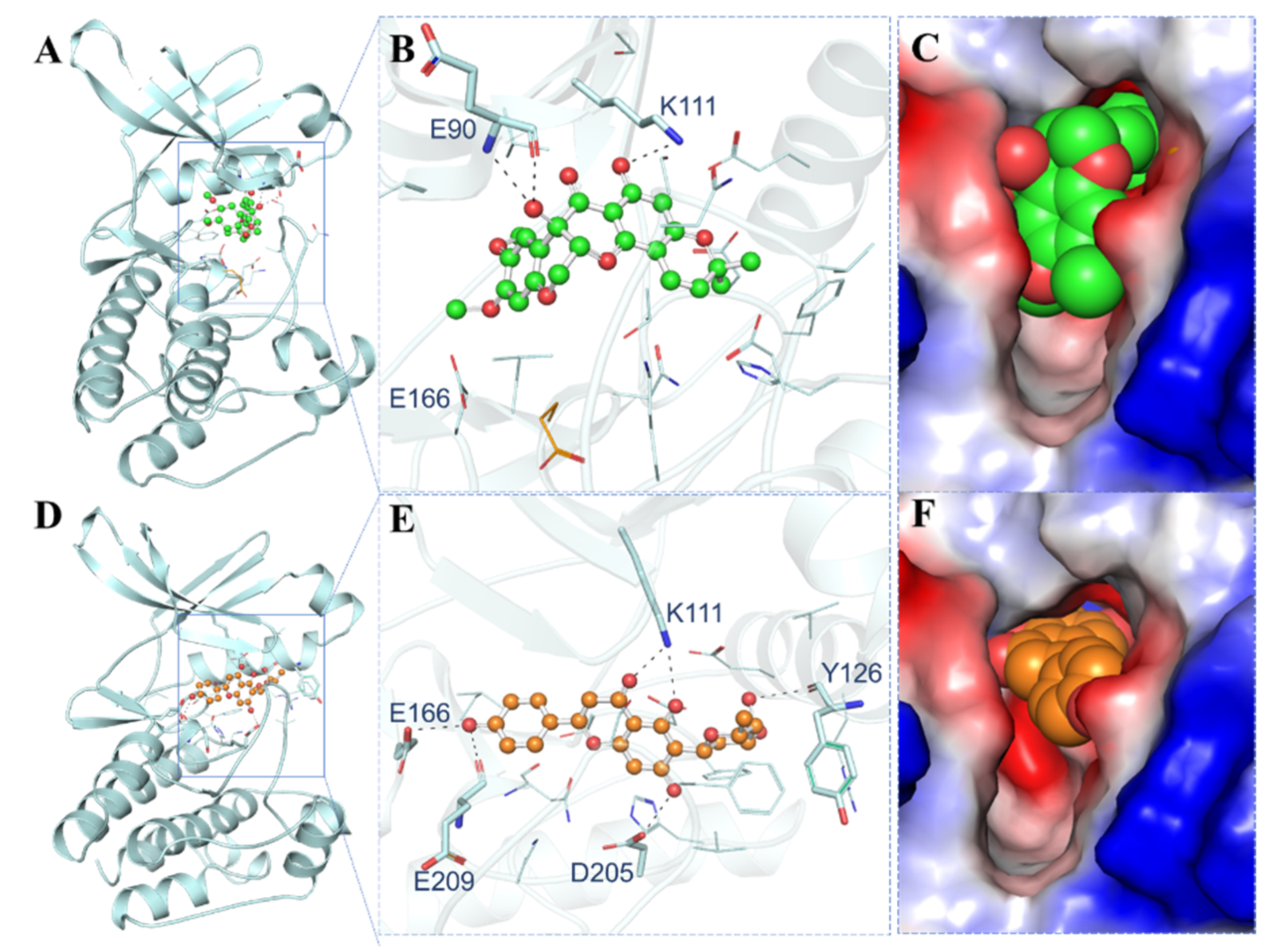
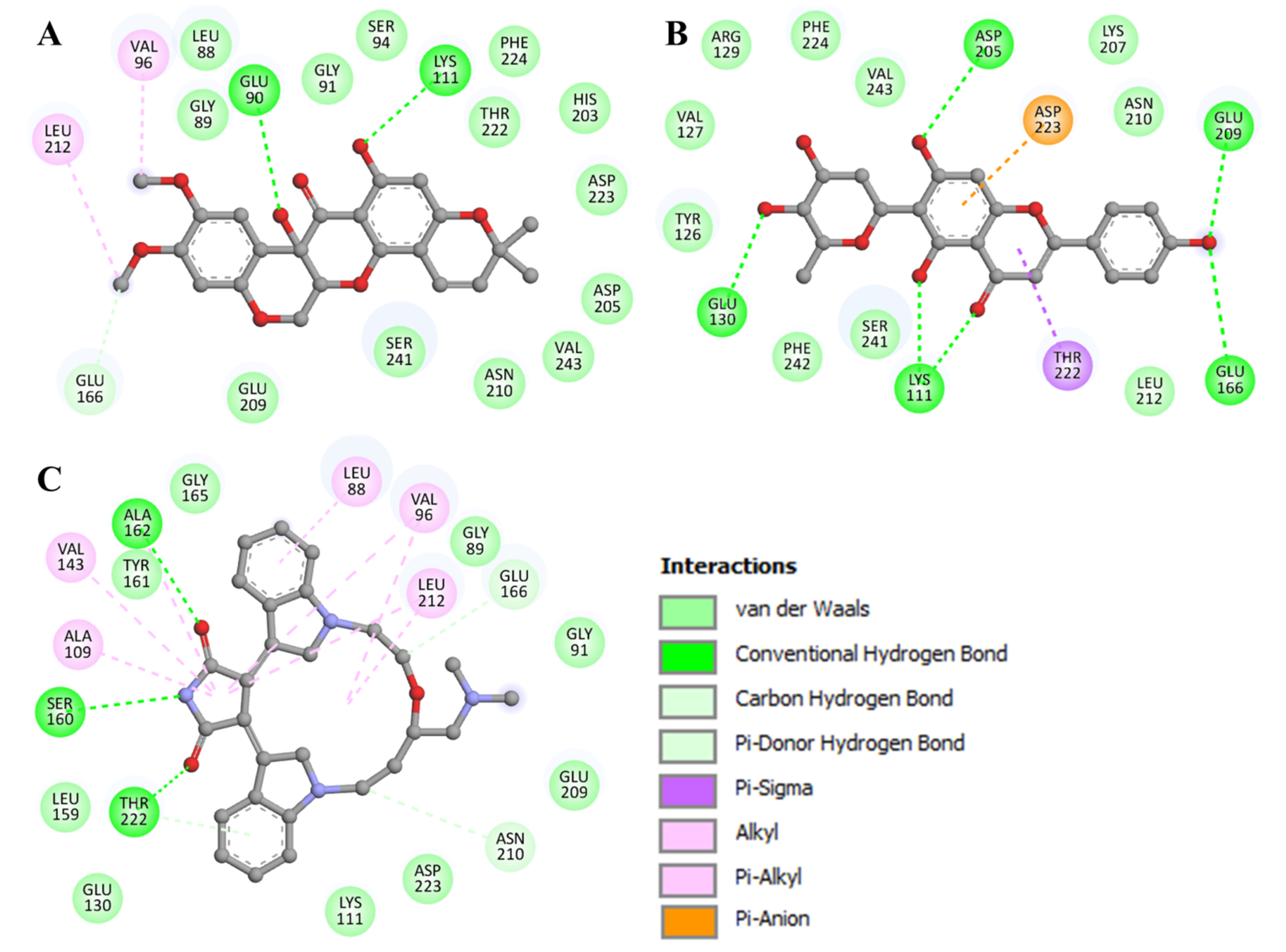
3.4. Interaction Analysis
3.5. MD Simulations
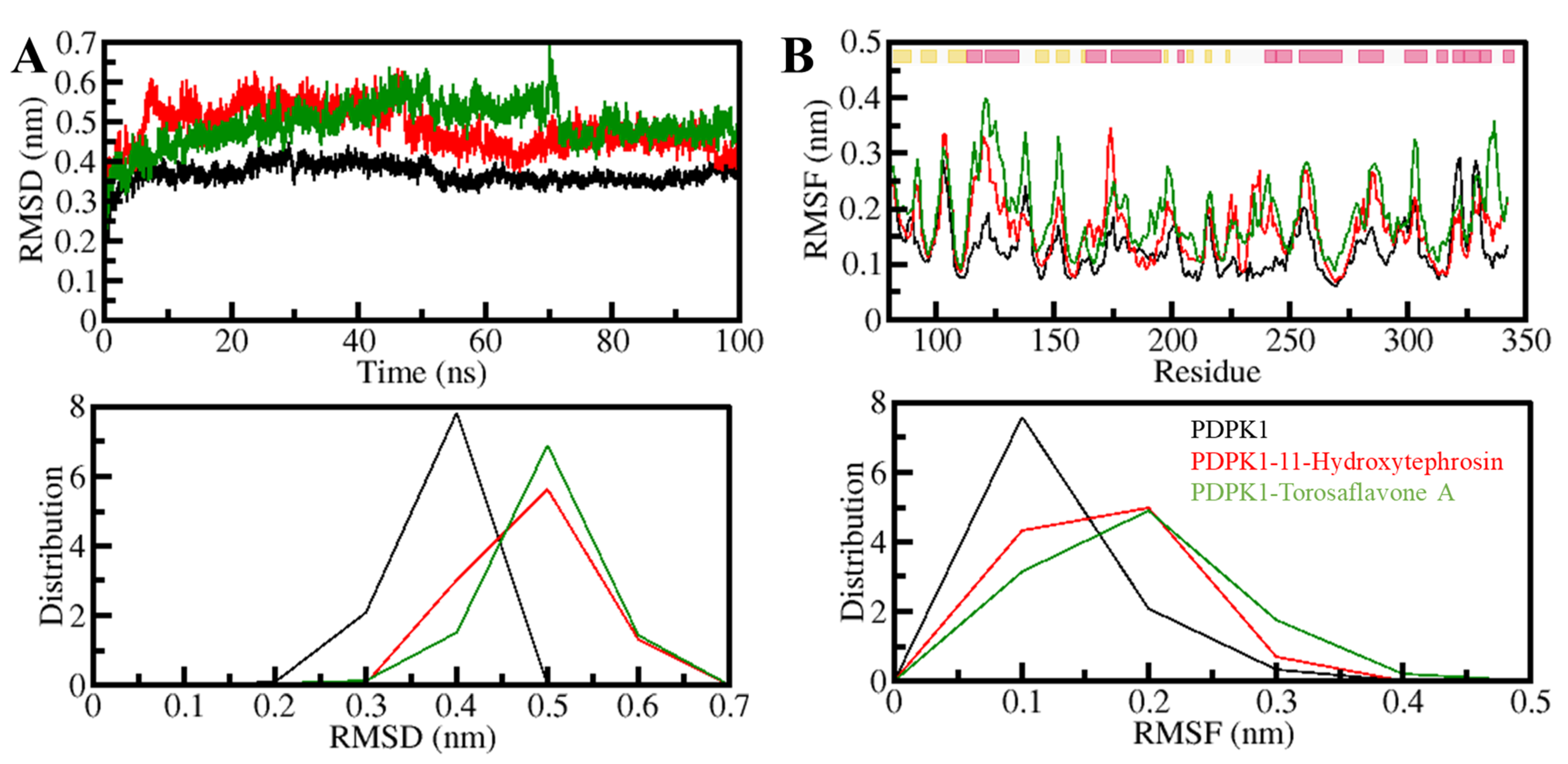
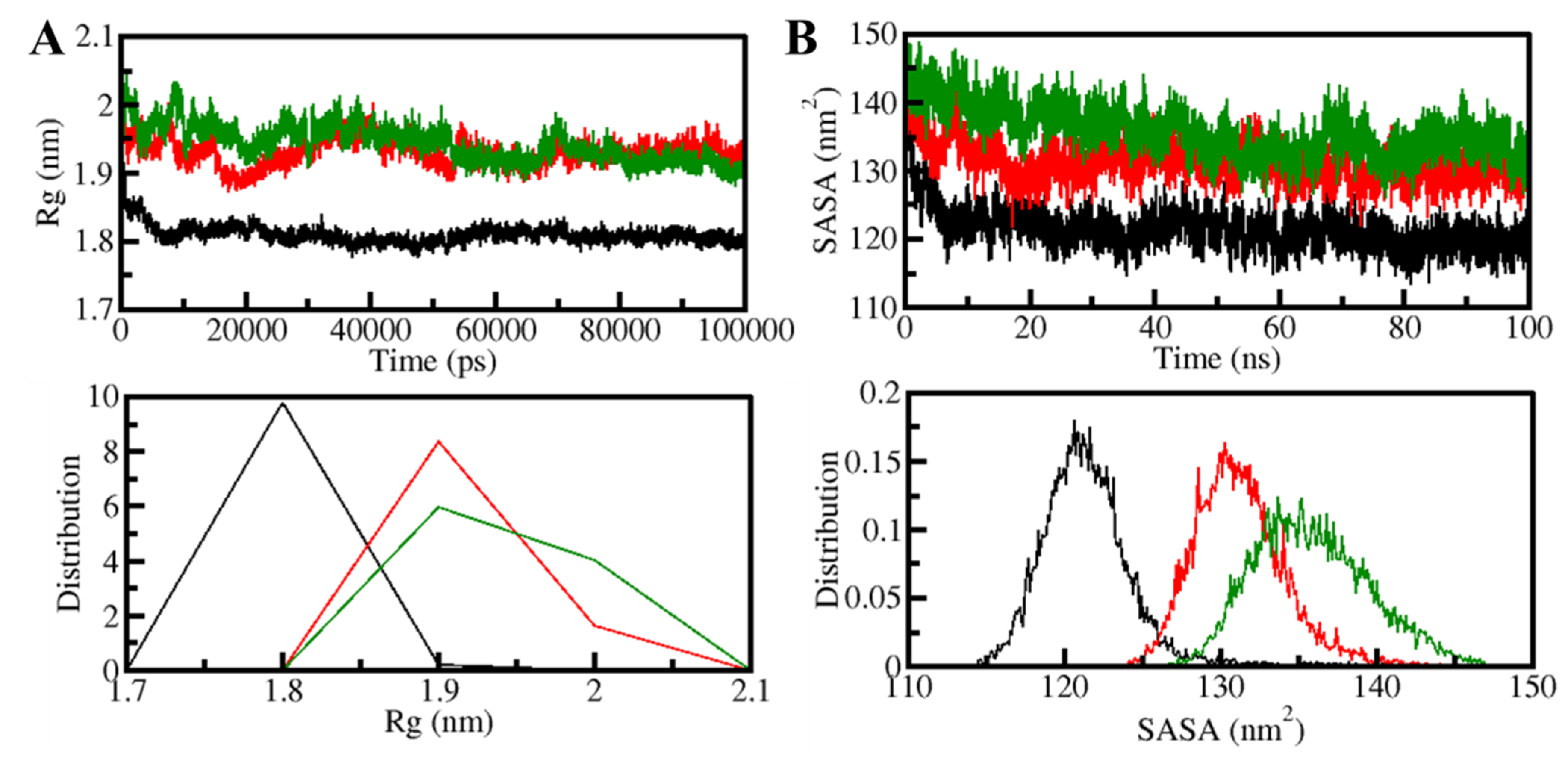
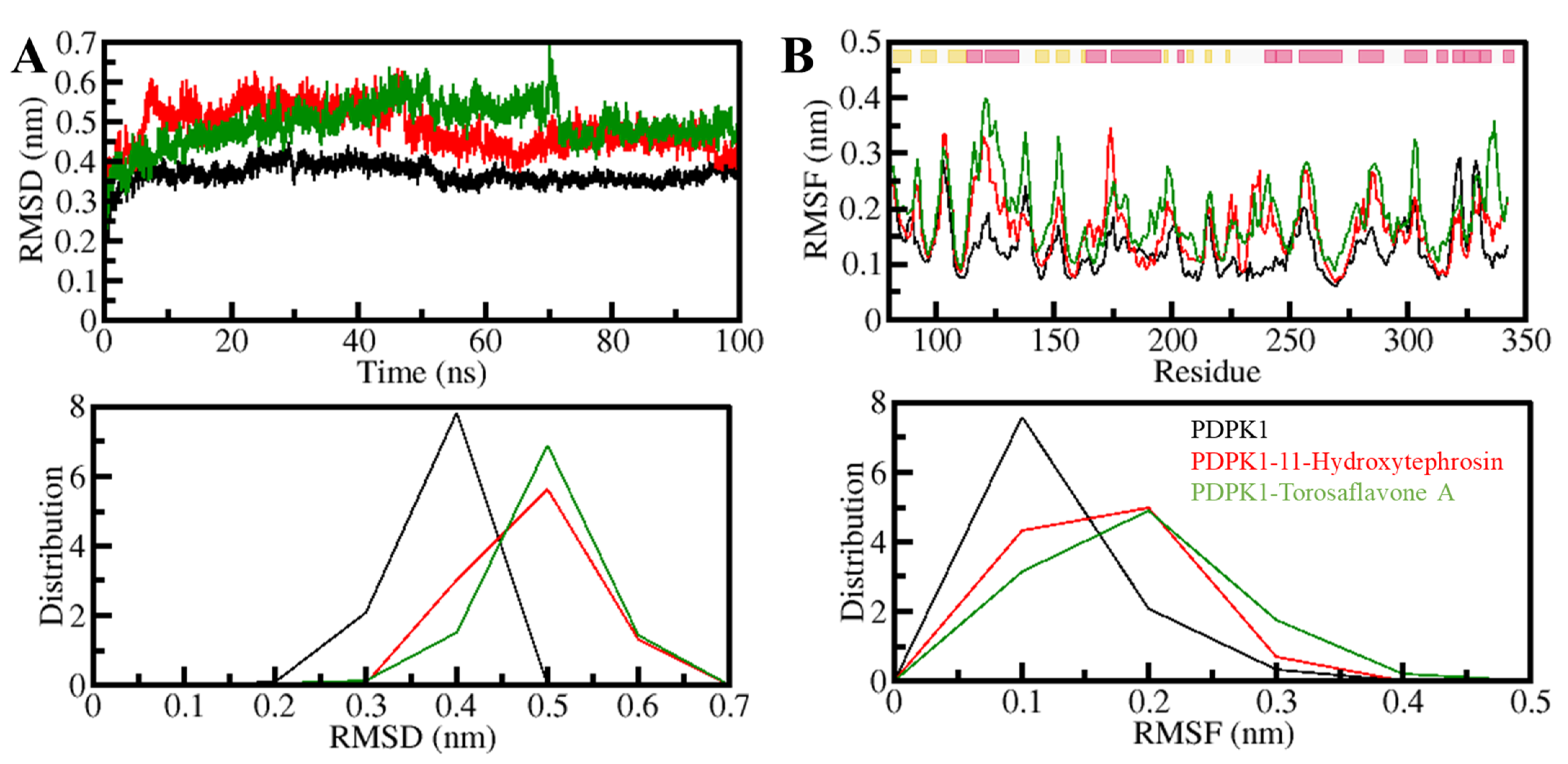
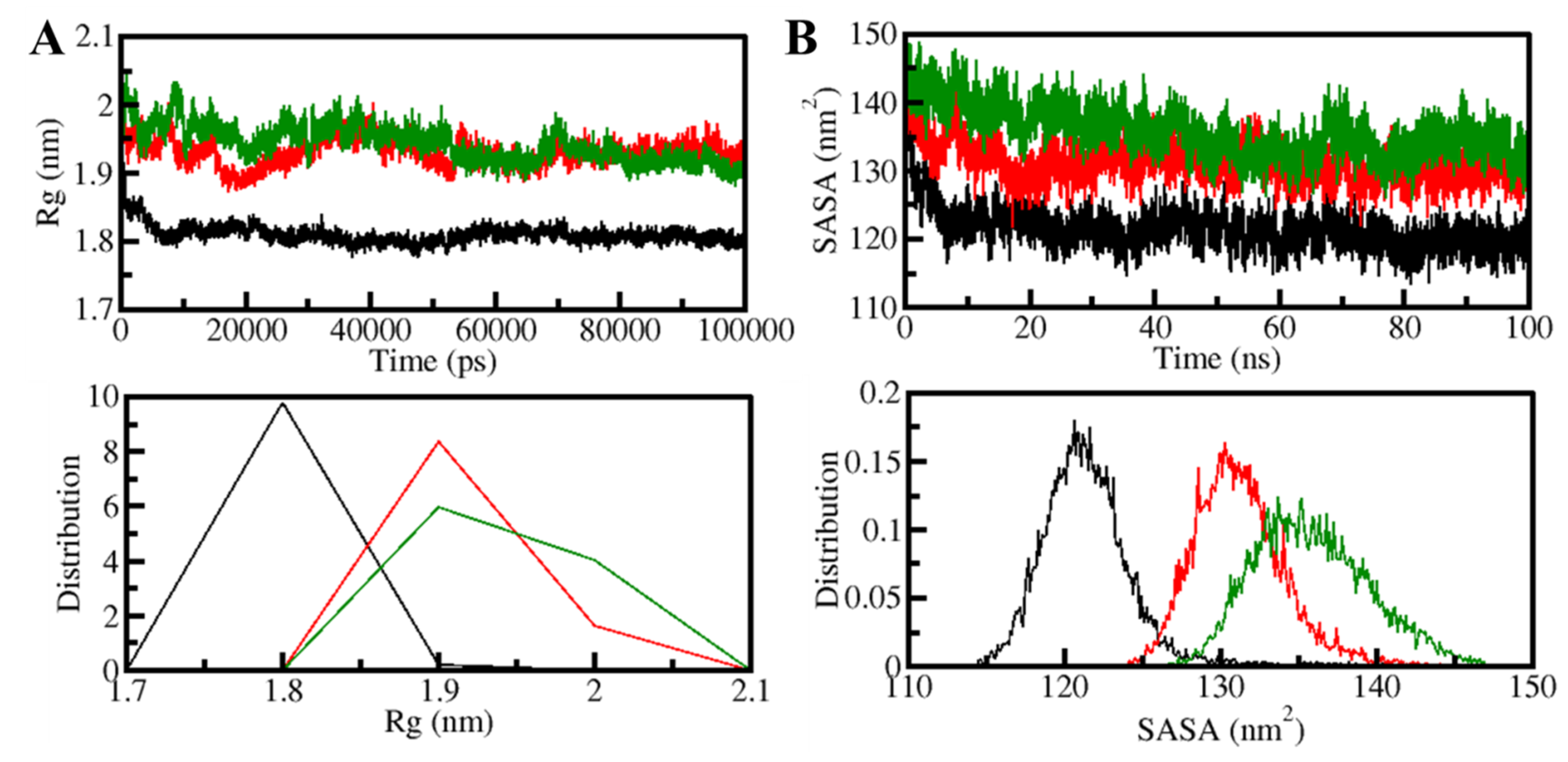
3.5.1. Structural Changes and Compactness
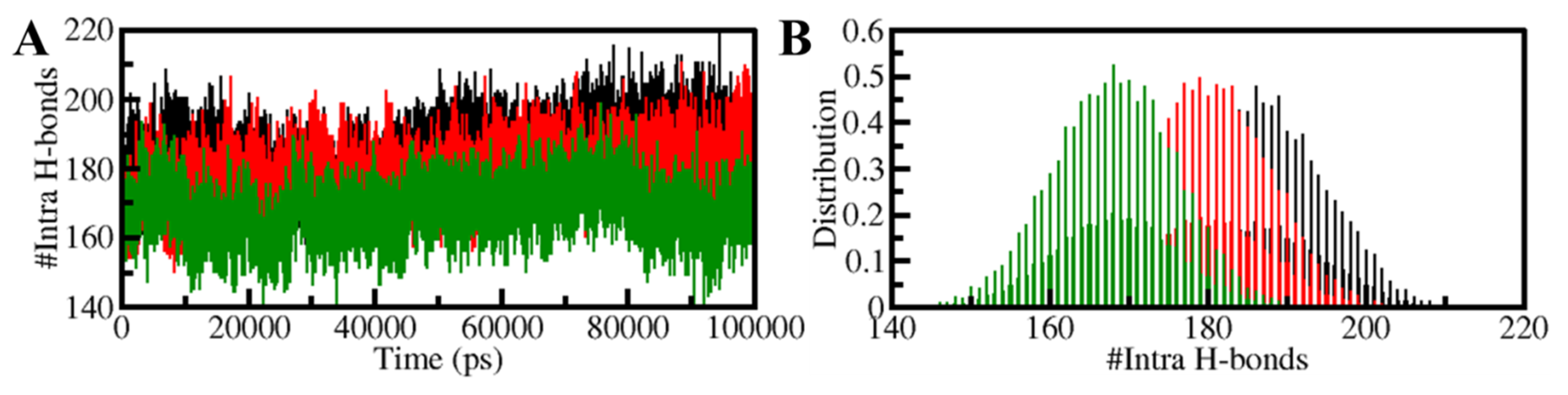
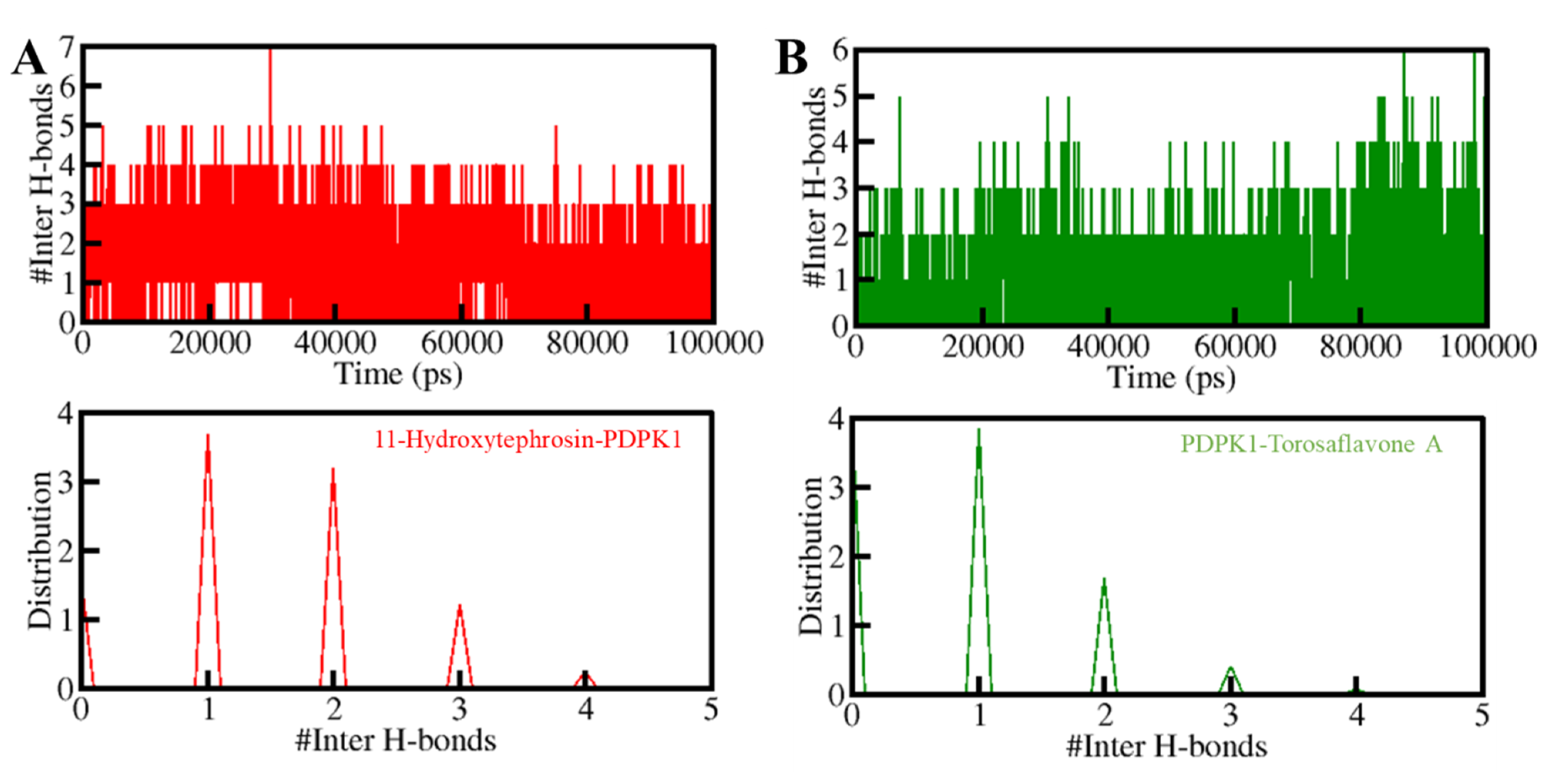
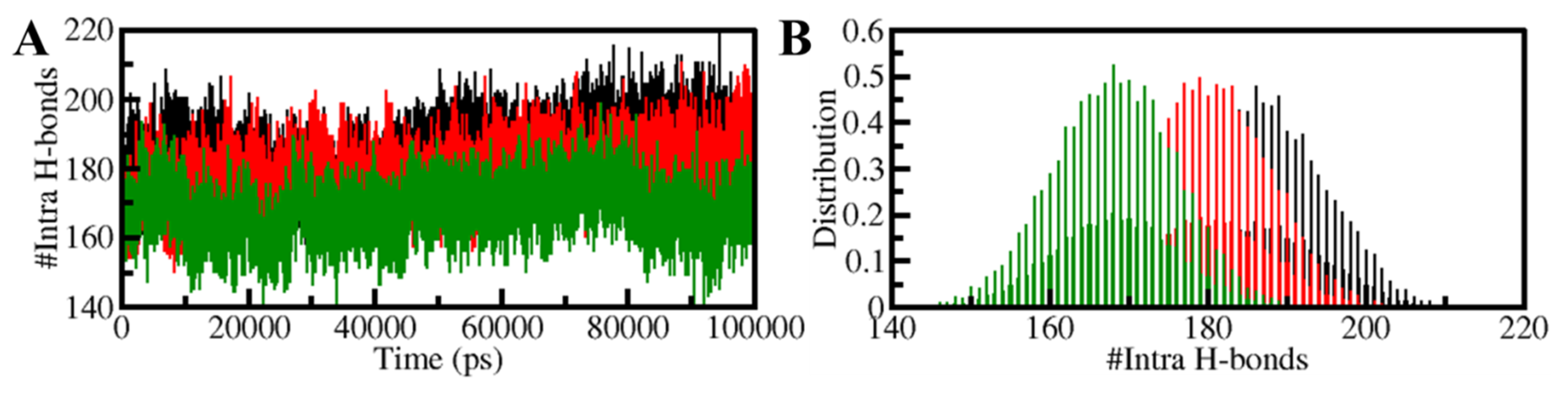
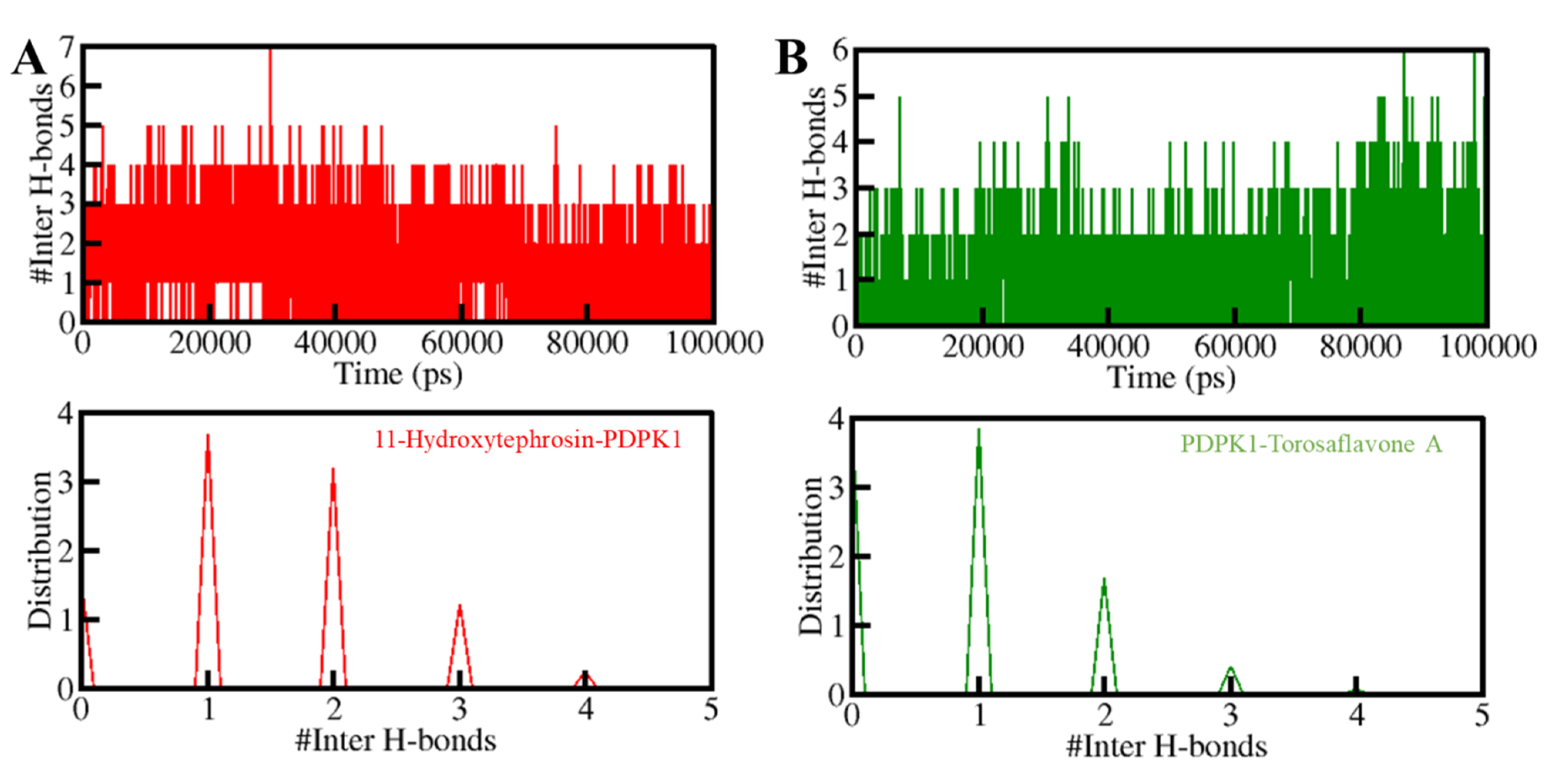
3.5.2. Dynamics of Hydrogen Bonds
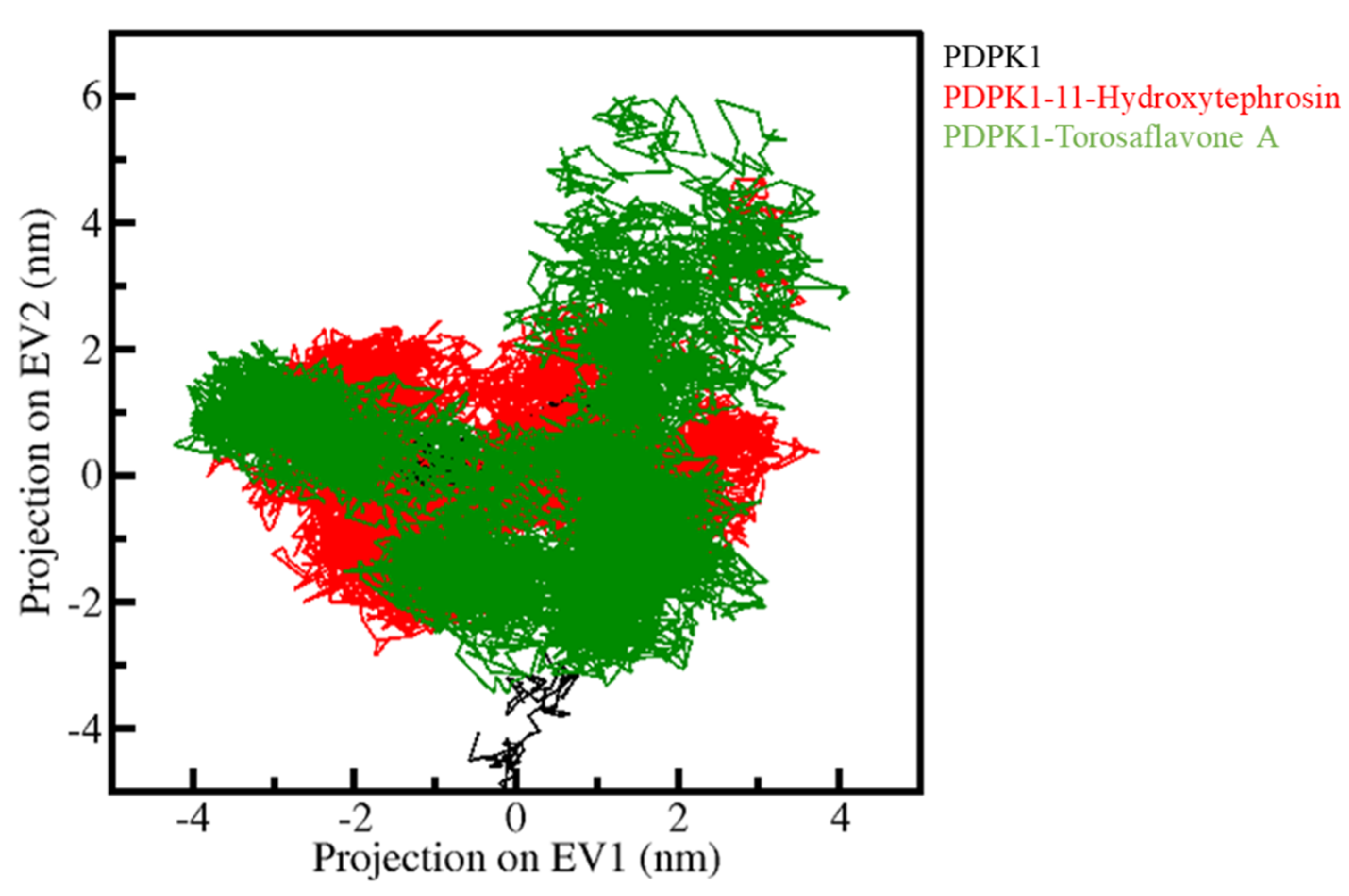
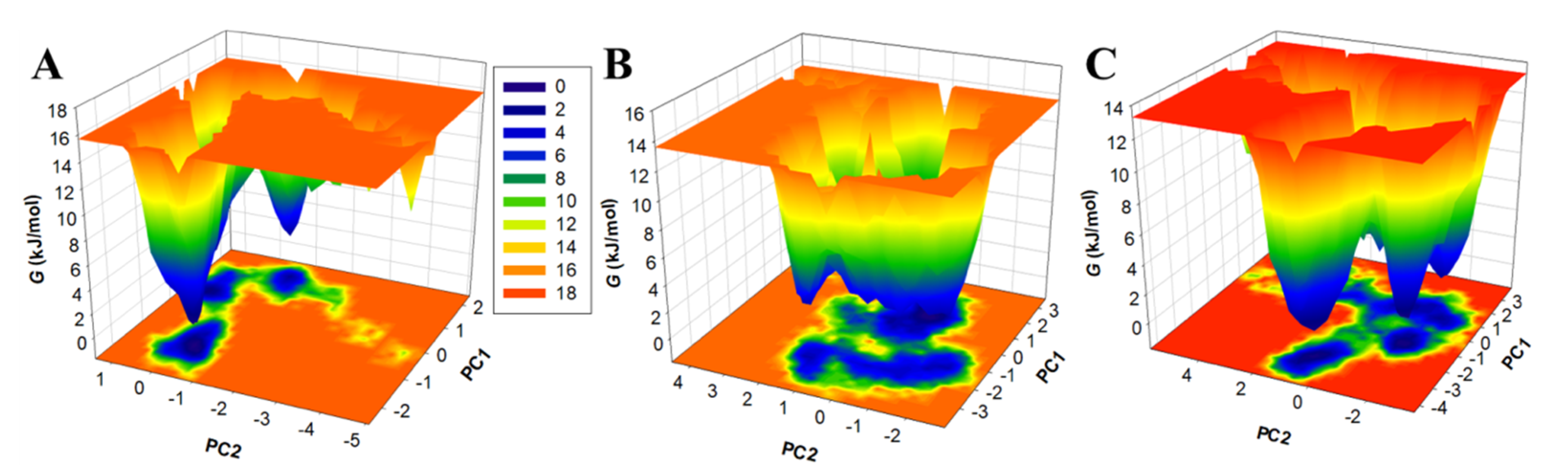
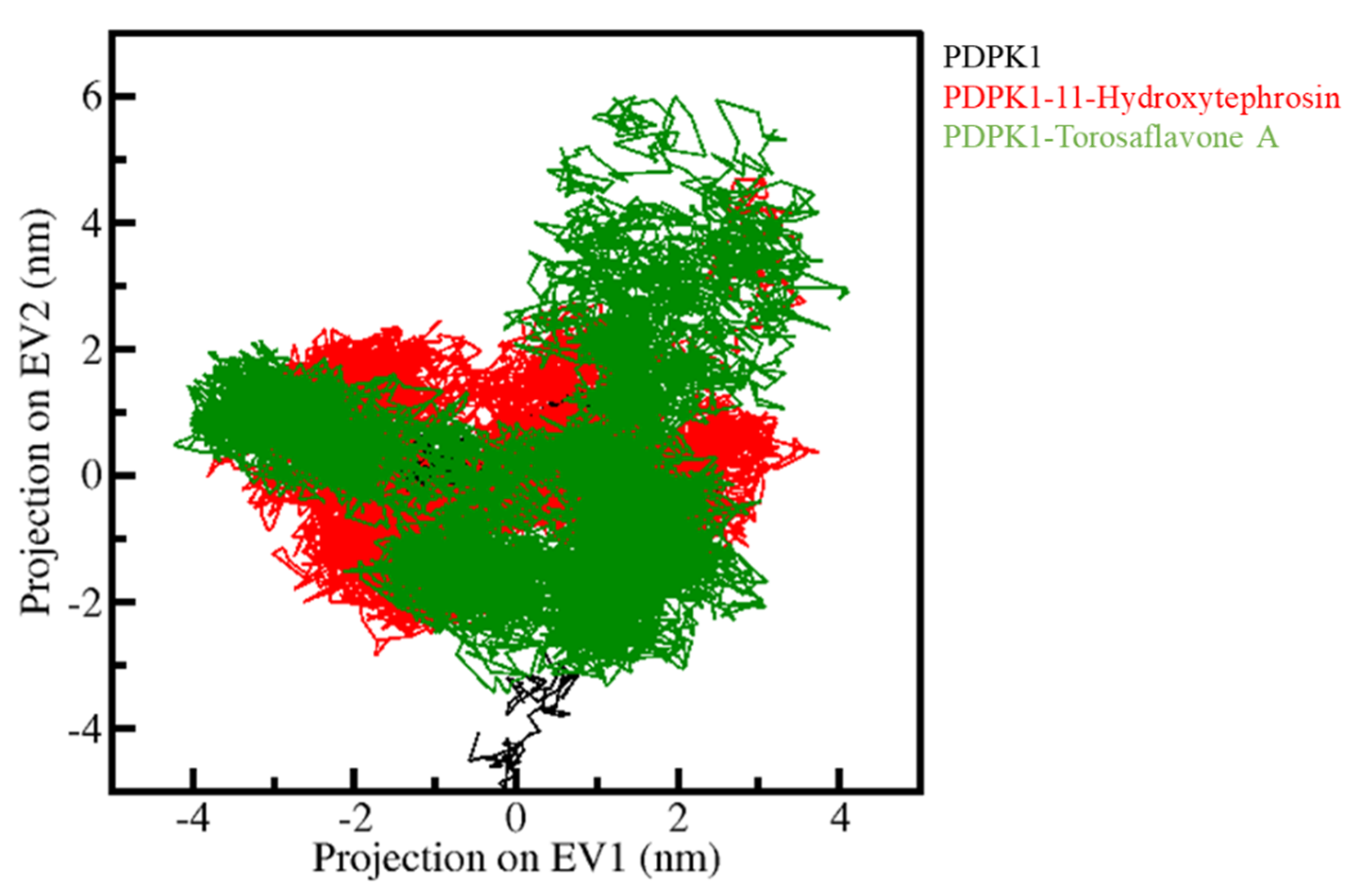
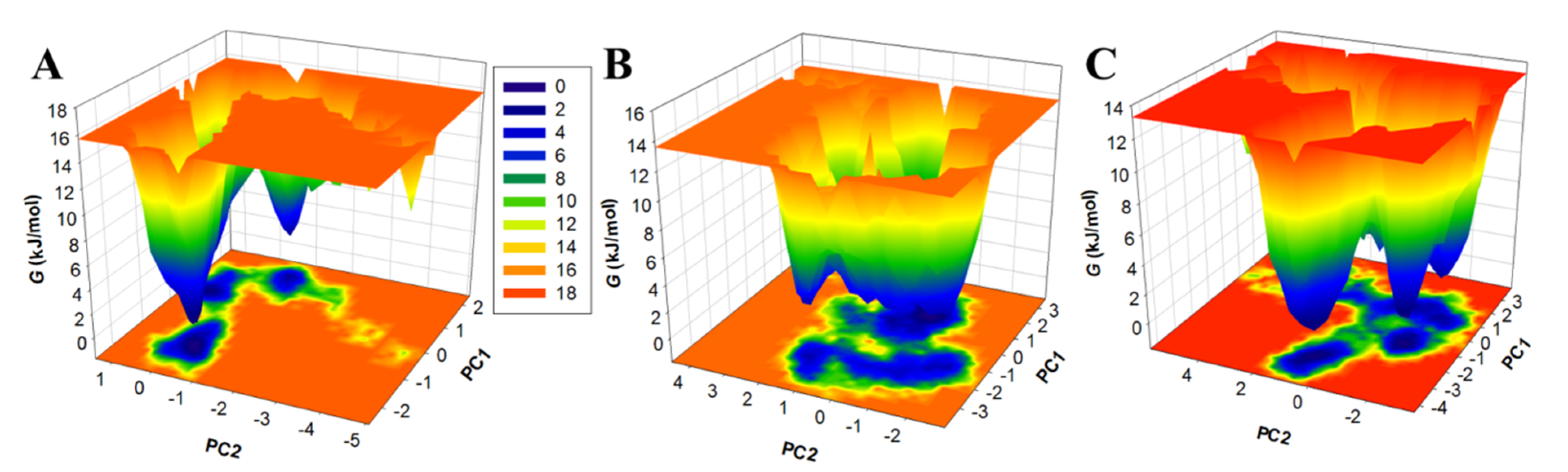
3.5.3. Principal Component Analysis and Free Energy Landscapes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emmanouilidi, A.; Fyffe, C.A.; Ferro, R.; Edling, C.E.; Capone, E.; Sestito, S.; Rapposelli, S.; Lattanzio, R.; Iacobelli, S.; Sala, G. Preclinical validation of 3-phosphoinositide-dependent protein kinase 1 inhibition in pancreatic cancer. J. Exp. Clin. Cancer Res. 2019, 38, 191. [Google Scholar] [CrossRef]
- Domrachev, B.; Singh, S.; Li, D.; Rudloff, U. Mini-review: PDPK1 (3-phosphoinositide dependent protein kinase-1), an emerging cancer stem cell target. J. Cancer Treat. Diagn. 2021, 5, 30. [Google Scholar] [CrossRef]
- Li, C.; Zhao, W.; Pan, X.; Li, X.; Yan, F.; Liu, S.; Feng, J.; Lu, J. LncRNA KTN1-AS1 promotes the progression of non-small cell lung cancer via sponging of miR-130a-5p and activation of PDPK1. Oncogene 2020, 39, 6157–6171. [Google Scholar] [CrossRef]
- Nalairndran, G.; Hassan Abdul Razack, A.; Mai, C.W.; Fei-Lei Chung, F.; Chan, K.K.; Hii, L.W.; Lim, W.M.; Chung, I.; Leong, C.O. Phosphoinositide-dependent Kinase-1 (PDPK1) regulates serum/glucocorticoid-regulated Kinase 3 (SGK3) for prostate cancer cell survival. J. Cell. Mol. Med. 2020, 24, 12188–12198. [Google Scholar] [CrossRef] [PubMed]
- Langlais, P.R.; Mandarino, L.J.; Garvey, W.T. Mechanisms of Insulin Signal Transduction. In International Textbook of Diabetes Mellitus; Wiley-Blackwell: Hoboken, NJ, USA, 2015; Volume 2, p. 163. [Google Scholar]
- Zhou, Y.; Zheng, X.; Xu, B.; Chen, L.; Wang, Q.; Deng, H.; Jiang, J. Circular RNA hsa_circ_0004015 regulates the proliferation, invasion, and TKI drug resistance of non-small cell lung cancer by miR-1183/PDPK1 signaling pathway. Biochem. Biophys. Res. Commun. 2019, 508, 527–535. [Google Scholar] [CrossRef]
- Alessi, D.R.; Deak, M.; Casamayor, A.; Caudwell, F.B.; Morrice, N.; Norman, D.G.; Gaffney, P.; Reese, C.B.; MacDougall, C.N.; Harbison, D. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 1997, 7, 776–789. [Google Scholar] [CrossRef]
- Seong, H.-A.; Jung, H.; Ichijo, H.; Ha, H. Reciprocal negative regulation of PDK1 and ASK1 signaling by direct interaction and phosphorylation. J. Biol. Chem. 2010, 285, 2397–2414. [Google Scholar] [CrossRef]
- Masters, T.A.; Calleja, V.; Armoogum, D.A.; Marsh, R.J.; Applebee, C.J.; Laguerre, M.; Bain, A.J.; Larijani, B. Regulation of 3-phosphoinositide–dependent protein kinase 1 activity by homodimerization in live cells. Sci. Signal. 2010, 3, ra78. [Google Scholar] [CrossRef]
- Barile, E.; De, S.K.; Pellecchia, M. PDK1 inhibitors. Pharm. Pat. Anal. 2012, 1, 145–163. [Google Scholar] [CrossRef]
- Komander, D.; Kular, G.S.; Schüttelkopf, A.W.; Deak, M.; Prakash, K.; Bain, J.; Elliott, M.; Garrido-Franco, M.; Kozikowski, A.P.; Alessi, D.R. Interactions of LY333531 and other bisindolyl maleimide inhibitors with PDK1. Structure 2004, 12, 215–226. [Google Scholar] [CrossRef]
- Connelly, K.A.; Kelly, D.J.; Zhang, Y.; Prior, D.L.; Advani, A.; Cox, A.J.; Thai, K.; Krum, H.; Gilbert, R.E. Inhibition of Protein Kinase C–β by Ruboxistaurin Preserves Cardiac Function and Reduces Extracellular Matrix Production in Diabetic Cardiomyopathy. Circ. Heart Fail. 2009, 2, 129–137. [Google Scholar] [CrossRef]
- Mohammad, T.; Shamsi, A.; Anwar, S.; Umair, M.; Hussain, A.; Rehman, M.T.; AlAjmi, M.F.; Islam, A.; Hassan, M.I. Identification of high-affinity inhibitors of SARS-CoV-2 main protease: Towards the development of effective COVID-19 therapy. Virus Res. 2020, 288, 198102. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, T.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. Identification and evaluation of bioactive natural products as potential inhibitors of human microtubule affinity-regulating kinase 4 (MARK4). J. Biomol. Struct. Dyn. 2019, 37, 1813–1829. [Google Scholar] [CrossRef]
- Amir, M.; Mohammad, T.; Prasad, K.; Hasan, G.M.; Kumar, V.; Dohare, R.; Islam, A.; Ahmad, F.; Imtaiyaz Hassan, M. Virtual high-throughput screening of natural compounds in-search of potential inhibitors for protection of telomeres 1 (POT1). J. Biomol. Struct. Dyn. 2020, 38, 4625–4634. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Mohammad, T.; Mathur, Y.; Hassan, M.I. InstaDock: A single-click graphical user interface for molecular docking-based virtual high-throughput screening. Brief. Bioinform. 2021, 22, bbaa279. [Google Scholar] [CrossRef]
- Studio, D. Discovery Studio; Version 2.1; Accelrys: San Diego, CA, USA, 2008. [Google Scholar]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.; Chand, R.B.; Aparna, S.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Janson, G.; Paiardini, A. PyMod 3: A complete suite for structural bioinformatics in PyMOL. Bioinformatics 2021, 37, 1471–1472. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Milne, G.W.; Nicklaus, M.C.; Driscoll, J.S.; Wang, S.; Zaharevitz, D. National Cancer Institute drug information system 3D database. J. Chem. Inf. Comput. Sci. 1994, 34, 1219–1224. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Chen, X.; Lin, Y.; Gilson, M.K. The binding database: Overview and user’s guide. Biopolym. Orig. Res. Biomol. 2001, 61, 127–141. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Modeling 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Yoo, C.; Shahlaei, M. The applications of PCA in QSAR studies: A case study on CCR5 antagonists. Chem. Biol. Drug Des. 2018, 91, 137–152. [Google Scholar] [CrossRef]
- Altis, A.; Otten, M.; Nguyen, P.H.; Hegger, R.; Stock, G. Construction of the free energy landscape of biomolecules via dihedral angle principal component analysis. J. Chem. Phys. 2008, 128, 06B620. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal component analysis for protein folding dynamics. J. Mol. Biol. 2009, 385, 312–329. [Google Scholar] [CrossRef]
- Khan, A.; Mohammad, T.; Shamsi, A.; Hussain, A.; Alajmi, M.F.; Husain, S.A.; Iqbal, M.A.; Hassan, M.I. Identification of plant-based hexokinase 2 inhibitors: Combined molecular docking and dynamics simulation studies. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef]
- Naqvi, A.A.; Mohammad, T.; Hasan, G.M.; Hassan, M. Advancements in docking and molecular dynamics simulations towards ligand-receptor interactions and structure-function relationships. Curr. Top. Med. Chem. 2018, 18, 1755–1768. [Google Scholar] [CrossRef]
- Dahiya, R.; Mohammad, T.; Roy, S.; Anwar, S.; Gupta, P.; Haque, A.; Khan, P.; Kazim, S.N.; Islam, A.; Ahmad, F. Investigation of inhibitory potential of quercetin to the pyruvate dehydrogenase kinase 3: Towards implications in anticancer therapy. Int. J. Biol. Macromol. 2019, 136, 1076–1085. [Google Scholar] [CrossRef]
- Mohammad, T.; Siddiqui, S.; Shamsi, A.; Alajmi, M.F.; Hussain, A.; Islam, A.; Ahmad, F.; Hassan, M. Virtual screening approach to identify high-affinity inhibitors of serum and glucocorticoid-regulated kinase 1 among bioactive natural products: Combined molecular docking and simulation studies. Molecules 2020, 25, 823. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Richmond, T.J. Solvent accessible surface area and excluded volume in proteins: Analytical equations for overlapping spheres and implications for the hydrophobic effect. J. Mol. Biol. 1984, 178, 63–89. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen bonds in proteins: Role and strength. eLS 2010. [Google Scholar] [CrossRef]
- Williams, M.; Ladbury, J. Hydrogen bonds in protein-ligand complexes. Methods Princ. Med. Chem. 2003, 19, 137. [Google Scholar]
- Fatima, S.; Mohammad, T.; Jairajpuri, D.S.; Rehman, M.T.; Hussain, A.; Samim, M.; Ahmad, F.J.; Alajmi, M.F.; Hassan, M.I. Identification and evaluation of glutathione conjugate gamma-l-glutamyl-l-cysteine for improved drug delivery to the brain. J. Biomol. Struct. Dyn. 2020, 38, 3610–3620. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V. Ligand binding free energy and kinetics calculation in 2020. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1455. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compound ID | Affinity (kcal/mol) |
|---|---|---|
| 1. | CID_24901683 | −11.6 |
| 2. | CID_3822 | −10.8 |
| 3. | CID_442851 | −10.5 |
| 4. | CID_101277405 | −10.4 |
| 5. | CID_44257658 | −10.3 |
| 6. | CID_13846202 | −10.2 |
| 7. | CID_4970 | −10.2 |
| 8. | CID_101746 | −10.2 |
| 9. | CID_5245667 | −10.1 |
| 10. | CID_102060338 | −9.9 |
| 11. | CID_44257628 | −9.9 |
| 12. | CID_73393 | −9.8 |
| 13. | CID_480799 | −9.8 |
| 14. | CID_102267534 | −9.8 |
| 15. | CID_120734 | −9.7 |
| 16. | CID_440583 | −9.7 |
| 17. | CID_196979 | −9.7 |
| 18. | CID_4437370 | −9.7 |
| 19. | CID_10925304 | −9.7 |
| 20. | CID_5318619 | −9.7 |
| 21. | CID_14033985 | −9.7 |
| 22. | CASID_53777-78-9 | −9.6 |
| 23. | CID_6770 | −9.6 |
| 24. | CASID_22296-77-1 | −9.6 |
| 25. | CID_124069 | −9.6 |
| 26. | CID_147329 | −9.6 |
| 27. | CID_124050 | −9.6 |
| 28. | CID_443716 | −9.6 |
| 29. | CID_197775 | −9.6 |
| 30. | CID_601058 | −9.6 |
| 31. | CID_641765 | −9.6 |
| 32. | CID_5281353 | −9.6 |
| 33. | CID_5316096 | −9.6 |
| 34. | CID_5281406 | −9.6 |
| 35. | CID_101277371 | −9.6 |
| 36. | CID_101667973 | −9.6 |
| 37. | CASID_13241-28-6 | −9.5 |
| 38. | CID_120698 | −9.5 |
| 39. | CID_114909 | −9.5 |
| 40. | CID_5459059 | −9.5 |
| 41. | CID_6510278 | −9.5 |
| 42. | CID_5281867 | −9.5 |
| 43. | CASID_74148-50-8 | −9.4 |
| 44. | CID_101595 | −9.4 |
| 45. | CID_146680 | −9.4 |
| 46. | CID_10336244 | −9.4 |
| 47. | CID_5281809 | −9.3 |
| 48. | CASID_94418-50-5 | −9.3 |
| 49. | CASID_64191-02-2 | −9.2 |
| 50. | CID_5154 | −9.2 |
| 51. | LY333531 | −8.3 |
| Compound ID | Compound Name | GI Absorption | Water Solubility (log mol/L) | BBB Permeability (log BB) | CYP2D6 Inhibitor | OCT2 Substrate | AMES |
|---|---|---|---|---|---|---|---|
| CID_5318619 | Isoononin | High | −3.39 | −1.27 | No | No | No |
| CID_44257658 | Torosaflavone A | High | −2.90 | −1.37 | No | No | No |
| CID_13846202 | 11-Hydroxytephrosin | High | −3.89 | −0.18 | No | No | No |
| LY333531 | Ruboxistaurin | High | −4.81 | −0.48 | No | No | Yes |
| Compound | Pa | Pi | Biological Activity |
|---|---|---|---|
| 11-Hydroxytephrosin | 0.934 | 0.004 | Antineoplastic |
| 0.904 | 0.003 | Antineoplastic (non-small cell lung cancer) | |
| 0.857 | 0.003 | Prostate cancer treatment | |
| 0.822 | 0.003 | Antineoplastic (ovarian cancer) | |
| 0.799 | 0.004 | Chemopreventive | |
| 0.775 | 0.014 | TP53 expression enhancer | |
| 0.654 | 0.020 | Apoptosis agonist | |
| 0.644 | 0.016 | Kinase inhibitor | |
| 0.542 | 0.005 | Antioxidant | |
| 0.540 | 0.015 | Antineoplastic (breast cancer) | |
| Torosaflavone A | 0.929 | 0.005 | Membrane integrity agonist |
| 0.889 | 0.006 | TP53 expression enhancer | |
| 0.860 | 0.006 | Antineoplastic | |
| 0.840 | 0.003 | Chemopreventive | |
| 0.832 | 0.003 | Cardioprotectant | |
| 0.827 | 0.005 | Kinase inhibitor | |
| 0.821 | 0.005 | Anticarcinogenic | |
| 0.800 | 0.004 | Hepatoprotectant | |
| 0.730 | 0.012 | Apoptosis agonist | |
| 0.725 | 0.009 | Antifungal | |
| LY333531 (ruboxistaurin) | 0.869 | 0.007 | Antineurotic |
| 0.823 | 0.009 | Antineoplastic | |
| 0.788 | 0.004 | Chemo preventive | |
| 0.745 | 0.011 | Apoptosis agonist | |
| 0.720 | 0.044 | Hepatoprotectant | |
| 0.710 | 0.012 | TP53 expression enhancer | |
| 0.707 | 0.010 | Kinase inhibitor | |
| 0.699 | 0.008 | Anticarcinogenic | |
| 0.638 | 0.004 | Antidiabetic | |
| 0.612 | 0.010 | Antioxidant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atiya, A.; Alhumaydhi, F.A.; Sharaf, S.E.; Al Abdulmonem, W.; Elasbali, A.M.; Al Enazi, M.M.; Shamsi, A.; Jawaid, T.; Alghamdi, B.S.; Hashem, A.M.; et al. Identification of 11-Hydroxytephrosin and Torosaflavone A as Potential Inhibitors of 3-Phosphoinositide-Dependent Protein Kinase 1 (PDPK1): Toward Anticancer Drug Discovery. Biology 2022, 11, 1230. https://doi.org/10.3390/biology11081230
Atiya A, Alhumaydhi FA, Sharaf SE, Al Abdulmonem W, Elasbali AM, Al Enazi MM, Shamsi A, Jawaid T, Alghamdi BS, Hashem AM, et al. Identification of 11-Hydroxytephrosin and Torosaflavone A as Potential Inhibitors of 3-Phosphoinositide-Dependent Protein Kinase 1 (PDPK1): Toward Anticancer Drug Discovery. Biology. 2022; 11(8):1230. https://doi.org/10.3390/biology11081230
Chicago/Turabian StyleAtiya, Akhtar, Fahad A. Alhumaydhi, Sharaf E. Sharaf, Waleed Al Abdulmonem, Abdelbaset Mohamed Elasbali, Maher M. Al Enazi, Anas Shamsi, Talha Jawaid, Badrah S. Alghamdi, Anwar M. Hashem, and et al. 2022. "Identification of 11-Hydroxytephrosin and Torosaflavone A as Potential Inhibitors of 3-Phosphoinositide-Dependent Protein Kinase 1 (PDPK1): Toward Anticancer Drug Discovery" Biology 11, no. 8: 1230. https://doi.org/10.3390/biology11081230
APA StyleAtiya, A., Alhumaydhi, F. A., Sharaf, S. E., Al Abdulmonem, W., Elasbali, A. M., Al Enazi, M. M., Shamsi, A., Jawaid, T., Alghamdi, B. S., Hashem, A. M., Ashraf, G. M., & Shahwan, M. (2022). Identification of 11-Hydroxytephrosin and Torosaflavone A as Potential Inhibitors of 3-Phosphoinositide-Dependent Protein Kinase 1 (PDPK1): Toward Anticancer Drug Discovery. Biology, 11(8), 1230. https://doi.org/10.3390/biology11081230