
Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and Genome Sequencing
2.3. SNP Calling
2.4. Population Genetic Structure
2.5. Genomic Diversity
2.6. Genome-Wide Selection
2.7. Functional Prediction Analysis
3. Results
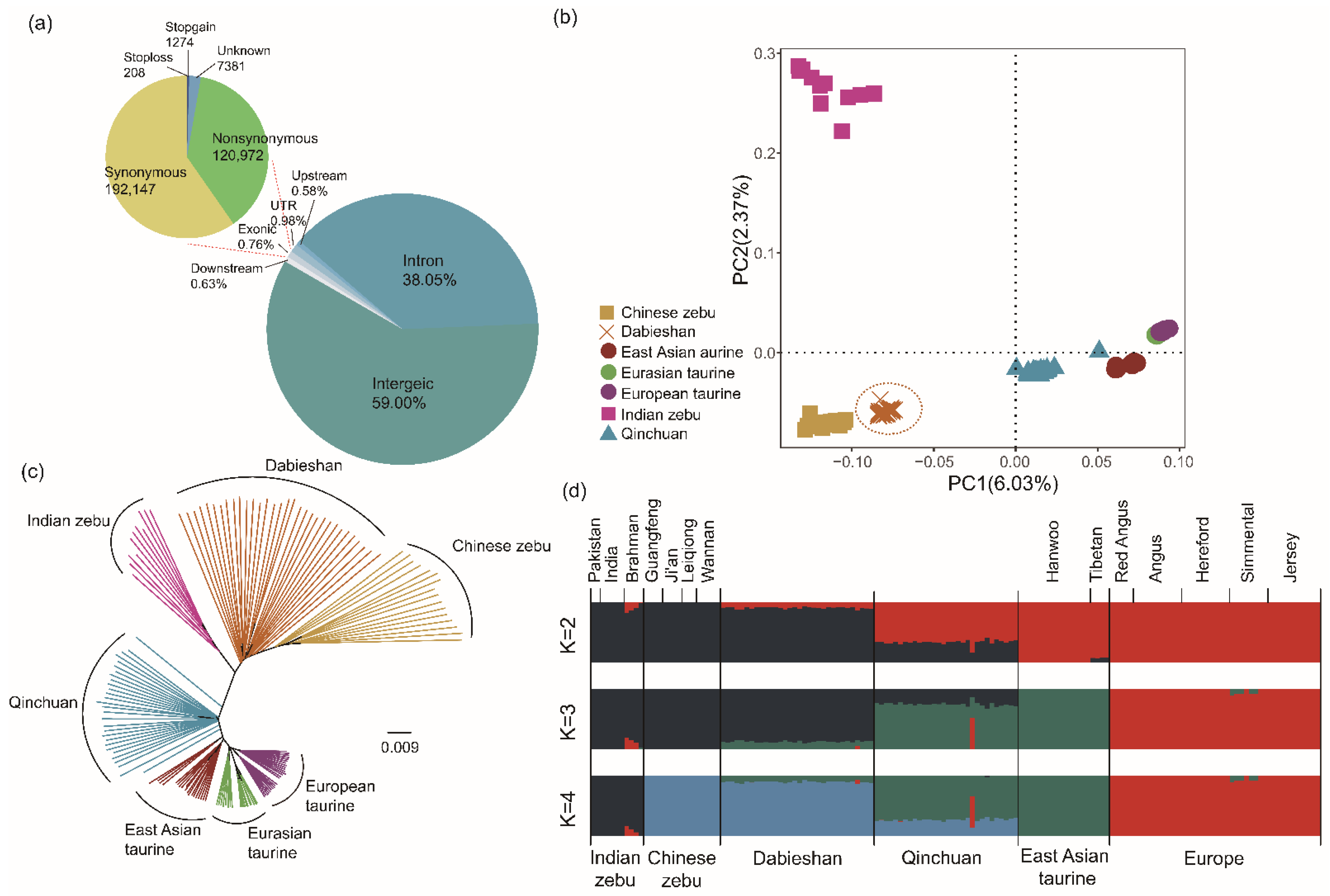
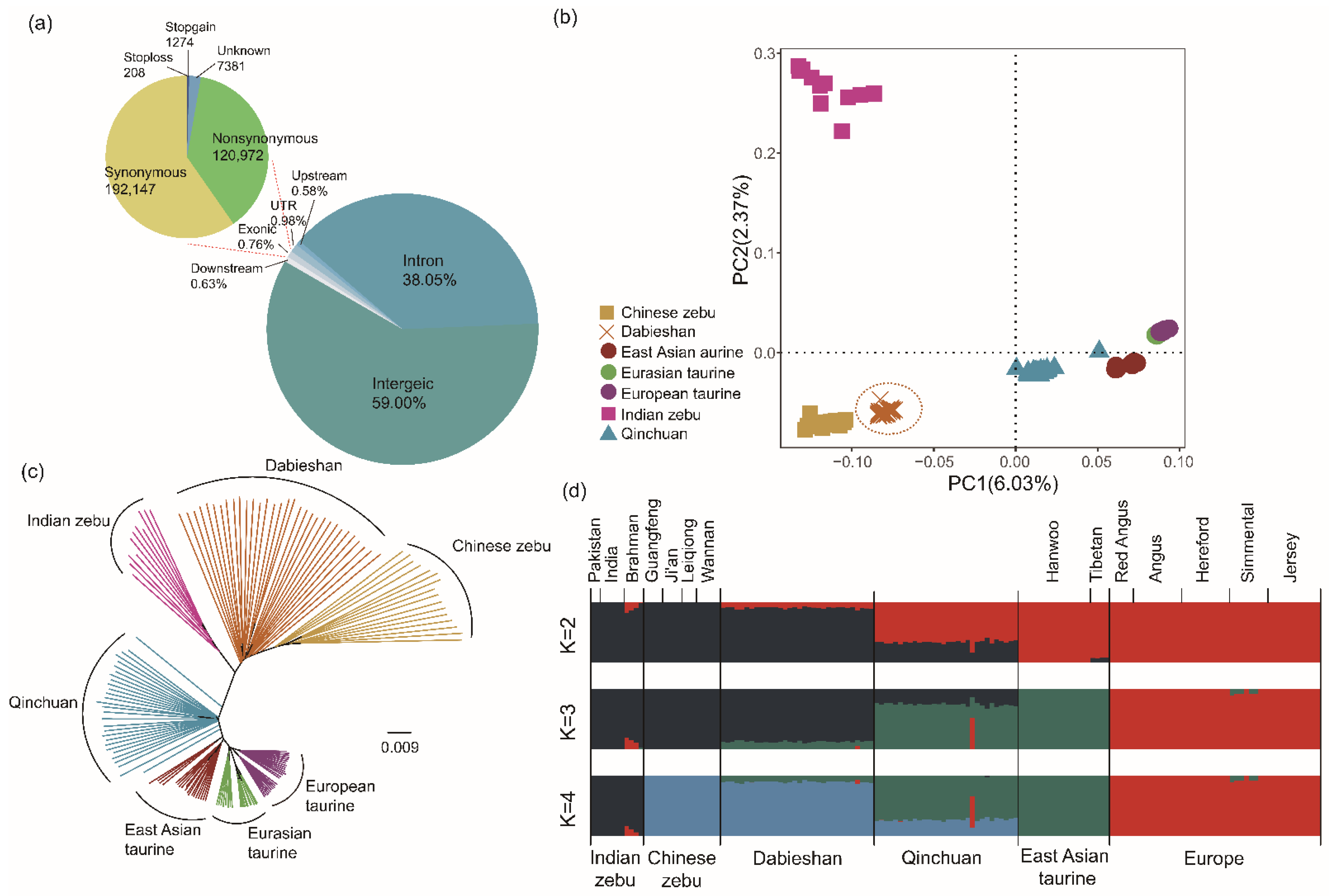
3.1. Whole-Genome Sequencing, Assembly, and Genetic Variation
3.2. Population Structure
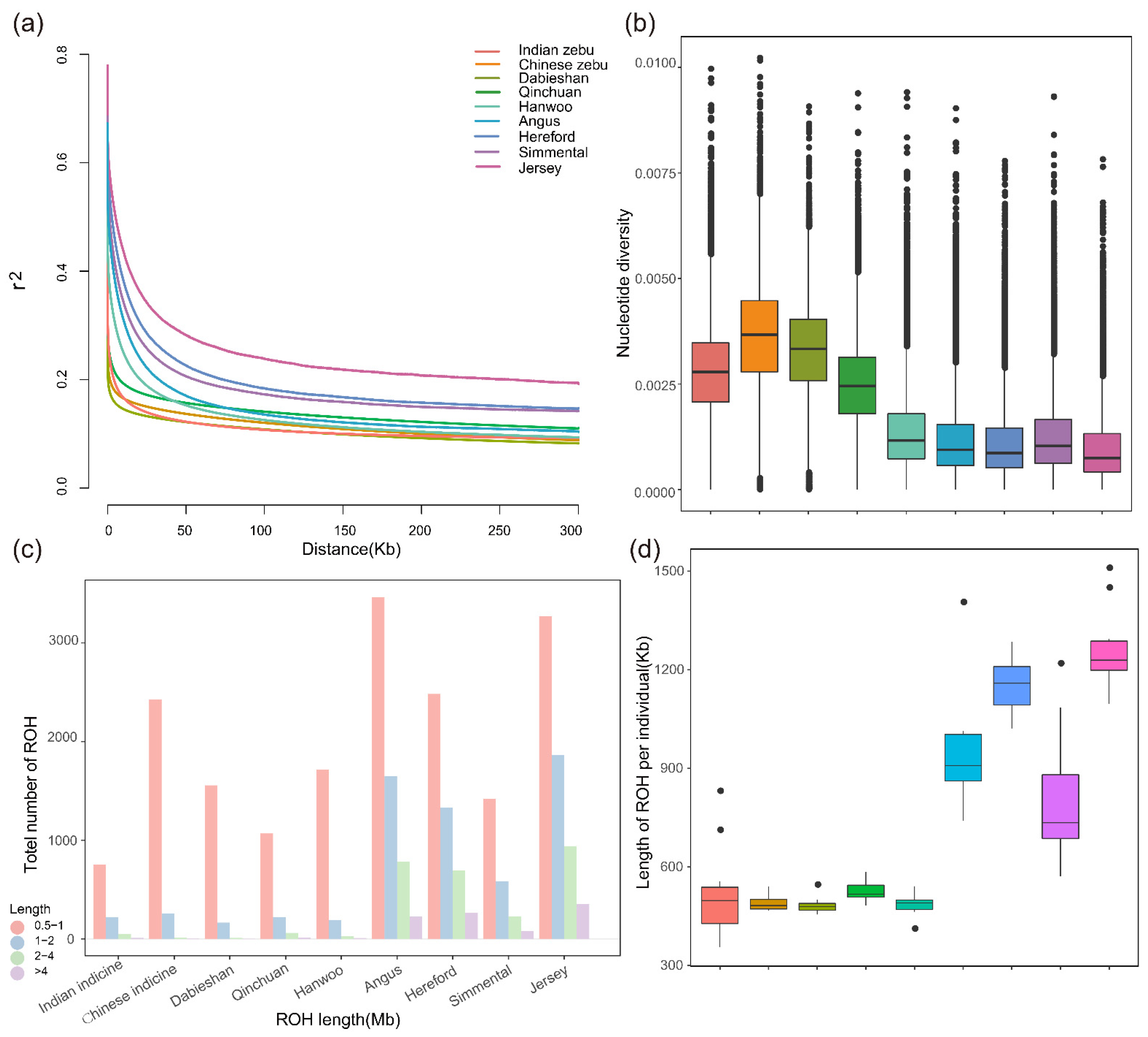
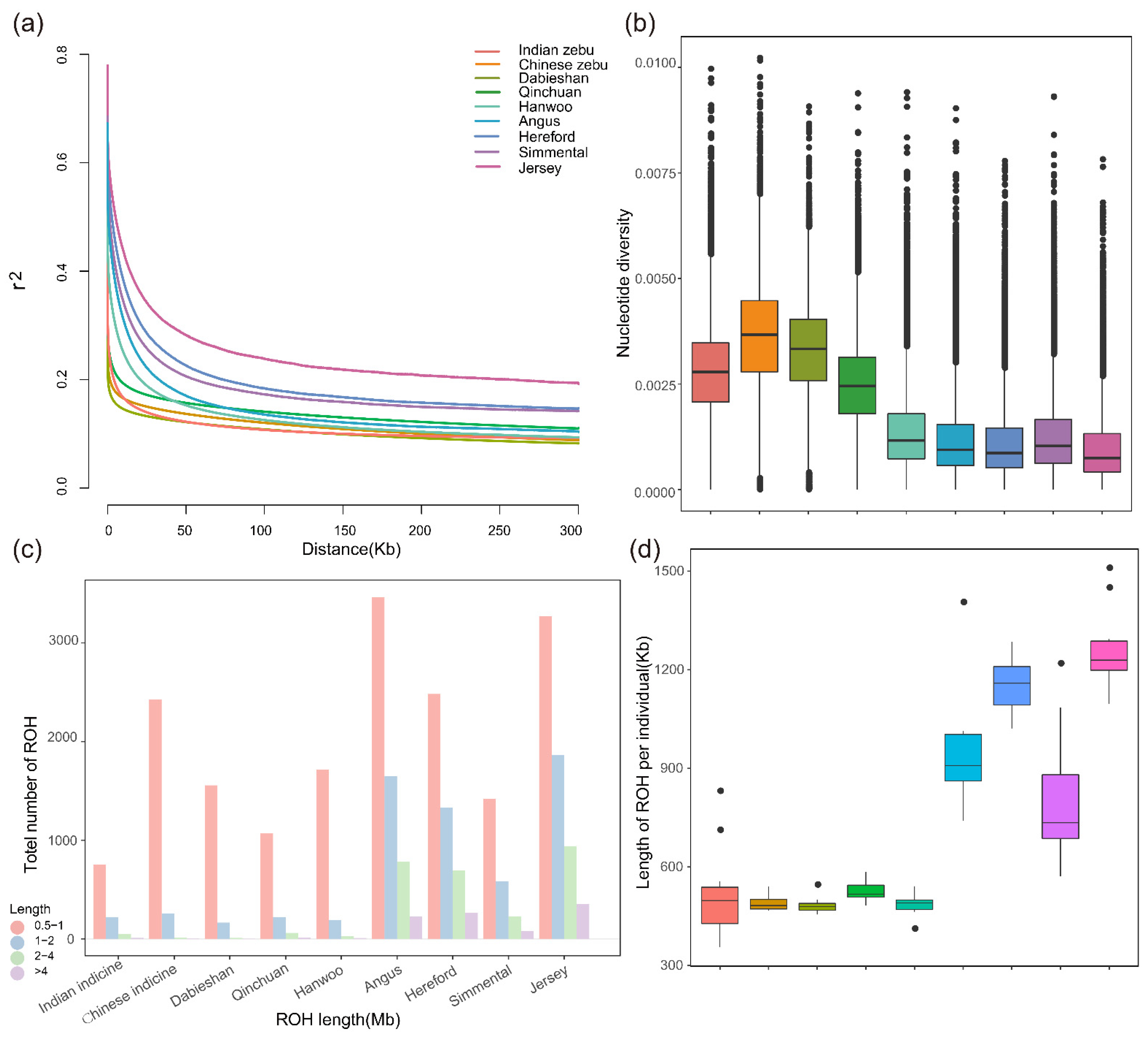
3.3. Patterns of Genomic Variation
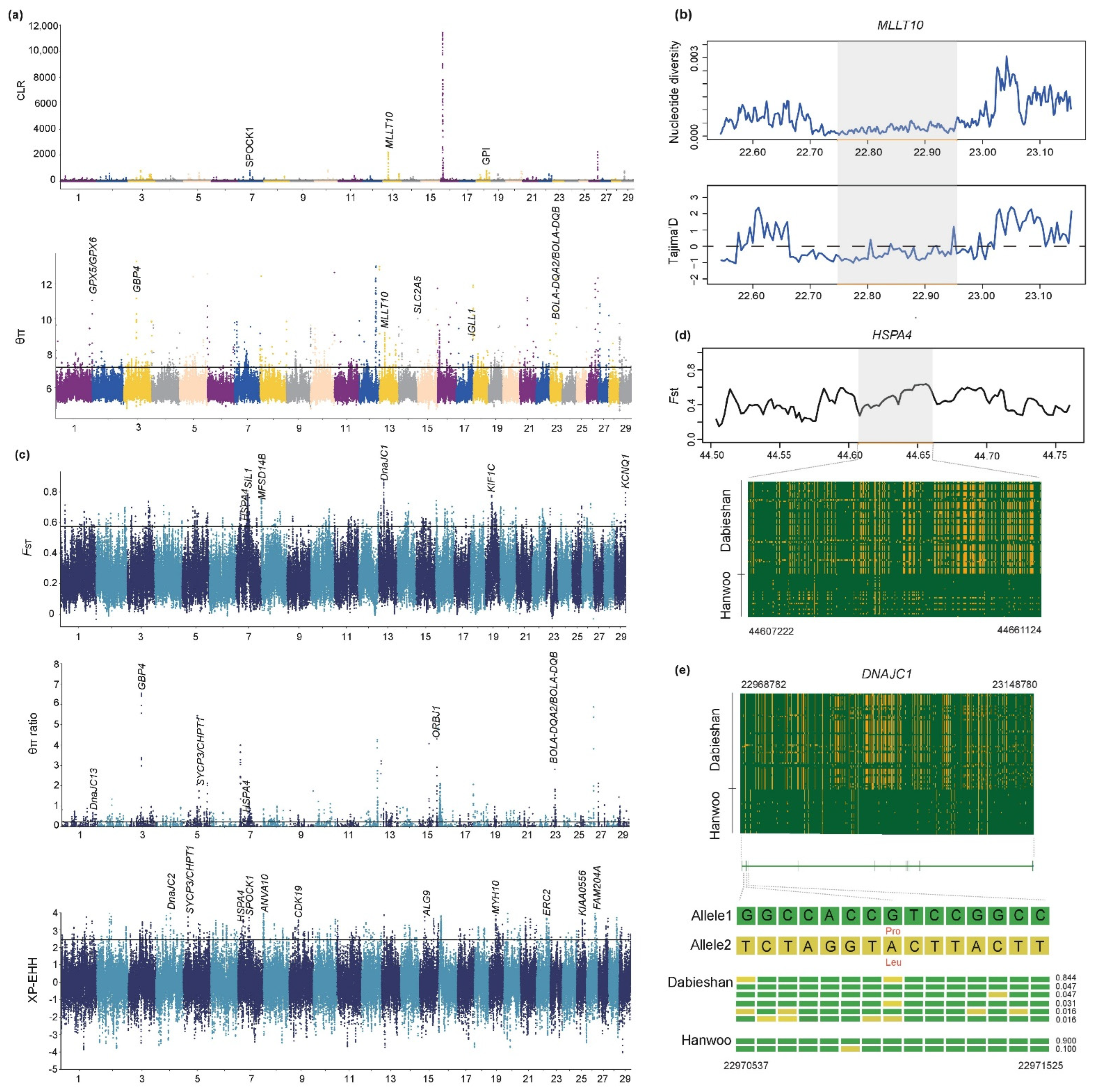
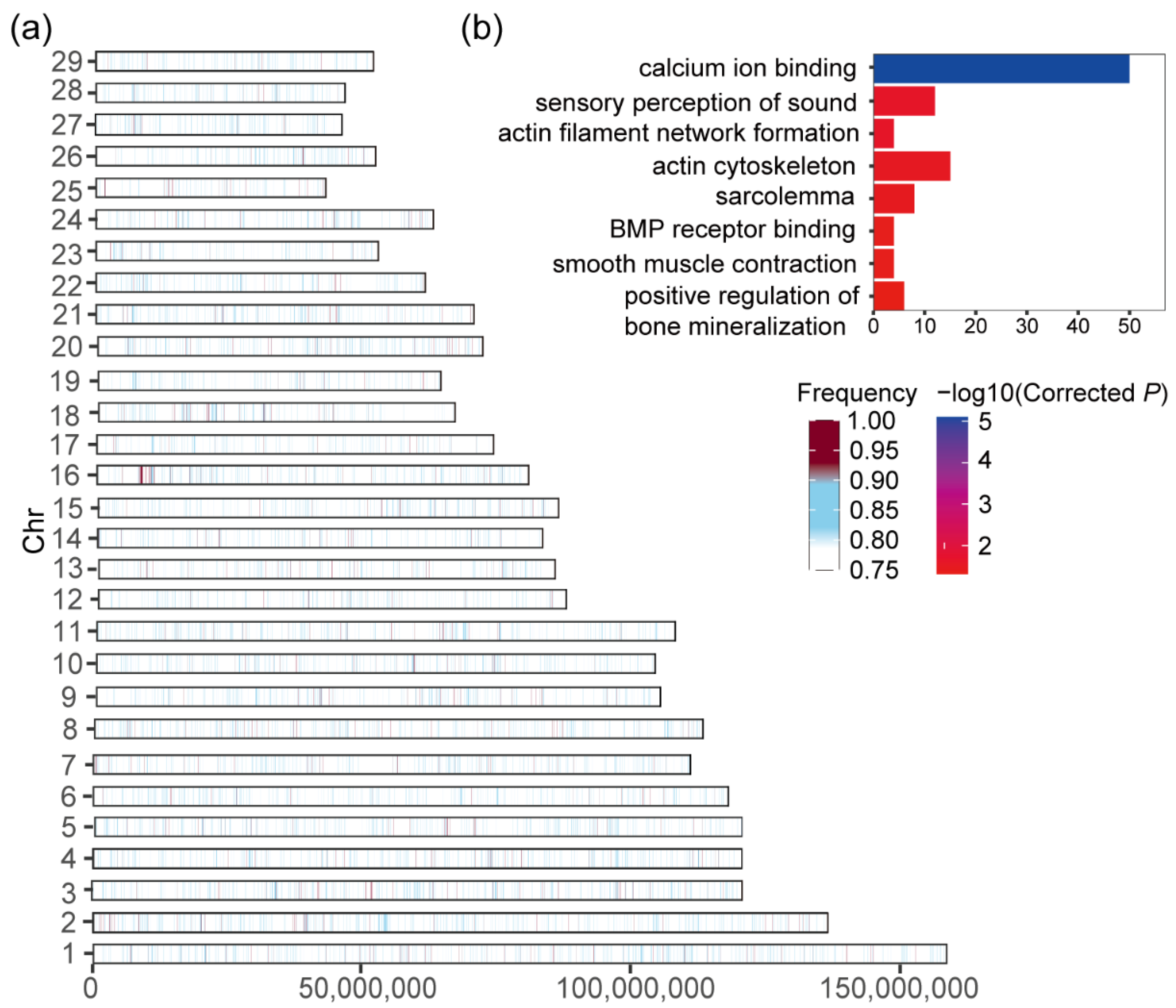
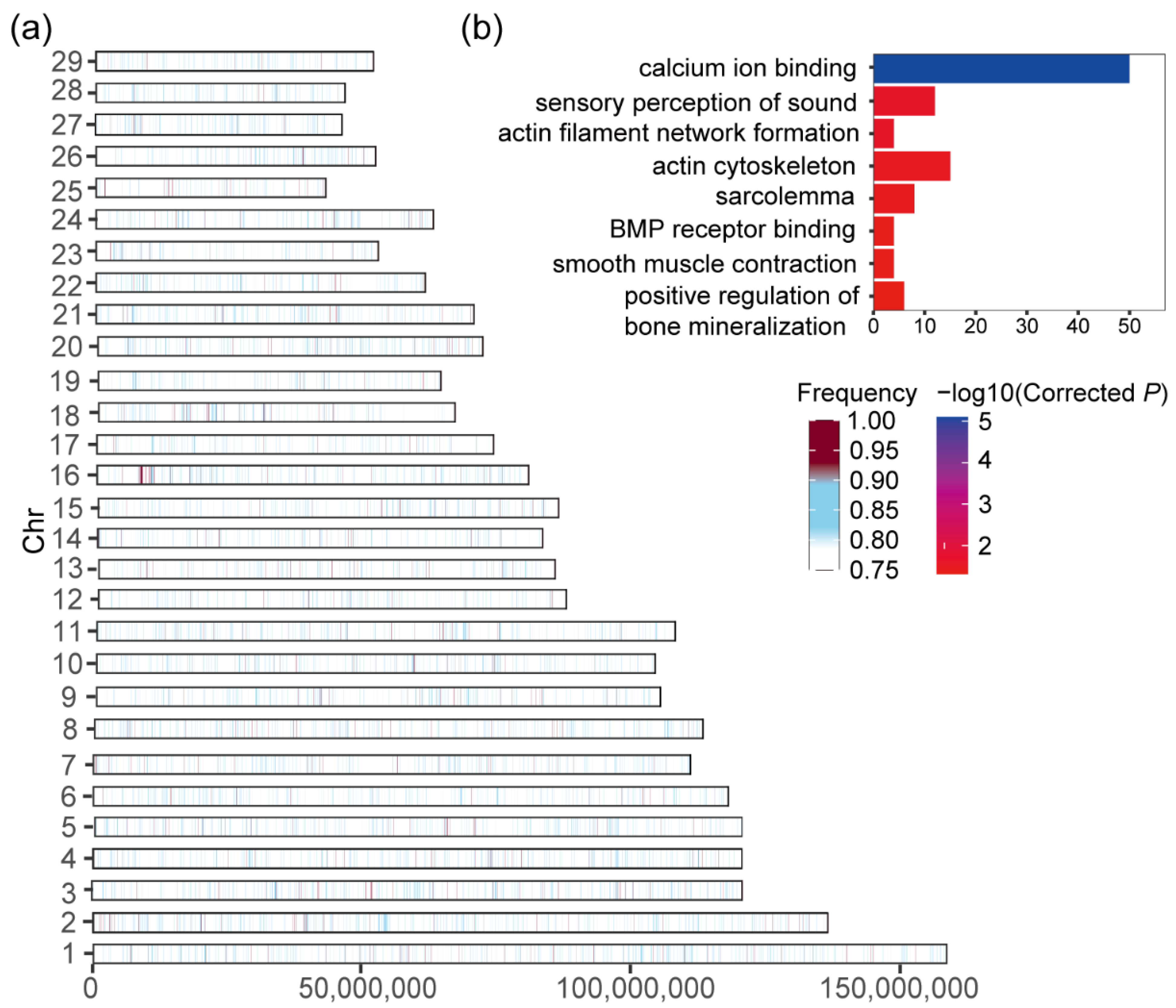
3.4. Candidate Genes under Selective Sweep
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hesse, B. The first steps of animal domestication. J. Ethnobiol. 2006, 26, 171–174. [Google Scholar] [CrossRef]
- Loftus, R.T.; MacHugh, D.E.; Bradley, D.G.; Sharp, P.M.; Cunningham, P. Evidence for two independent domestications of cattle. Proc. Natl. Acad. Sci. USA 1994, 91, 2757–2761. [Google Scholar] [CrossRef] [PubMed]
- Felius, M. On the Breeds of Cattle: Their History, Classification and Conservation, 2nd ed.; Utrecht University: Utrecht, The Netherlands, 2016; ISBN 978-90-393-6471-0. [Google Scholar]
- Upadhyay, M.; Bortoluzzi, C.; Barbato, M.; Ajmone-Marsan, P.; Colli, L.; Ginja, C.; Sonstegard, T.S.; Bosse, M.; Lenstra, J.A.; Groenen, M.; et al. Deciphering the patterns of genetic admixture and diversity in southern European cattle using genome-wide SNPs. Evol. Appl. 2019, 12, 951–963. [Google Scholar] [CrossRef] [PubMed]
- China National Commission of Animal Genetic Resources. Animal Genetic Resources in China Bovines; Chinese Agricultural Press: Beijing, China, 2011. (In Chinese)
- Xia, X.; Qu, K.; Zhang, G.; Jia, Y.; Ma, Z.; Zhao, X.; Huang, Y.; Chen, H.; Huang, B.; Lei, C. Comprehensive analysis of the mitochondrial DNA diversity in Chinese cattle. Anim. Genet. 2019, 50, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Yao, Y.; Li, C.; Zhang, F.; Qu, K.; Chen, H.; Huang, B.; Lei, C. Genetic diversity of Chinese cattle revealed by Y-SNP and Y-STR markers. Anim. Genet. 2019, 50, 64–69. [Google Scholar] [CrossRef]
- Kim, J.; Hanotte, O.; Mwai, O.A.; Dessie, T.; Bashir, S.; Diallo, B.; Agaba, M.; Kim, K.; Kwak, W.; Sung, S.; et al. The genome landscape of indigenous African cattle. Genome Biol. 2017, 18, 34. [Google Scholar] [CrossRef]
- Shen, J.; Hanif, Q.; Cao, Y.; Yu, Y.; Lei, C.; Zhang, G.; Zhao, Y. Whole Genome Scan and Selection Signatures for Climate Adaption in Yanbian Cattle. Front. Genet. 2020, 11, 94. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2012. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, l.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef]
- DeGiorgio, M.; Huber, C.D.; Hubisz, M.J.; Hellmann, I.; Nielsen, R. SweepFinder2: Increased sensitivity, robustness and flexibility. Bioinformatics 2016, 32, 1895–1897. [Google Scholar] [CrossRef]
- Hudson, R.R.; Slatkin, M.; Maddison, W.P. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583–589. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef]
- Maples, B.K.; Gravel, S.; Kenny, E.E.; Bustamante, C.D. RFMix: A discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 2013, 93, 278–288. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Lachance, J.; Vernot, B.; Elbers, C.C.; Ferwerda, B.; Froment, A.; Bodo, J.M.; Lema, G.; Fu, W.; Nyambo, T.B.; Rebbeck, T.R.; et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 2012, 150, 457–469. [Google Scholar] [CrossRef]
- Choi, J.W.; Liao, X.; Park, S.; Jeon, H.J.; Chung, W.H.; Stothard, P.; Park, Y.S.; Lee, J.K.; Lee, K.T.; Kim, S.H.; et al. Massively parallel sequencing of Chikso (Korean brindle cattle) to discover genome-wide SNPs and InDels. Mol. Cells 2013, 36, 203–211. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, K.; Zhang, D.; Zhao, Z.; Huang, J.; Zhou, L.; Feng, M.; Shi, J.; Wei, H.; Li, L.; et al. GPx6 is involved in the in vitro induced capacitation and acrosome reaction in porcine sperm. Theriogenology 2020, 156, 107–115. [Google Scholar] [CrossRef]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Heaney, A.P. Regulation of adipose differentiation by fructose and GluT5. Mol. Endocrinol. 2012, 26, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Diniz, W.J.; da Rosa, K.O.; Tizioto, P.C.; Mourão, G.B.; de Oliveira, P.S.; de Souza, M.M.; Regitano, L.C. FABP1 and SLC2A5 expression levels affect feed efficiency-related traits. Agri Gene 2020, 15, 100100. [Google Scholar] [CrossRef]
- Ekman, A.; Ilves, M.; Iivanainen, A. B lymphopoiesis is characterized by pre-B cell marker gene expression in fetal cattle and declines in adults. Dev. Comp. Immunol. 2012, 37, 39–49. [Google Scholar] [CrossRef]
- Fries, R.; Hediger, R.; Stranzinger, G. Tentative chromosomal localization of the bovine major histocompatibility complex by in situ hybridization. Anim. Genet. 1986, 17, 287–294. [Google Scholar] [CrossRef]
- Spooner, R.L.; Leveziel, H.; Grosclaude, F.; Oliver, R.A.; Vaiman, M. Evidence for a possible major histocompatibility complex (BLA) in cattle. J. Immunogenet. 1978, 5, 325–346. [Google Scholar] [CrossRef]
- Behl, J.D.; Verma, N.K.; Tyagi, N.; Mishra, P.; Behl, R.; Joshi, B.K. The major histocompatibility complex in bovines: A review. ISRN Vet. Sci. 2012, 2012, 872710. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, Z.; Xu, J.; Qu, K.; Lyv, S.; Wang, X.; Cai, C.; Li, Z.; Wang, E.; Xie, J.; et al. The Association of the Copy Number Variation of the MLLT10 Gene with Growth Traits of Chinese Cattle. Animals 2020, 10, 250. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, J.; Lee, T.; Son, J.K.; Yoon, H.B.; Baek, K.S.; Jeong, J.Y.; Cho, Y.M.; Lee, K.T.; Yang, B.C.; et al. Deciphering the genetic blueprint behind Holstein milk proteins and production. Genome Biol. Evol. 2014, 6, 1366–1374. [Google Scholar] [CrossRef]
- Chevalier, M.; Rhee, H.; Elguindi, E.C.; Blond, S.Y. Interaction of murine BiP/GRP78 with the DnaJ homologue MTJ1. J. Biol. Chem. 2000, 275, 19620–19627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahi, C.; Kominek, J.; Ziegelhoffer, T.; Yu, H.Y.; Baranowski, M.; Marszalek, J.; Craig, E.A. Sequential duplications of an ancient member of the DnaJ-family expanded the functional chaperone network in the eukaryotic cytosol. Mol. Biol. Evol. 2013, 30, 985–998. [Google Scholar] [CrossRef]
- Beckham, J.T.; Mackanos, M.A.; Crooke, C.; Takahashi, T.; O’Connell-Rodwell, C.; Contag, C.H.; Jansen, E.D. Assessment of cellular response to thermal laser injury through bioluminescence imaging of heat shock protein 70. Photochem. Photobiol. 2004, 79, 76–85. [Google Scholar] [CrossRef]
- Basiricò, L.; Morera, P.; Primi, V.; Lacetera, N.; Nardone, A.; Bernabucci, U. Cellular thermotolerance is associated with heat shock protein 70.1 genetic polymorphisms in Holstein lactating cows. Cell Stress Chaperones 2011, 16, 441–448. [Google Scholar] [CrossRef]
- Trigo, B.B.; Utsunomiya, A.; Fortunato, A.; Milanesi, M.; Torrecilha, R.; Lamb, H.; Nguyen, L.; Ross, E.M.; Hayes, B.; Padula, R.; et al. Variants at the ASIP locus contribute to coat color darkening in Nellore cattle. Genet. Sel. Evol. GSE 2021, 53, 40. [Google Scholar] [CrossRef]
- Chen, Q.; Shen, J.; Hanif, Q.; Chen, N.; Huang, Y.; Dang, R.; Lan, X.; Chen, H.; Lei, C. Whole genome analyses revealed genomic difference between European taurine and East Asian taurine. J. Anim. Breed. Genet. 2021, 138, 56–68. [Google Scholar] [CrossRef]
- Zhang, F.; Qu, K.; Chen, N.; Hanif, Q.; Jia, Y.; Huang, Y.; Dang, R.; Zhang, J.; Lan, X.; Chen, H.; et al. Genome-Wide SNPs and InDels Characteristics of Three Chinese Cattle Breeds. Animal 2019, 9, 596. [Google Scholar] [CrossRef]
- Singh, A.K.; Liu, W.; Zakari, S.; Wu, J.; Yang, B.; Jiang, X.J.; Zhu, X.; Zou, X.; Zhang, W.; Chen, C.; et al. A global review of rubber plantations: Impacts on ecosystem functions, mitigations, future directions, and policies for sustainable cultivation. Sci. Total Environ. 2021, 796, 148948. [Google Scholar] [CrossRef]
- Pausch, H.; Kölle, S.; Wurmser, C.; Schwarzenbacher, H.; Emmerling, R.; Jansen, S.; Trottmann, M.; Fuerst, C.; Götz, K.U.; Fries, R. A nonsense mutation in TMEM95 encoding a nondescript transmembrane protein causes idiopathic male subfertility in cattle. PLoS Genet. 2014, 10, e1004044. [Google Scholar] [CrossRef]
- Kadri, N.K.; Sahana, G.; Charlier, C.; Iso-Touru, T.; Guldbrandtsen, B.; Karim, L.; Nielsen, U.S.; Panitz, F.; Aamand, G.P.; Schulman, N.; et al. A 660-Kb deletion with antagonistic effects on fertility and milk production segregates at high frequency in Nordic Red cattle: Additional evidence for the common occurrence of balancing selection in livestock. PLoS Genet. 2014, 10, e1004049. [Google Scholar] [CrossRef]
- Pausch, H.; Ammermüller, S.; Wurmser, C.; Hamann, H.; Tetens, J.; Drögemüller, C.; Fries, R. A nonsense mutation in the COL7A1 gene causes epidermolysis bullosa in Vorderwald cattle. BMC Genet. 2016, 17, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signer-Hasler, H.; Burren, A.; Neuditschko, M.; Frischknecht, M.; Garrick, D.; Stricker, C.; Gredler, B.; Bapst, B.; Flury, C. Population structure and genomic inbreeding in nine Swiss dairy cattle populations. Genet. Sel. Evol. GSE 2017, 49, 83. [Google Scholar] [CrossRef] [PubMed]
- Rothammer, S.; Seichter, D.; Förster, M.; Medugorac, I. A genome-wide scan for signatures of differential artificial selection in ten cattle breeds. BMC Genom. 2013, 14, 908. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.; Baccari, F., Jr.; Titto, E.A.; Almeida, J.A. Effect of thermal stress on physiological parameters, feed intake and plasma thyroid hormones concentration in Alentejana, Mertolenga, Frisian and Limousine cattle breeds. Int. J. Biometeorol. 2008, 52, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Berman, A. Nychthemeral and seasonal patterns of thermoregulation in cattle. Aust. J. Agric. Res. 1968, 19, 648–657. [Google Scholar] [CrossRef]
- Pilla-Moffett, D.; Barber, M.F.; Taylor, G.A.; Coers, J. Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J. Mol. Biol. 2016, 428, 3495–3513. [Google Scholar] [CrossRef] [PubMed]
- Chabory, E.; Damon, C.; Lenoir, A.; Henry-Berger, J.; Vernet, P.; Cadet, R.; Saez, F.; Drevet, J.R. Mammalian glutathione peroxidases control acquisition and maintenance of spermatozoa integrity. J. Anim. Sci. 2010, 88, 1321–1331. [Google Scholar] [CrossRef]
- Sherman, E.L.; Nkrumah, J.D.; Moore, S.S. Whole genome single nucleotide polymorphism associations with feed intake and feed efficiency in beef cattle. J. Anim. Sci. 2010, 88, 16–22. [Google Scholar] [CrossRef]
- Wang, K.; Liu, D.; Hernandez-Sanchez, J.; Chen, J.; Liu, C.; Wu, Z.; Fang, M.; Li, N. Genome Wide Association Analysis Reveals New Production Trait Genes in a Male Duroc Population. PLoS ONE 2015, 10, e0139207. [Google Scholar] [CrossRef]
- Taye, M.; Kim, J.; Yoon, S.H.; Lee, W.; Hanotte, O.; Dessie, T.; Kemp, S.; Mwai, O.A.; Caetano-Anolles, K.; Cho, S.; et al. Whole genome scan reveals the genetic signature of African Ankole cattle breed and potential for higher quality beef. BMC Genet. 2017, 18, 11. [Google Scholar] [CrossRef]
- Winbanks, C.E.; Chen, J.L.; Qian, H.; Liu, Y.; Bernardo, B.C.; Beyer, C.; Watt, K.I.; Thomson, R.E.; Connor, T.; Turner, B.J.; et al. The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J. Cell Biol. 2013, 203, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Calhabeu, F.; Morgan, J.E.; Katagiri, T.; Amthor, H.; Zammit, P.S. BMP signalling permits population expansion by preventing premature myogenic differentiation in muscle satellite cells. Cell Death Differ. 2011, 18, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Gozo, M.C.; Aspuria, P.J.; Cheon, D.J.; Walts, A.E.; Berel, D.; Miura, N.; Karlan, B.Y.; Orsulic, S. Foxc2 induces Wnt4 and Bmp4 expression during muscle regeneration and osteogenesis. Cell Death Differ. 2013, 20, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Santos Silva, D.; Fonseca, L.; Magalhães, A.; Muniz, M.; Baldi, F.; Ferro, J.A.; Chardulo, L.; Pinheiro, D.G.; Albuquerque, L.G. Transcriptome profiling of muscle in Nelore cattle phenotypically divergent for the ribeye muscle area. Genomics 2020, 112, 1257–1263. [Google Scholar] [CrossRef]
- Taniguchi, M.; Guan, L.L.; Zhang, B.; Dodson, M.V.; Okine, E.; Moore, S.S. Gene expression patterns of bovine perimuscular preadipocytes during adipogenesis. Biochem. Biophys. Res. Commun. 2008, 366, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Karpe, F.; Fielding, B.A.; Macdonald, I.A.; Coppack, S.W. Integrative physiology of human adipose tissue. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2003, 27, 875–888. [Google Scholar] [CrossRef]
- Liang, R.R.; Zhu, H.; Mao, Y.W.; Zhang, Y.M.; Zhu, L.X.; Cornforth, D.; Wang, R.H.; Meng, X.Y.; Luo, X. Tenderness and sensory attributes of the longissimus lumborum muscles with different quality grades from Chinese fattened yellow crossbred steers. Meat Sci. 2016, 112, 52–57. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, X.; Zhao, S.; Xiang, W.; Jin, H.; Chen, N.; Lei, C.; Jia, Y.; Xu, L. Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology 2022, 11, 1327. https://doi.org/10.3390/biology11091327
Guan X, Zhao S, Xiang W, Jin H, Chen N, Lei C, Jia Y, Xu L. Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology. 2022; 11(9):1327. https://doi.org/10.3390/biology11091327
Chicago/Turabian StyleGuan, Xiwen, Shuanping Zhao, Weixuan Xiang, Hai Jin, Ningbo Chen, Chuzhao Lei, Yutang Jia, and Lei Xu. 2022. "Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing" Biology 11, no. 9: 1327. https://doi.org/10.3390/biology11091327
APA StyleGuan, X., Zhao, S., Xiang, W., Jin, H., Chen, N., Lei, C., Jia, Y., & Xu, L. (2022). Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology, 11(9), 1327. https://doi.org/10.3390/biology11091327