Simple Summary
Even though in monogenic diseases a mutation will lead to a “classic” manifestation, many disorders exhibit great clinical variability that could be due to modifying genes also called minor genes. Fabry disease (FD) is an X-linked inborn error resulting from the deficient or absent activity of alpha-galactosidase A (α-GAL) enzyme, that leads to deposits of globotriaosylceramide. With our proprietary software SNPclinic v.1.0, we analyzed 110 single nucleotide polymorphisms (SNPs) in the proximal promoter of 14 genes that could be modifying the phenotype of FD. We found seven regulatory-SNP (rSNPs) in three genes (IL10, TGFB1 and EDN1) in five cell lines relevant to FD (cardiac myocytes, cardiac fibroblasts, astrocytes-cerebellar, endothelial cells and T helper cells 1-TH1). Each SNP was confirmed as a true rSNP in public eQTL databases and prediction of variants was suggested by additional software. The two proposed rSNPs in IL10, could explain components for the regulation of active B cells that influence the fibrosis process. The three predicted rSNPs in TGFB1, could act in apoptosis-autophagy regulation. The two putative rSNPs in EDN1, putatively regulate chronic inflammation. The rSNPs described here could act to modulate Fabry’s clinical phenotype so we propose that IL10, TGFB1 and EDN1 be considered genetic modifiers in FD.
Abstract
Even though a mutation in monogenic diseases leads to a “classic” manifestation, many disorders exhibit great clinical variability that could be due to modifying genes also called minor genes. Fabry disease (FD) is an X-linked inborn error resulting from the deficient or absent activity of alpha-galactosidase A (α-GAL) enzyme, that leads to deposits of globotriaosylceramide. With our proprietary software SNPclinic v.1.0, we analyzed 110 single nucleotide polymorphisms (SNPs) in the proximal promoter of 14 genes that could modify the FD phenotype FD. We found seven regulatory-SNP (rSNPs) in three genes (IL10, TGFB1 and EDN1) in five cell lines relevant to FD (Cardiac myocytes and fibroblasts, Astrocytes-cerebellar, endothelial cells and T helper cells 1-TH1). Each SNP was confirmed as a true rSNP in public eQTL databases, and additional software suggested the prediction of variants. The two proposed rSNPs in IL10, could explain components for the regulation of active B cells that influence the fibrosis process. The three predicted rSNPs in TGFB1, could act in apoptosis-autophagy regulation. The two putative rSNPs in EDN1, putatively regulate chronic inflammation. The seven rSNPs described here could act to modulate Fabry’s clinical phenotype so we propose that IL10, TGFB1 and EDN1 be considered minor genes in FD.
Keywords:
Fabry disease; promoter; regulatory SNPs; modifying genes; clinical variability; IL10; TGFB1; EDN1 1. Introduction
In monogenic diseases a mutation leads to a “classic” manifestation, but many disorders exhibit a great clinical or phenotypical variability that cannot be explained only by the mutation in the major gene. This variability could be due to modifying or minor genes that modulate the disease’s phenotype []. Such genes could act directly on the gene product causing the disease or indirectly on alternative etiopathogenic pathways []. Therefore, the role those genetic variants play in each disease can be in the phenotypic expression, severity, or even the age of onset in patients []. When genetic interactions occur, such as combinations between mutations in different genes, an unexpected phenotype may occur, which would differ from the effects of the individual mutant phenotypes []. The effect of the combination of mutations has been studied in the cardiomyopathy called left ventricular non-compaction (LVNC). In a family of three children with early-onset cardiomyopathy, the complete exome was sequenced and the presence of three variants in the MKL2, MYH7 and NKX2 genes was found, the variants in the first two genes mentioned were inherited from the asymptomatic affected father and the rare variant in NKX2 from the unaffected mother. The evaluation of functional consequences in vivo in murine models led to the conclusion that NKX2 acted as a modifier gene []. It is important to emphasize that the effect may not only be towards an increase in clinical features but also a suppression effect by interaction can also occur, where some modifying gene may suppress the expected phenotype []. Both effects have been seen in rare Mendelian diseases, such as lysosomal diseases, but much remains to be deciphered.
Fabry disease (FD) (OMIM #301500) [] is an X-linked inborn error of glycosphingolipid catabolism resulting from the deficient or absent activity of the lysosomal enzyme alpha-galactosidase A (α-GAL). The functional modification of the enzyme causes the accumulation of complex sphingolipids, especially globotriaosylceramide (Gb3) [].
Depending on the enzyme activity, two main phenotypes in FD have been recognized: the multisystemic subtype with a classic phenotype since childhood due to scarce or null α-GAL activity, and the late-onset subtype which usually begins in adulthood with failure of organs such as the heart, kidney or brain and results from the partial activity of α-GAL [].
Vascular endothelium cells are the main affected, it has been observed that excessive intracellular Gb3 induces oxidative stress and expression of cell adhesion molecules in endothelial cells []. The exact location of these deposits has been identified in GLA KO mouse [] in which the accumulation of Gb3 occurs in the lipid rafts of the cell membranes []. The deposits lead to pathogenic cascades that culminate in an inflammatory response in any tissue []. This complex process requires the participation of inflammatory cytokines (IFNγ, TNF-α), chemokines, effector enzymes as well as anti-inflammatory cytokines (IL-10, IL-4, and IL-13) that counteract any chronic inflammation. A dysfunctional autophagy pathway that could contribute to the pathological process has been found in cultured renal cells, fibroblasts, and lymphocytes of patients with FD []. The presence of cytokine IL-10, highly expressed in FD is also a negative regulator of autophagy [,].
Fibrosis has been found in the organs mainly affected by FD, at the renal level it has been seen that the progression goes from podocyte injury to the generation of fibrosis, at the cardiac level, fibrosis can be found even in the early stages of cardiomyopathy []. In studies with a fibrosis-mimicking device, when TGF-β1 is administered to fibroblasts, it induces a change in several cytokines and reflects the fibrotic process, accompanied by cellular responses such as epithelial-mesenchymal transition (EMT) [].
Another key component that can directly affect both renal and cardiac levels is vascular function. Rohard et al. [] analyzed polymorphisms in NOS3, a gene with the function of producing nitric oxide (NO), finding that the genotypes of this gene could partly explain the variability of cardiac phenotypes in FD.
For this reason, we decided to analyze in this article genes related to interleukins, fibrosis-sclerosis, renal disease and endothelial or vascular disease that could be modifying the clinical characteristics of FD, to find, through our proprietary software SNPclinic, regulatory variants with clinical significance.
2. Materials and Methods
2.1. Input for the SNPClinic v.1.0
For this study, 14 genes were selected, related either directly to the injury of an organ affected by FD or indirectly, these were selected through PubMed []. By using our own SNPClinic software v.1.0 [], we first generated in silico human proximal (2 Kb from transcriptional start site) pseudo-promoters comprising all common SNPs (MAF > 1%) in the averaged world population (code = ALL) according to the 1000 Genomes Project for each of the 14 modifying genes in FD (Table 1). Subsequently, to assess chromatin accessibility, we obtained DNAseI- HUP data from five FD/ modulator genes cell lines from ENCODE project, namely Hcm (human cardiac myocytes), Hcf (primary human cardiac fibroblasts), Hac (astrocytes-cerebellar), Huvec (umbilical vein endothelial cells), TH1 (Immunophenotype of T-helper 1 lymphocytes).

Table 1.
List of the genes and their total number of common SNPs in proximal promoters that were analyzed with SNPClinic v.1.0 software.
Transcription factor Position Frequency Matrices (PFMs) were obtained from the JASPAR [] database. Finally in this step, we obtained chromosome coordinates, biallelic alleles, DNAse-HUP accessibilities, and PFMs as input data.
2.2. Prediction of Regulatory SNPs (rSNPs) with SNPclinic v.1.0
We followed the method of Flores Saiffe et al. [], where SNPClinic scans base-per-base the proximal promoter for each gene for each of the two DNA strands calculating the local DNA affinity to each of the 396 human transcription factors from the JASPAR Database. In this way, SNPClinic explores the overlap of chromatin accessibility between the DNase HUP-1 of the cell lines mentioned in Table 1 and the common SNPs. The outputs of SNP Clinic v.1.0 are exact transcription factors (TF) binding sequence, transcription factor binding sites (TFBS) strand (coding/+, non-coding/−), altered TFBS, a relative binding score for the Major Allele (RBSM), RBS for the Minor Allele (RBSm), affinity impact (%), homotypic redundancy (HR), Homotypic redundance Weight Factor (HWF) and the Functional Impact Factor (FIF).
2.3. Confirmatory Predictions for the Reported rSNPs
To evaluate the functionality of the variants predicted by SNPclinic, the scoring and prediction of the GWAVA [] and FunSeq2 [] tools were used. To obtain a prioritization score for functional variants, the probability score for an SNP to be an eQTL was used, using the DeepSEA [] software.
Finally, free access databases were searched for the effect of SNPs on expression, the databases were Ensembl [], GTEX databases [], Expression Atlas [], and ENCODE [].
3. Results and Discussion
In this work, the functional prediction as rSNP of 110 Common SNPs was carried out using the SNPclinic software. The classification of the true rSNPs was based on the TFBSs altered by the presence of each SNP. Through the calculated percentage of the binding affinity impact and considering the homotypic redundancy, it was obtained for the TF-SNP association, in contrast with other less sensitive methods which are mainly based on p-value filtering and/or ranking. Of the analyzed genes, only IL10, TGFB1 and EDN1 were obtained with the presence of rSNPs and altering TFBS (Table 2, and Figure 1).
Table 2.
Prediction of rSNPs with SNPClinic v.1.0 software. SNPClinic outputs include cell line specificity, altered TFBS and quantitative ranking according to transcriptional relevance with the Functional Impact Factor (FIF).
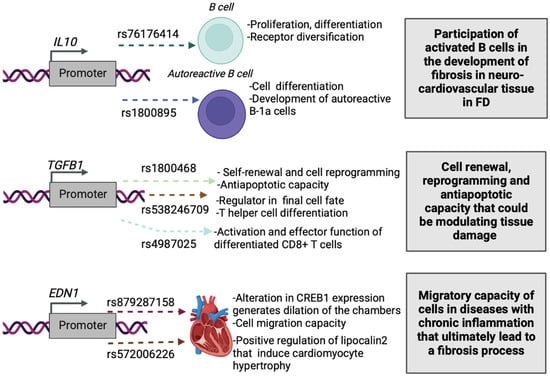
Figure 1.
Proposed impact of the SNPClinic-predicted rSNPs in FD (“ALL” population from 1000 Genomes Project). Figure was designed in Biorender (https://biorender.com/, accessed on 15 March 2022).
The participation of interleukins has been seen in the pathogenic process of many diseases, within these we decided toselect the main cytokines reported to be elevated or altered in the FD process, such as TNF-α, IL-10, IL-1a, IL-1b, and IL-6. In FD it has been seen that there may be variations according to sex, and management stage such as enzyme replacement therapy that can modify interleukins levels. Serum increased levels of TNF-a have been found in women with FD. However, in men with FD, TNF-α and IL-6 are increased compared to healthy controls []. The proinflammatory state determined by these interleukins has also been determined by other workgroups, Biancini et al. concluded that even the levels of globotriaosylceramide (Gb3) are positively correlated with plasma levels of IL-6 []. The accumulation of Gb3 also has a close relationship with TNF, since TNF can increase its accumulation and be related to the pain crises present in FD, corroborating an in vitro model with peripheral blood mononuclear cells (PBMC) of male patients, who have higher expressions of TNF and IL-1β []. Clinical severity in FD patients, measured by the Mainz Severity Score Index (MSSI), has also been correlated with serum IL-6 and TNF-a levels, regardless of gender [].
Of the included interleukins, we found two rSNPs in IL10, in the Hcm, Hac, and TH1 cell lines. The altered TFBS in this region were SPIB, BATF: JUN, USF2, MLX, BHLHE41, BHLHE23 and BHLHE22. Of these transcription factors it has been seen that the implication of the transcription factor SpiB is related to PU.1, together they have a participation in proliferation, survival, as well as the regulation of components of the signaling pathway of different points of differentiation of B cells, but above all, it is important to emphasize its participation in the pathways that allow the detection and response to environmental signals in B cells []. This could mean a key point in FD, since a decrease in fractions of memory B cells (CD20+/CD27+) has been seen in patients compared to healthy controls, but not a difference in the expression of the CD20 marker [].
Among the other altered TFs important for differentiation are the helix-loop-helix (HLH) class, fulfilling functions of cell development and differentiation, within these USF1 and USF2 members of the bHLHZIP family are present according to the type of cell as well as their state of differentiation []. Other members of the basic-helix-loop-helix (bHLH) family, such as the class I bHLH proteins, E47, E12, Heb, and E2-2, interact with promoters of the pre-B-cell-specific gene for their activation, along with the activity of the early B -cell factor (EBF) []. Another transcription factor of the bHLH family with functions in immune cells is BHLHE41, which has been seen to participate in the control of the development of autoreactive B-1a cells, presenting in these cells a greater expression in contrast with other members of the family such as BHLHE40 [], so this family can cover different roles in development depending on the cell.
BATF was another TFBS altered by variants in IL10 and has been studied in various murine models, finding its key role as a regulator in class switch recombination, one of the mechanisms in which it performs this is by controlling the expression of Activation-induced cytidine deaminase (AID) []. AID is a member of the APOBEC family of cytidine deaminases, which facilitates receptor diversification on B cells []. All these TFs found by the SNPclinic support the function of IL-10 since it is known that in an autocrine manner can promote the differentiation of activated B cells, towards cells that secrete IgM and IgG [], these TF may be part of the necessary components to fulfill this function. It is important to emphasize that we obtained these TFs in cardiac tissue and cerebellar astrocytes; at the cardiac level, the essential participation of activated B cells in the development of fibrosis after an ischemic event has already been reported, since they maintain the local inflammatory process with the expression of cytokines such as TGF-1β, IL-1β, IL-6, TNF-α and promote the expression of myocardial collagen []. A tissue where we mainly find altered TFBS is in the TH1 immunological profile that could be related to the inflammatory component and production of cytokines that the activated B cells originate, being this cell necessary for the development of neuro-cardiovascular lesions observed in FD.
Of the analyzed genes that play a role in the development of fibrosis and sclerosis, 3 rSNPs were obtained in TGFB1, which has been extensively studied in the development of fibrosis, participating in a feed-forward loop, where after being activated by integrins, favors the production of collagen in fibroblasts []. The first variant obtained was rs1800468, altering the TFBS of ESRRB, GMEB2, and CREB. ESRRB has been found functionally linked to the protein factor Ncoa3, which together can bind to RNApol2 complexes, for transcription activation, leading to cellular changes of self-renewal and reprogramming []. The self-renewal role of TGFB1 has been studied for the development of different types of cancer since especially in late stages it can act as a tumor promoter []. The function of TF GMEB2 has been studied in conjunction with GMEB1, with a marked antiapoptotic capacity. In an in vitro study, it has been seen that the expression of GMEB-1 and GMEB-2, increase 2.82 ± 1.14 and 2.57 ± 0.31 times, respectively, after stimulating peripheral blood mononuclear cells (PBMC) with IL-12 []. cAMP-responsive element-binding protein (CREB) has been associated with cell proliferation and survival functions []. CREB overexpression can protect cells from apoptosis even in the presence of induced endoplasmic reticulum stress [].
The relationship between the process of apoptosis and the development of fibrosis has been seen in different models, Chung et al., 2021 [] developed a mouse model with FD, analyzing the reaction of the kidneys to profibrotic or inflammatory stimuli secondary to ureteral obstruction, found that autophagy is altered in FD, leading to renal apoptosis and fibrosis formation. There are multiple studies on the relationship between these two processes, some that support that autophagy can promote cell death and others that, on the contrary, prevent death as the destination of the cell, some authors believe that perhaps the relationship is more than the autophagy only modifies the time of cell death []. It has been seen that TGFB1 can increase the expression of sphingosine kinase 1 (SK1), which has been implicated in cell proliferation processes. Following SK1 activation, it can protect cells from death by inducing autophagy [].
The second variant of TGFB1 was rs538246709, with alteration of the TFBS for LBX1, GATA3, and ZEB1, in both Huvec and TH1 cell lines. LBX1 has been related as a key factor in the differentiation of embryonic dorsal neurons, called class B neurons [], a key regulator in the transcription and final cell fate of interneurons is the Corl1 factor, acting as a co-repressor of Lbx1 [].
LBX1 is also necessary for the correct migration of myogenic progenitors in the extremities, but this function needs the somite-derived endothelial cells, which influence the migratory behavior []. This could explain why we obtained rs538246709 as a putative variant in the Huvec cell line (Umbilical vein endothelial cells). The role of GATA3 has been seen particularly in key elements of T helper cell differentiation. The elimination of GATA3 in mice leads to different events in TH2 cells, where it produces changes in the expression of 623 genes, some specific for the immunophenotype such as Il4, Il5, Il10, Il13, and Il1rl1, on the other hand, it slightly increases TH1-specific genes such as Tbx21, Fasl, and Il12rb2 []. ZEB1 is also required in late stages for T cell differentiation, where it enhances the inhibitory effect of TGF-B1 on CD4+ T cells [].
The third variant of TGFB1 was rs4987025, with alteration of the TFBS BATF::JUN and SMAD3. A necessary component for a correct activation and effector function of differentiated CD8+ T cells is the participation of Batf::Jun, T-bet, SREBP2, and AP-1, it has been verified that they have enrichment for the mentioned TF motifs []. The relationship of CD8+ T cells with the rest of TGFB1-altered TFBS could be explained by their known interaction to perpetuate inflammation and the development of fibrosis. Their participation in the fibrotic process after influenza infection has been seen, where CD8+ resident memory T cells play a key role in the respiratory tract of aged hosts, depending on TGF-β signals [].
Within the altered TFBS in the TGFB1 gene promoter, we found several key elements in cell renewal, reprogramming, and antiapoptotic capacity that could ultimately be modulating tissue damage, which has been a well-studied component in FD patients. In FD nephropathy, the proximal renal tubular cell is the main producer of TGF-β 1, which initiates profibrotic changes and induces cellular differentiation of renal cells into myofibroblasts, ultimately leading to renal cell apoptosis []. Therefore, the altered transcription factors reported here could be essential elements to modulate the fibrotic processes that occur in FD.
Of the genes analyzed with participation in Endothelial/Vascular disease, the involvement of the Endothelin-1 gene (EDN1) is notable, where it induces deregulation in vasoconstriction and vasodilatation events, and has been widely seen in the literature [,]. The rs5370 variant of EDN1 located in exon 5, is associated with the risk of pulmonary arterial hypertension, a disease characterized by increased pulmonary vascular resistance []. Due to this essential relationship in the regulation of the vessels, we analyze the promoter region, finding two variants as putatively regulatory rs879287158 and rs572006226. The TFBS for CREB1 was altered in both variants, in the cardiac myocytes, cardiac fibroblasts, astrocytes, endothelial cells, and TH1 cell lines, so it could be fulfilling an essential role in the function of EDN1. The participation of the TF cyclic AMP response element (CRE)-binding protein (CREB) has been seen in models of transgenic mice, when a dominant-negative expression of this TF occurs, generates dilation of the chambers and thinning of ventricular walls, reflecting features of dilated cardiomyopathy []. It is known that the distribution of expression in cardiac tissue of the members of the CREB family is different according to the type of cell, where there is an expression of CREB exclusively in fibroblasts and of CREM in myocytes []. Interestingly, we found this altered TF with FIF score without any difference between myocytes and cardiac fibroblasts.
There is also a relationship between EDN1 with the formation of dilated cardiomyopathy, in murine models with an expression of Edn1 that progressively decreases, produces a deterioration of cardiac function, with increased plasma volumes, and finally causes dilated cardiomyopathy []. Therefore, CREB1 could be directly influenced in the development of dilated cardiomyopathy already reported for EDN1. In Fabry disease, the main cardiac manifestation is left ventricular hypertrophy (LVH) [], although there are also reports of manifestations such as congestive heart failure such as dilated cardiomyopathy [].
Another of the transcription factors that we found altered in different cell lines was TCF3, with changes in both cardiac fibroblasts and endothelial cells. TCF3 has been related to cell migration capacity, this effect is only seen by TFC4 and not by the rest of the members of the Lef/Tcf family. The mechanisms by which it induces this effect are due to the positive regulation of lipocalin2 (LCN2) []. LCN2, on the other hand, can induce cardiomyocyte hypertrophy. When it is overexpressed, it can even decrease the number of cells and reduce cell mitosis in cardiac tissue []. The function of LCN2 has also been seen to induce fibrosis, mainly in human alcoholic hepatitis, where overexpression correlates with portal hypertension and the degree of fibrosis. Specifically, in human hepatic stellate cells, LCN2 can increase the expression of EDN1 mediated by HIF1A [].
Participation in cell migration by EDN1 was confirmed in a model of hepatocarcinogenesis, which is a process that evolves from inflammation, cirrhosis, fibrosis, to finally, liver carcinoma. EDN1 can stimulate the expression of cell cycle genes, cell proliferation, and migration, these functions may be mediated by the activation of the AKT signaling pathway []. The migratory capacity of cells could be playing an essential role in diseases with chronic inflammation that ultimately lead to a fibrosis process, with TCF3 perhaps being the preliminary component for the development of this process. Regarding the rest of the altered TFBS, NOTO, HOXD12, and USF1, we did not find a relationship with the cellular migratory capacity or in the fibrosis process.
The rest of the genes analyzed, interleukins, the participants in fibrosis processes, vascular and renal disease, without rSNP results, could be due to the lack of relevance in the cell lines affected by FD, or because they could be having a secondary regulation.
Of the rSNPs proposed by SNPclinic, we corroborate it with other software such as GWAVA, where we find similar results, with a final prediction of deleterious, in 70% of the rSNPs. In addition, to corroborate not only its classification as a deleterious variant but also the regulation in the expression, the probability score of being an eQTL was obtained for each variant, obtaining a score equivalent to that given by the other tools. Finally, the effect of the variants in different tissues was evaluated, see Table 3.
Table 3.
Functional confirmation of putative rSNPs in other databases, first the classification of variants was carried out using the tools GWAVA and Funseq2, secondly, a score was obtained in DeepSea, for each SNP the probability of contributing to the expression as eQTL was found. Finally, the effect of each SNP on tissue-dependent expression was obtained in the ENSEMBL, GTEx, EBI-EMBL and ENCODE databases. Confirmed true rSNPs (firstly predicted by SNPclinic v.1.0) are shown in bold numbers.
The participation of the genes analyzed in this article reflects the growing interest in the study of variants in modifying genes, which can influence as enhancers or suppressors of the severity of diseases and in most cases may not result in any phenotype by themselves []. Gaucher disease (GD) has been an example where it has been continuously analyzed how modifying genes, epigenetics, and other factors can lose the clear limits between simple and complex inheritance []. Durán et al., 2021 found 271 Single Nucleotide Variants (SNVs) within nine genes associated with the hepatic activity of the enzyme β-glucocerebrosidase that causes GD, which could act as modifying the activity of the enzyme []. In Charcot-Marie-tooth disease type 1A (CMT1A), mutations in the LITAF gene can predispose to the appearance of CMT1A for up to 13 years, in contrast with those who do not present the variant []. Other genes have also been associated with CMT1A, such as SIPA1L2, which may increase the severity of anterior tibial muscle weakness, leading to foot drop []. In other archetypal monogenic disorders, a great clinical heterogeneity has been seen, as is the case of β-thalassemia, in which factors can even improve the clinical phenotype, as is the case of variants in the BCL11A gene []. Some groups have focused on the importance of genetic background, in diseases such as PMM2-CDG the most common disorder of glycosylation (CDG), finding possible disease-modifying genes []. One problem facing the identification of genetic modifiers is that the vast majority are based on small portions of the phenotypic variability, such as individual families []. We propose to consider the component of modifier genes in the clinical presentation of FD, similar to what has already been proposed for other diseases, so that the IL10, TGFB1, and EDN1 genes could explain part of the phenotypic heterogeneity.
4. Conclusions
In conclusion, we found seven rSNPs in three genes that could partially explain the variations in the phenotype of patients with FD, these are involved in processes such as proliferation, survival, and state of differentiation of B cells. Once activated, B cells could favor chronic inflammation that leads to fibrosis. Fibrosis is an essential component in the apoptosis-autophagy regulation axis.
The rSNPs proposed here could modulate the clinical phenotype of Fabry disease, so we propose that IL10, TGFB1 and EDN1 genes be considered modifier/minor genes in FD, in charge of regulating its neuro-cardiovascular variant.
Author Contributions
A.V.R.R.; investigation, interpretation of data, visualization, writing—original draft preparation. L.E.F.; interpretation of data, editing and revision. E.P.M.d.O.; resources, interpretation of data, editing and revision. All authors have read and agreed to the published version of the manuscript.
Funding
AVRR is a CONACYT Fellow from the Doctorate Program in Human Genetics (Guadalajara University/IMSS-CONACYT).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
1. The datasets analyzed during the current study are available in the Ensembl Genome Browser 102, 2020. https://www.ensembl.org/index.html. Accessed: 15 March 2022. 2. The datasets analyzed during the current study are available in the Genotype-Tissue Expression (GTEx) Consortium, GTEx Project, 2021. https://gtexportal.org/home/GTEx/. Accessed: 15 March 2022. 3. The datasets analyzed during the current study are available in the European Molecular Biology Laboratory-European Bio-informatics Institute, Gene Expression Atlas, 2021. https://www.ebi.ac.uk/gxa/home. Accessed: 15 March 2022. 4. The datasets analyzed during the current study are available in the ENCODE portal. https://www.encodeproject.org. Accessed: 15 March 2022.
Conflicts of Interest
The authors declare that the research was carried out in the absence of any commercial or financial relationship that could be interpreted as a potential conflict of interest.
References
- Génin, E.; Feingold, J.; Clerget-Darpoux, F. Identifying modifier genes of monogenic disease: Strategies and difficulties. Hum. Genet. 2008, 124, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.D.; Nadeau, J.H. From Peas to Disease: Modifier Genes, Network Resilience, and the Genetics of Health. Am. J. Hum. Genet. 2017, 101, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Arning, L. The search for modifier genes in Huntington disease—Multifactorial aspects of a monogenic disorder. Mol. Cell. Probes 2016, 30, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, E.; VanderSluis, B.; Wang, W.; Tan, G.; Deshpande, R.; Chen, Y.; Usaj, M.; Balint, A.; Mattiazzi Usaj, M.; van Leeuwen, J.; et al. Systematic analysis of complex genetic interactions. Science 2018, 360, eaao1729. [Google Scholar] [CrossRef]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef]
- Van Leeuwen, J.; Pons, C.; Boone, C.; Andrews, B.J. Mechanisms of suppression: The wiring of genetic resilience. Bioessays 2017, 39, 1700042. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). 6 March 2020. Available online: https://omim.org/ (accessed on 1 February 2022).
- Csányi, B.; Hategan, L.; Nagy, V.; Obál, I.; Varga, E.T.; Borbás, J.; Tringer, A.; Eichler, S.; Forster, T.; Rolfs, A.; et al. Identification of a Novel GLA Gene Mutation, p.Ile239Met, in Fabry Disease With a Predominant Cardiac Phenotype. Int. Heart. J. 2017, 58, 454–458. [Google Scholar] [CrossRef][Green Version]
- Hsu, T.R.; Hung, S.C.; Chang, F.P.; Yu, W.C.; Sung, S.H.; Hsu, C.L.; Dzhagalov, I.; Yang, C.F.; Chu, T.H.; Lee, H.J.; et al. Later Onset Fabry Disease, Cardiac Damage Progress in Silence: Experience with a Highly Prevalent Mutation. J. Am. Coll. Cardiol. 2016, 68, 2554–2563. [Google Scholar] [CrossRef]
- Shen, J.S.; Meng, X.L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef]
- Ohshima, T.; Murray, G.J.; Swaim, W.D.; Longenecker, G.; Quirk, J.M.; Cardarelli, C.O.; Sugimoto, Y.; Pastan, I.; Gottesman, M.M.; Brady, R.O.; et al. alpha-Galactosidase A deficient mice: A model of Fabry disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2540–2544. [Google Scholar] [CrossRef]
- Shu, L.; Shayman, J.A. Caveolin-associated Accumulation of Globotriaosylceramide in the Vascular Endothelium of α-Galactosidase A Null Mice. J. Biol. Chem. 2007, 282, 20960–20967. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell. Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Chévrier, M.; Brakch, N.; Céline, L.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Annie Laquerrière, A.L.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef]
- Safyan, R.; Whybra, C.; Beck, M.; Elstein, D.; Altarescu, G. An association study of inflammatory cytokine gene polymorphisms in Fabry disease. Eur. Cytokine Netw. 2006, 17, 271–275. [Google Scholar]
- Park, H.J.; Lee, S.J.; Kim, S.H.; Han, J.; Bae, J.; Kim, S.J.; Park, C.G.; Chun, T. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol. Immunol. 2011, 48, 720–727. [Google Scholar] [CrossRef]
- Weidemann, F.; Sanchez-Niño, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef]
- Hwang, S.H.; Lee, Y.M.; Choi, Y.; Son, H.E.; Ryu, J.Y.; Na, K.Y.; Chin, H.J.; Jeon, N.L.; Kim, S. Role of Human Primary Renal Fibroblast in TGF-β1-Mediated Fibrosis-Mimicking Devices. Int. J. Mol. Sci. 2021, 22, 10758. [Google Scholar] [CrossRef]
- Rohard, I.; Schaefer, E.; Kampmann, C.; Beck, M.; Gal, A. Association between polymorphisms of endothelial nitric oxide synthase gene (NOS3) and left posterior wall thickness (LPWT) of the heart in Fabry disease. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), S349–S356. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information (NCBI). Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 7 January 2022).
- Flores Saiffe Farías, A.; Jaime Herrera López, E.; Moreno Vázquez, C.J.; Li, W.; Prado Montes de Oca, E. Predicting functional regulatory SNPs in the human antimicrobial peptide genes DEFB1 and CAMP in tuberculosis and HIV/AIDS. Comput. Biol. Chem. 2015, 59 Pt A, 117–125. [Google Scholar] [CrossRef]
- Mathelier, A.; Fornes, O.; Arenillas, D.J.; Chen, C.Y.; Denay, G.; Lee, J.; Shi, W.; Shyr, C.; Tan, G.; Worsley-Hunt, R.; et al. JASPAR 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016, 44, D110–D115. [Google Scholar] [CrossRef]
- Ritchie, G.R.; Dunham, I.; Zeggini, E.; Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods 2014, 11, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, Z.; Lou, S.; Bedford, J.; Mu, X.J.; Yip, K.Y.; Khurana, E.; Gerstein, M. FunSeq2: A framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014, 15, 480. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.; George, N.; et al. Expression Atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016, 44, D726–D732. [Google Scholar] [CrossRef]
- Ruiz Ramírez, A.V.; Flores-Saiffe Farías, A.; Chávez Álvarez, R.D.C.; Prado Montes de Oca, E. Predicted regulatory SNPs reveal potential drug targets and novel companion diagnostics in psoriasis. J. Transl. Autoimmun. 2021, 4, 100096. [Google Scholar] [CrossRef]
- Rosa Neto, N.S.; Bento, J.C.B.; Caparbo, V.F.; Pereira, R.M.R. Increased Serum Interleukin-6 and Tumor Necrosis Factor Alpha Levels in Fabry Disease: Correlation with Disease Burden. Clinics 2021, 76, e2643. [Google Scholar] [CrossRef]
- Biancini, G.B.; Vanzin, C.S.; Rodrigues, D.B.; Deon, M.; Ribas, G.S.; Barschak, A.G.; Manfredini, V.; Netto, C.B.; Jardim, L.B.; Giugliani, R.; et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim. Biophys. Acta 2012, 1822, 226–232. [Google Scholar] [CrossRef]
- Üçeyler, N.; Urlaub, D.; Mayer, C.; Uehlein, S.; Held, M.; Sommer, C. Tumor necrosis factor-α links heat and inflammation with Fabry pain. Mol. Genet. Metab. 2019, 127, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Tellier, J.; Liao, Y.; Trezise, S.; Light, A.; O’Donnell, K.; Garrett-Sinha, L.A.; Shi, W.; Tarlinton, D.M.; Nutt, S.L. Environmental sensing by mature B cells is controlled by the transcription factors PU.1 and SpiB. Nat. Commun. 2017, 8, 1426. [Google Scholar] [CrossRef] [PubMed]
- Limgala, R.P.; Jennelle, T.; Plassmeyer, M.; Boutin, M.; Lavoie, P.; Abaoui, M.; Auray-Blais, C.; Nedd, K.; Alpan, O.; Goker-Alpan, O. Altered immune phenotypes in subjects with Fabry disease and responses to switching from agalsidase alfa to agalsidase beta. Am. J. Transl. Res. 2019, 11, 1683–1696. [Google Scholar] [PubMed]
- Anantharaman, A.; Lin, I.J.; Barrow, J.; Liang, S.Y.; Masannat, J.; Strouboulis, J.; Huang, S.; Bungert, J. Role of helix-loop-helix proteins during differentiation of erythroid cells. Mol. Cell. Biol. 2011, 31, 1332–1343. [Google Scholar] [CrossRef]
- Sigvardsson, M. Overlapping expression of early B-cell factor and basic helix-loop-helix proteins as a mechanism to dictate B-lineage-specific activity of the lambda5 promoter. Mol. Cell. Biol. 2000, 20, 3640–3654. [Google Scholar] [CrossRef]
- Kreslavsky, T.; Vilagos, B.; Tagoh, H.; Poliakova, D.K.; Schwickert, T.A.; Wöhner, M.; Jaritz, M.; Weiss, S.; Taneja, R.; Rossner, M.J.; et al. Essential role for the transcription factor Bhlhe41 in regulating the development, self-renewal and BCR repertoire of B-1a cells. Nat. Immunol. 2017, 18, 442–455. [Google Scholar] [CrossRef]
- Ise, W.; Kohyama, M.; Schraml, B.U.; Zhang, T.; Schwer, B.; Basu, U.; Alt, F.W.; Tang, J.; Oltz, E.M.; Murphy, T.L.; et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011, 12, 536–543. [Google Scholar] [CrossRef]
- Mechtcheriakova, D.; Svoboda, M.; Meshcheryakova, A.; Jensen-Jarolim, E. Activation-induced cytidine deaminase (AID) linking immunity, chronic inflammation, and cancer. Cancer Immunol. Immunother. 2012, 61, 1591–1598. [Google Scholar] [CrossRef]
- Heine, G.; Drozdenko, G.; Grün, J.R.; Chang, H.D.; Radbruch, A.; Worm, M. Autocrine IL-10 promotes human B-cell differentiation into IgM- or IgG-secreting plasmablasts. Eur. J. Immunol. 2014, 44, 1615–1621. [Google Scholar] [CrossRef]
- Mo, F.; Luo, Y.; Yan, Y.; Li, J.; Lai, S.; Wu, W. Are activated B cells involved in the process of myocardial fibrosis after acute myocardial infarction? An in vivo experiment. BMC Cardiovasc. Disord. 2021, 21, 5. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Lavial, F.; Ng, J.H.; Kumar, V.; Tomaz, R.A.; Martin, N.; Yeo, J.C.; Gil, J.; Prabhakar, S.; Ng, H.H.; et al. Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev. 2012, 26, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Woosley, A.N.; Dalton, A.C.; Hussey, G.S.; Howley, B.V.; Mohanty, B.K.; Grelet, S.; Dincman, T.; Bloos, S.; Olsen, S.K.; Howe, P.H. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene 2019, 38, 3794–3811. [Google Scholar] [CrossRef]
- Kawabe, K.; Lindsay, D.; Braitch, M.; Fahey, A.J.; Showe, L.; Constantinescu, C.S. IL-12 inhibits glucocorticoid-induced T cell apoptosis by inducing GMEB1 and activating PI3K/Akt pathway. Immunobiology 2012, 217, 118–123. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Balogh, A.; Németh, M.; Koloszár, I.; Markó, L.; Przybyl, L.; Jinno, K.; Szigeti, C.; Heffer, M.; Gebhardt, M.; Szeberényi, J.; et al. Overexpression of CREB protein protects from tunicamycin-induced apoptosis in various rat cell types. Apoptosis 2014, 19, 1080–1098. [Google Scholar] [CrossRef]
- Chung, S.; Son, M.; Chae, Y.; Oh, S.; Koh, E.S.; Kim, Y.K.; Shin, S.J.; Park, C.W.; Jung, S.C.; Kim, H.S. Fabry disease exacerbates renal interstitial fibrosis after unilateral ureteral obstruction via impaired autophagy and enhanced apoptosis. Kidney Res. Clin. Pract. 2021, 40, 208–219. [Google Scholar] [CrossRef]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef]
- Du, C.; Ren, Y.; Yao, F.; Duan, J.; Zhao, H.; Du, Y.; Xiao, X.; Duan, H.; Shi, Y. Sphingosine kinase 1 protects renal tubular epithelial cells from renal fibrosis via induction of autophagy. Int. J. Biochem. Cell Biol. 2017, 90, 17–28. [Google Scholar] [CrossRef]
- Müller, T.; Brohmann, H.; Pierani, A.; Heppenstall, P.A.; Lewin, G.R.; Jessell, T.M.; Birchmeier, C. The homeodomain factor lbx1 distinguishes two major programs of neuronal differentiation in the dorsal spinal cord. Neuron 2002, 34, 551–562. [Google Scholar] [CrossRef]
- Mizuhara, E.; Nakatani, T.; Minaki, Y.; Sakamoto, Y.; Ono, Y. Corl1, a novel neuronal lineage-specific transcriptional corepressor for the homeodomain transcription factor Lbx1. J. Biol. Chem. 2005, 280, 3645–3655. [Google Scholar] [CrossRef] [PubMed]
- Mayeuf-Louchart, A.; Montarras, D.; Bodin, C.; Kume, T.; Vincent, S.D.; Buckingham, M. Endothelial cell specification in the somite is compromised in Pax3-positive progenitors of Foxc1/2 conditional mutants, with loss of forelimb myogenesis. Development 2016, 143, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Abraham, B.J.; Yagi, R.; Jothi, R.; Cui, K.; Sharma, S.; Narlikar, L.; Northrup, D.L.; Tang, Q.; Paul, W.E.; et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity 2011, 35, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, S.; Yamazaki, S.; Nakauchi, H.; Morishita, K. Downregulation of ZEB1 and overexpression of Smad7 contribute to resistance to TGF-β1-mediated growth suppression in adult T-cell leukemia/lymphoma. Oncogene 2010, 29, 4157–4169. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhang, K.; Milner, J.J.; Toma, C.; Chen, R.; Scott-Browne, J.P.; Pereira, R.M.; Crotty, S.; Chang, J.T.; Pipkin, M.E.; et al. Epigenetic landscapes reveal transcription factors that regulate CD8(+) T cell differentiation. Nat. Immunol. 2017, 18, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Goplen, N.P.; Wu, Y.; Son, Y.M.; Li, C.; Wang, Z.; Cheon, I.S.; Jiang, L.; Zhu, B.; Ayasoufi, K.; Chini, E.N.; et al. Tissue-resident CD8(+) T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci. Immunol. 2020, 5, eabc4557. [Google Scholar] [CrossRef]
- Rozenfeld, P.A.; de Los Angeles Bolla, M.; Quieto, P.; Pisani, A.; Feriozzi, S.; Neuman, P.; Bondar, C. Pathogenesis of Fabry nephropathy: The pathways leading to fibrosis. Mol. Genet. Metab. 2020, 129, 132–141. [Google Scholar] [CrossRef]
- Lago-Docampo, M.; Solarat, C.; Méndez-Martínez, L.; Baloira, A.; Valverde, D. Common Variation in EDN1 Regulatory Regions Highlights the Role of PPARγ as a Key Regulator of Endothelin in vitro. Front. Cardiovasc. Med. 2022, 9, 823133. [Google Scholar] [CrossRef]
- Li, J.; Yin, W.; Liu, M.S.; Mao, L.J.; Wang, X.H. Potential correlation between EDN1 gene polymorphisms with preeclampsia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1602–1608. [Google Scholar]
- Jiao, Y.R.; Wang, W.; Lei, P.C.; Jia, H.P.; Dong, J.; Gou, Y.Q.; Chen, C.L.; Cao, J.; Wang, Y.F.; Zhu, Y.K. 5-HTT, BMPR2, EDN1, ENG, KCNA5 gene polymorphisms and susceptibility to pulmonary arterial hypertension: A meta-analysis. Gene 2019, 680, 34–42. [Google Scholar] [CrossRef]
- Fentzke, R.C.; Korcarz, C.E.; Shroff, S.G.; Lin, H.; Leiden, J.M.; Lang, R.M. The left ventricular stress-velocity relation in transgenic mice expressing a dominant negative CREB transgene in the heart. J. Am. Soc. Echocardiogr. 2001, 14, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Husse, B.; Isenberg, G. CREB expression in cardiac fibroblasts and CREM expression in ventricular myocytes. Biochem. Biophys. Res. Commun. 2005, 334, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, C.K.; Grant, R.; Hagaman, J.R.; Hiller, S.; Li, F.; Xu, L.; Chang, A.S.; Madden, V.J.; Bagnell, C.R.; Rojas, M.; et al. Endothelin-1 critically influences cardiac function via superoxide-MMP9 cascade. Proc. Natl. Acad. Sci. USA 2015, 112, 5141–5146. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef]
- Igawa, O.; Miake, J.; Hisatome, I. Ventricular tachycardias and dilated cardiomyopathy caused by Fabry disease. Pacing Clin. Electrophysiol. 2005, 28, 1142–1143. [Google Scholar] [CrossRef]
- Miao, Q.; Ku, A.T.; Nishino, Y.; Howard, J.M.; Rao, A.S.; Shaver, T.M.; Garcia, G.E.; Le, D.N.; Karlin, K.L.; Westbrook, T.F.; et al. Tcf3 promotes cell migration and wound repair through regulation of lipocalin 2. Nat. Commun. 2014, 5, 4088. [Google Scholar] [CrossRef]
- Marques, F.Z.; Prestes, P.R.; Byars, S.G.; Ritchie, S.C.; Würtz, P.; Patel, S.K.; Booth, S.A.; Rana, I.; Minoda, Y.; Berzins, S.P.; et al. Experimental and Human Evidence for Lipocalin-2 (Neutrophil Gelatinase-Associated Lipocalin [NGAL]) in the Development of Cardiac Hypertrophy and heart failure. J. Am. Heart Assoc. 2017, 6, e005971. [Google Scholar] [CrossRef]
- Chen, J.; Argemi, J.; Odena, G.; Xu, M.J.; Cai, Y.; Massey, V.; Parrish, A.; Vadigepalli, R.; Altamirano, J.; Cabezas, J.; et al. Hepatic lipocalin 2 promotes liver fibrosis and portal hypertension. Sci. Rep. 2020, 10, 15558. [Google Scholar] [CrossRef]
- Lu, J.W.; Liao, C.Y.; Yang, W.Y.; Lin, Y.M.; Jin, S.L.; Wang, H.D.; Yuh, C.H. Overexpression of endothelin 1 triggers hepatocarcinogenesis in zebrafish and promotes cell proliferation and migration through the AKT pathway. PLoS ONE 2014, 9, e85318. [Google Scholar] [CrossRef]
- Rahit, K.; Tarailo-Graovac, M. Genetic Modifiers and Rare Mendelian Disease. Genes 2020, 11, 239. [Google Scholar] [CrossRef]
- Ryan, E.; Seehra, G.K.; Sidransky, E. Mutations, modifiers and epigenetics in Gaucher disease: Blurred boundaries between simple and complex disorders. Mol. Genet. Metab. 2019, 128, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Durán, A.; Rebolledo-Jaramillo, B.; Olguin, V.; Rojas-Herrera, M.; Las Heras, M.; Calderón, J.F.; Zanlungo, S.; Priestman, D.A.; Platt, F.M.; Klein, A.D. Identification of genetic modifiers of murine hepatic β-glucocerebrosidase activity. Biochem. Biophys. Rep. 2021, 28, 101105. [Google Scholar] [CrossRef] [PubMed]
- Sinkiewicz-Darol, E.; Lacerda, A.F.; Kostera-Pruszczyk, A.; Potulska-Chromik, A.; Sokołowska, B.; Kabzińska, D.; Brunetti, C.R.; Hausmanowa-Petrusewicz, I.; Kochański, A. The LITAF/SIMPLE I92V sequence variant results in an earlier age of onset of CMT1A/HNPP diseases. Neurogenetics 2015, 16, 27–32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tao, F.; Beecham, G.W.; Rebelo, A.P.; Svaren, J.; Blanton, S.H.; Moran, J.J.; Lopez-Anido, C.; Morrow, J.M.; Abreu, L.; Rizzo, D.; et al. Variation in SIPA1L2 is correlated with phenotype modification in Charcot-Marie-Tooth disease type 1A. Ann. Neurol. 2019, 85, 316–330. [Google Scholar] [CrossRef]
- Mettananda, S.; Higgs, D.R. Molecular Basis and Genetic Modifiers of Thalassemia. Hematol./Oncol. Clin. N. Am. 2018, 32, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cimmaruta, C.; Monticelli, M.; Riccio, G.; Hay Mele, B.; Cubellis, M.V.; Andreotti, G. The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers. Int. J. Mol. Sci. 2018, 19, 2218. [Google Scholar] [CrossRef] [PubMed]
- Bis-Brewer, D.M.; Fazal, S.; Züchner, S. Genetic modifiers and non-Mendelian aspects of CMT. Brain Res. 2020, 1726, 146459. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).