
Differential Proteomic Analysis of Listeria monocytogenes during High-Pressure Processing
 , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Culture Conditions
2.2. High-Pressure Processing Conditions
2.3. Whole-Protein Extraction for Proteomic Analysis
2.4. Mass Spectrometric Analysis
2.4.1. In-Solution Digestion
2.4.2. Liquid Chromatography Coupled Tandem Mass Spectrometry (LC–MS/MS)
2.5. Data Analysis and Protein Network Construction
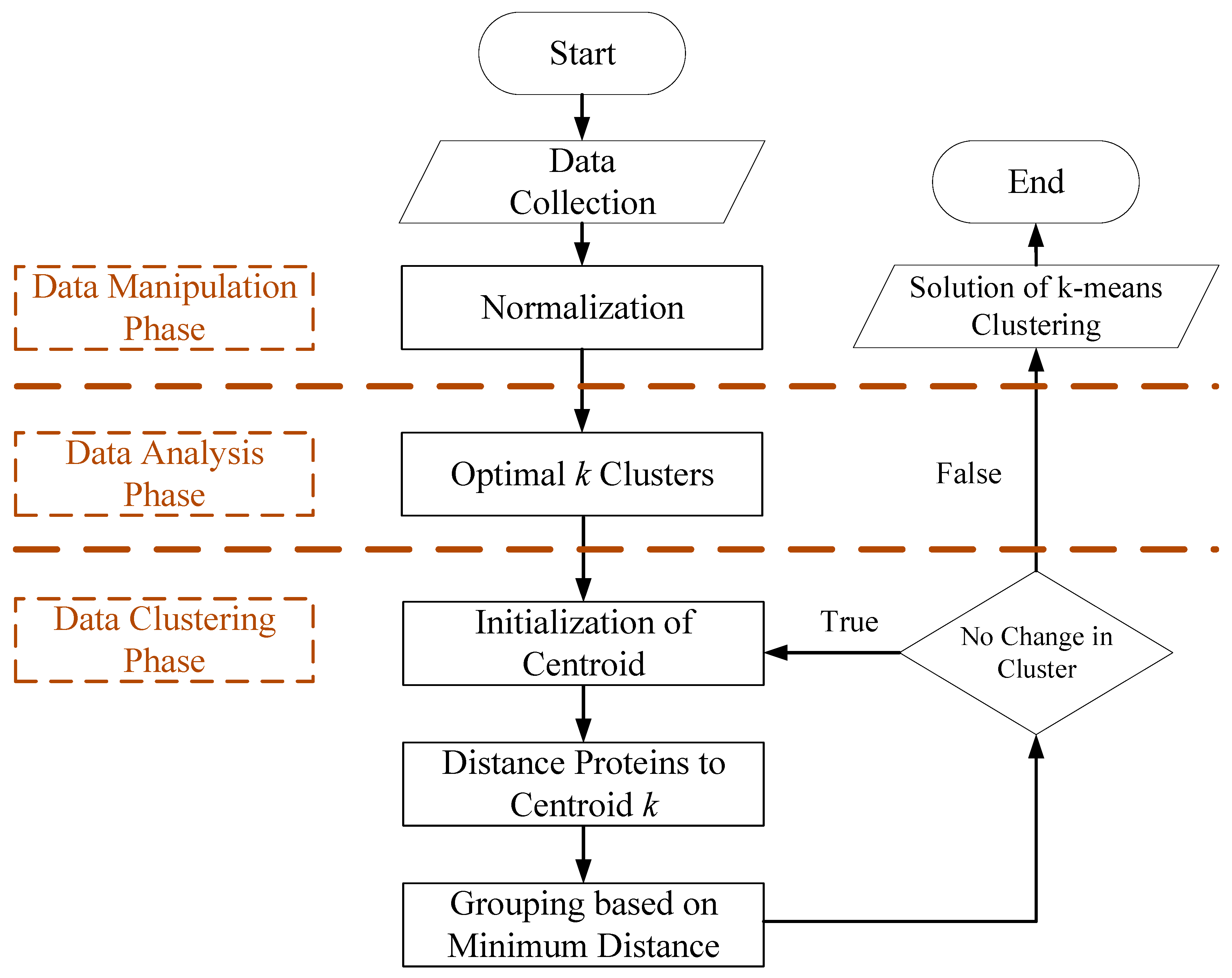
2.6. Machine Learning of Grouping Proteins
2.6.1. The Data Manipulation Phase
2.6.2. The Data Analysis Phase
- (i)
- To cluster observed proteins. Varying the total number of clusters from k = 1, 2, …, K, and giving within-dispersion measures Wk, where k = 1, 2, …, K. E(Wk) is the expected value of total variation within-group variance.
- (ii)
- To generate B reference datasets and to cluster each one, giving the within-dispersion measure Wkb, where b = 1, 2, …, B and k = 1, 2, …, K. The Gap Statistic, Gap(k), is estimated using Formula (1)
- (iii)
- To compute the standard deviation (Sd(Wk)) and the standard error (sk). The standard deviation (Sd(Wk)) and standard error (sk) are described using Formulae (2) and (3).
- (iv)
- To choose the optimal number of clusters. Hence, k* is estimated as the smallest k, such that Gap(k) ≥ Gap(k + 1) − sk+1.
2.6.3. The Data Clustering Phase Is Responsible for Setting Clusters and Separate Proteins
- (i)
- Initialization of centroid.
- (ii)
- Group proteins to centroid k* based on minimum distance.
- (iii)
- Update centroids.
- (1)
- 9G: log2 (H2-1/C-1), log2 (H2-2/C-2), log2 (H2-3/C-3), log2 (H3-1/C-1), log2 (H3-2/C-2), log2 (H3-3/C-3), log2 (H4-1/C-1), log2 (H4-2/C-2) and log2 (H4-3/C-3).
- (2)
- 9GE: [log2 (H2-1/C-1)]/2, [log2 (H2-2/C-2)]/2, [log2 (H2-3/C-3)]/2, [log2 (H3-1/C-1)]/3, [log2 (H3-2/C-2)]/3, [log2 (H3-3/C-3)]/3, [log2 (H4-1/C-1)]/4, [log2 (H4-2/C-2)]/4 and [log2 (H4-3/C-3)]/4. Herein E stand for equivalence, and 9GE mimic log2 (fold change) under 100 MPa equally for each group.
- (3)
- 3G: mean values of log2 (H2/C), log2 (H3/C), and log2 (H4/C).
- (4)
- 3GE: mean values of [log2 (H2/C)]/2, [log2 (H3/C)]/3, and [log2 (H4/C)]/4.
3. Results
3.1. The Influence of HPP on L. monocytogenes Viability
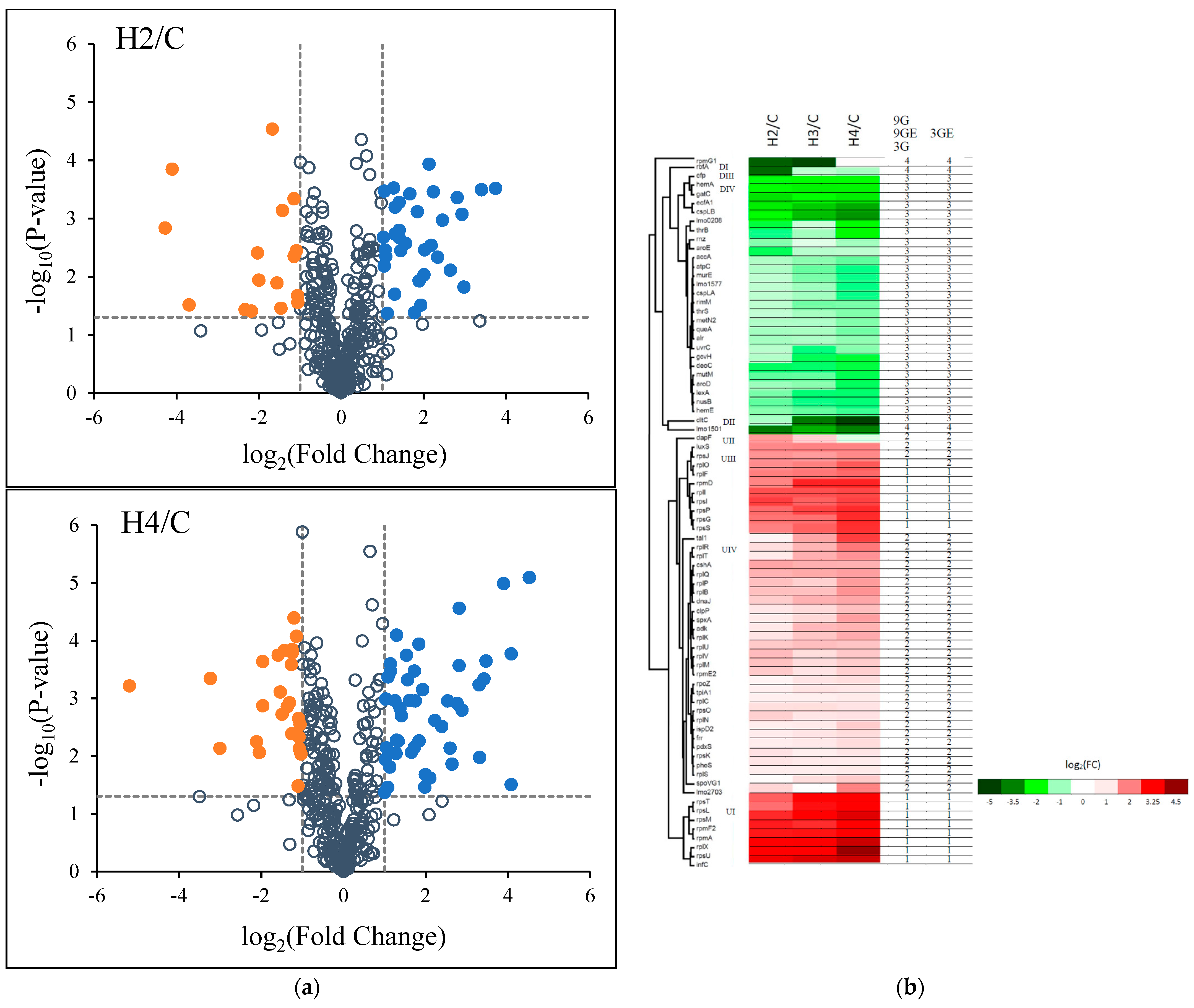
3.2. The Influence of HPP on Differential Expression of L. monocytogenes Proteomes
3.3. Machine Learning of Grouping DEPs
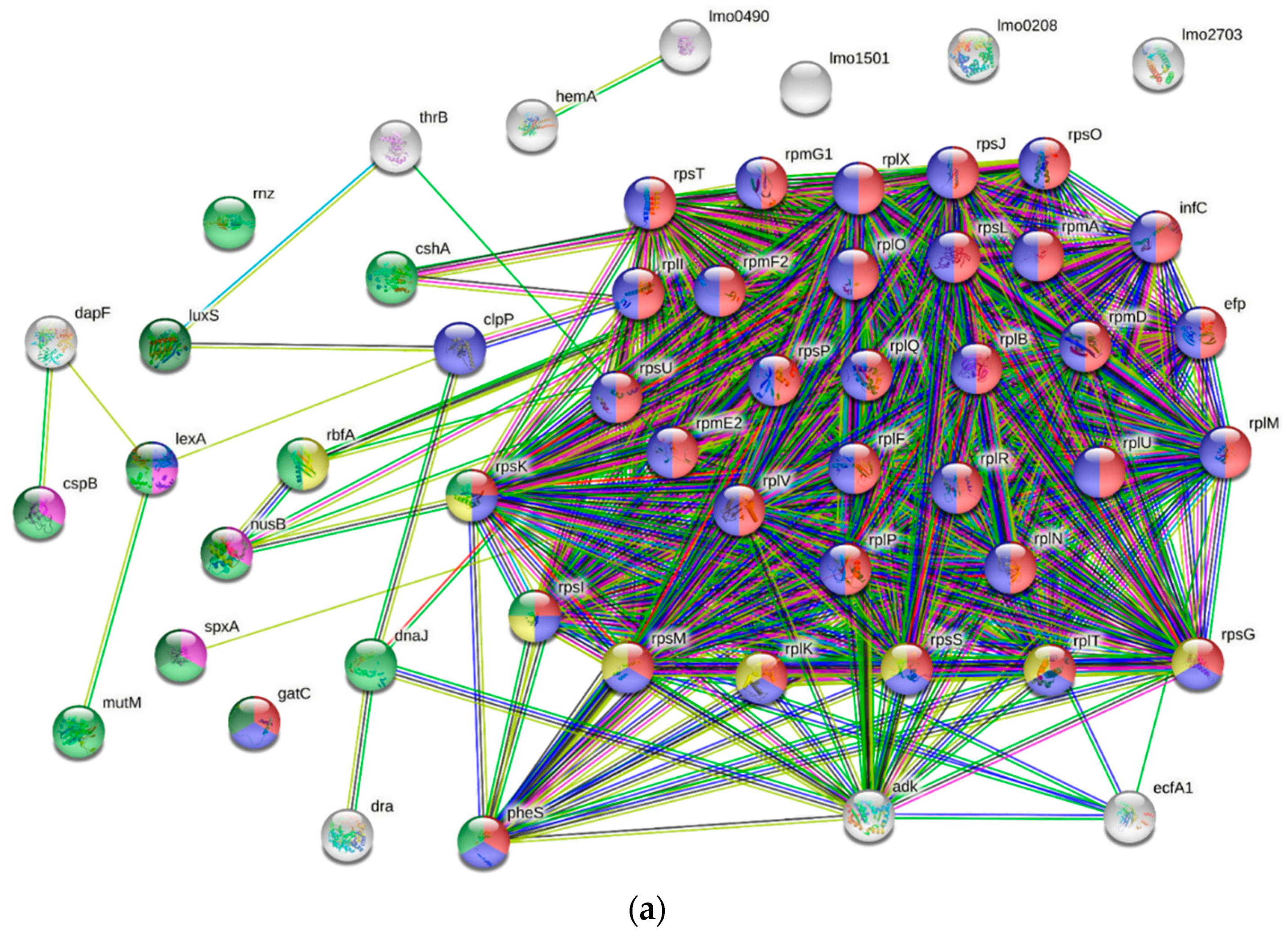
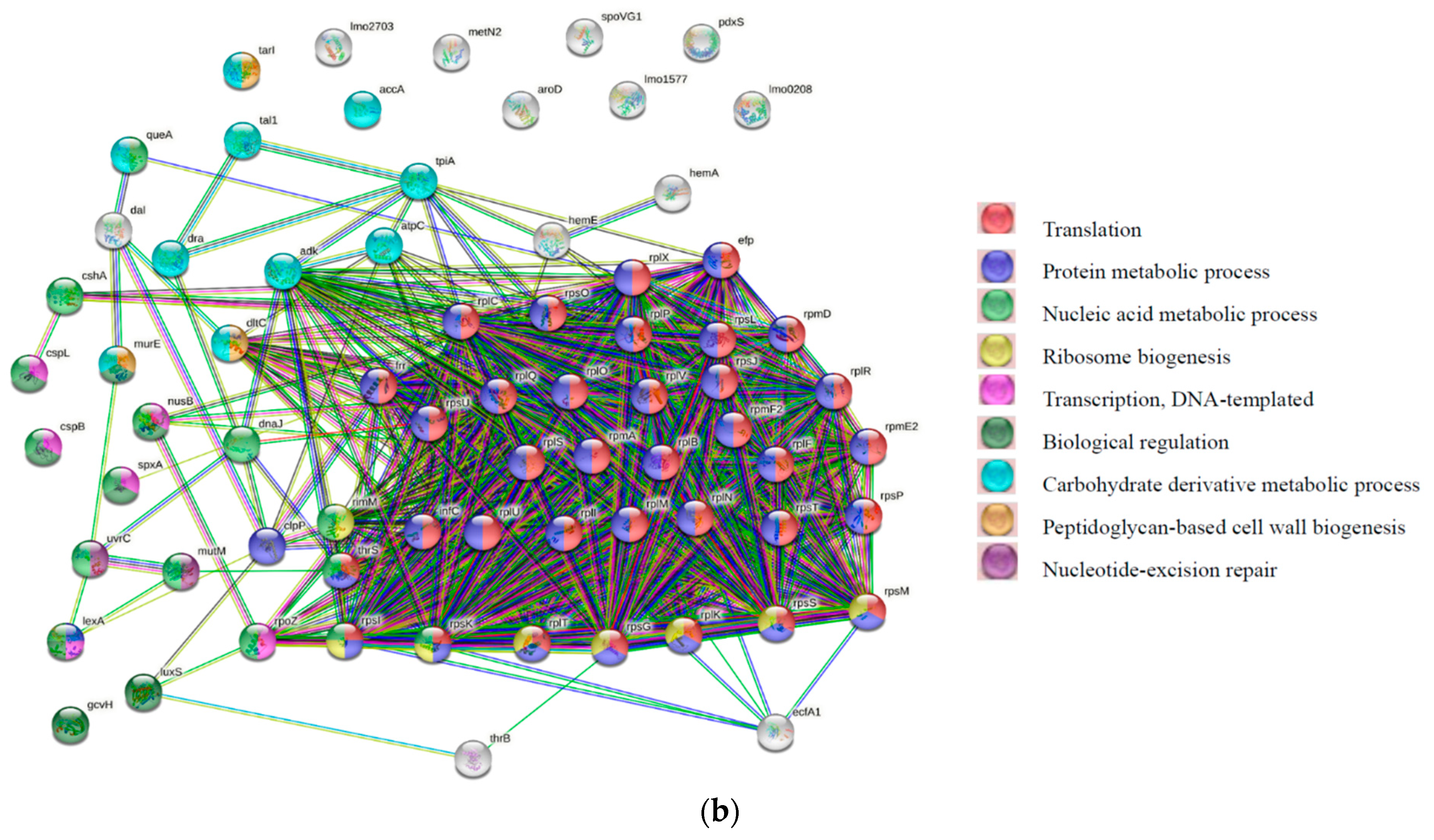
3.4. GO and COGs Enrichment Analysis of HPP-Induced DEPs
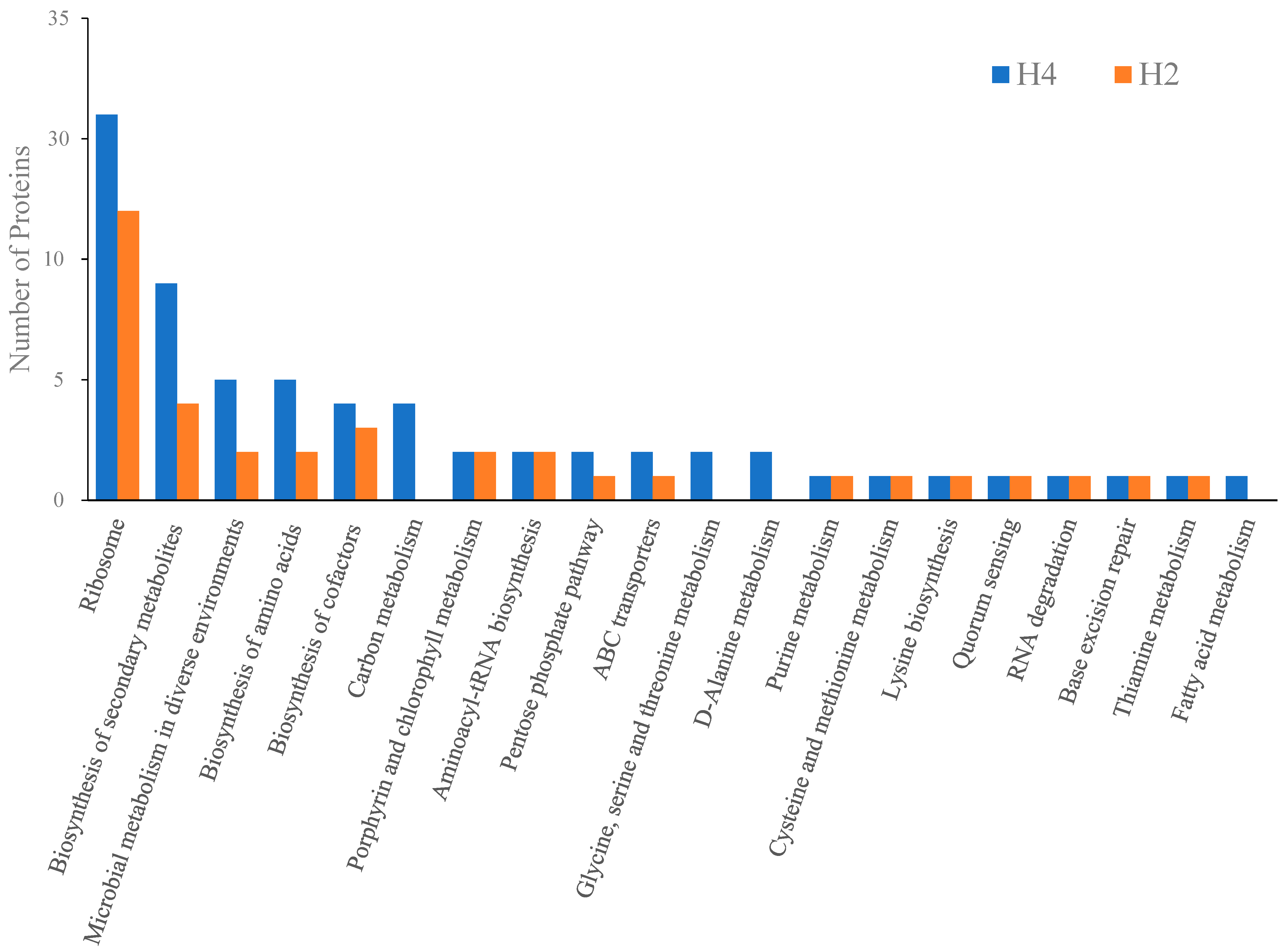
3.5. KEGG Pathway Analysis of HPP-Induced DEPs
4. Discussion
4.1. Translational Regulation
4.2. HPP Promoted Translation Initiation and Retarded Ribosome Biogenesis
4.3. HPP Response-Associated Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention (CDC). Listeria (Listeriosis). 2020. Available online: https://www.cdc.gov/listeria/prevention.html (accessed on 31 August 2020).
- Buchanan, R.L.; Gorris, L.G.M.; Hayman, M.M.; Jackson, T.C.; Whiting, R.C. A review of Listeria monocytogenes: An updated on outbreaks, virulence, dose-response, ecology, and risk assessments. Food Control 2017, 75, 1–13. [Google Scholar] [CrossRef]
- European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC). The European Union one health 2018 zoonoses report. EFSA J. 2019, 17, 5926. [Google Scholar]
- Daher, D.; Le Gourrierec, S.; Pérez Lamela, C. Effect of high pressure processing on the microbial inactivation in fruit preparation and other vegetable based beverages. Agriculture 2017, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Lorido, L.; Estevez, M.; Ventanas, J.; Ventanas, S. Comparative study between Serrano and Iberian dry-cured hams in relation to the application of high hydrostatic pressure and temporal sensory perceptions. LWT Food Sci. Technol. 2015, 64, 1234–1242. [Google Scholar] [CrossRef]
- Meloni, D. High-hydrostatic-pressure (HHP) processing technology as a novel control method for Listeria monocytogenes occurrence in Mediterranean-Style dry-fermented sausages. Foods 2019, 8, 672. [Google Scholar] [CrossRef] [Green Version]
- Rode, T.M.; Rotabakk, B.T. Extending shelf life of desalted cod by high pressure processing. Innov. Food Sci. Emerg. Technol. 2021, 69, 102476. [Google Scholar] [CrossRef]
- Lin, C.S.; Lee, Y.C.; Kung, H.F.; Cheng, Q.L.; Ou, T.Y.; Chang, S.K.C.; Tsai, Y.H. Inactivation of microbial loads and retardation of quality loss in Asian hard clam (Meretrix lusoria) using high-hydrostatic-pressure processing during refrigerated storage. Food Control 2022, 133, 108583. [Google Scholar] [CrossRef]
- Chen, T.Y.; Kuo, S.H.; Chen, S.T.; Hwang, D.F. Differential proteomics to explore the inhibitory effects of acidic, slightly acidic electrolysed water and sodium hypochlorite solution on Vibrio parahaemolyticus. Food Chem. 2016, 194, 529–537. [Google Scholar] [CrossRef] [Green Version]
- Al Shweiki, M.R.; Mönchgesang, S.; Mojovsky, P.; Thieme, D.; Trutschel, D.; Hoehenwarter, W. Assessment of label-free quantitation in discovery proteomics and impact of technological factors and natural variability of protein abundance. J. Proteome Res. 2017, 16, 1410–1424. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.P.; Bittencourt, C.R.; Ross, T. Differential gene expression of Listeria monocytogenes during high hydrostatic pressure processing. Microbiology 2008, 154, 462–475. [Google Scholar] [CrossRef] [Green Version]
- Duru, I.C.; Bucur, F.I.; Andreevskaya, M.; Nikparvar, B.; Ylinen, A.; Grigore-Gurgu, L.; Rode, T.M.; Crauwels, P.; Laine, P.; Paulin, L.; et al. High-pressure processing-induced transcriptome response during recovery of Listeria monocytogenes. BMC Genom. 2021, 22, 117. [Google Scholar] [CrossRef]
- Wang, L.; Xia, Q.; Li, Y. Label free-based proteomic analysis of proteins in Bacillus cereus spores regulated by high pressure processing and slightly acidic electrolyzed water treatment. Food Control 2018, 90, 392–400. [Google Scholar] [CrossRef]
- Chen, G.W.; Chen, Y.A.; Chang, H.Y.; Huang, T.C.; Chen, T.Y. Combined impact of high-pressure processing and slightly acidic electrolysed water on Listeria monocytogenes proteomes. Food Res. Int. 2021, 147, 110494. [Google Scholar] [CrossRef] [PubMed]
- Bièche, C.; de Lamballerie, M.; Chevret, D.; Federighi, M.; Tresse, O. Dynamic proteomic changes in Campylobacter jejuni 81–176 after high pressure shock and subsequent recovery. J. Proteom. 2012, 75, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Drews, O.; Weiss, W.; Reil, G.; Parlar, H.; Wait, R.; Görg, A. High pressure effects step-wise altered protein expression in Lactobacillus sanfranciscensis. Proteomics 2002, 2, 765–774. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R.; Walther, G.; Hastie, T. Estimating the number of clusters in a data Set via the Gap statistic. J. R. Stat. Soc. Ser. B 2001, 63, 411–423. [Google Scholar] [CrossRef]
- Ferreira, M.; Almeida, A.; Delgadillo, I.; Saraiva, J.; Cunha, Â. Susceptibility of Listeria monocytogenes to high pressure processing; A review. Food Rev. Int. 2016, 32, 377–399. [Google Scholar] [CrossRef]
- Huang, H.W.; Lung, H.M.; Yang, B.B.; Wang, C.Y. Responses of microorganisms to high hydrostatic pressure processing. Food Control 2014, 40, 250–259. [Google Scholar] [CrossRef]
- Nguyen, H.T.M.; Akanuma, G.; Hoa, T.T.M.; Nakai, Y.; Kimura, K.; Yamamoto, K.; Inaoka, T. Ribosome reconstruction during recovery from high-hydrostatic-pressure induced injury in Bacillus subtilis. Appl. Environ. Microbiol. 2020, 86, e01640-19. [Google Scholar] [CrossRef] [PubMed]
- Niven, G.W.; Miles, C.A.; Mackey, B.M. The effects of hydrostatic pressure on ribosome conformation in Escherichia coli: An in vivo study using differential scanning calorimetry. Microbiology 1999, 145, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimatsu, K.; Inaoka, T.; Nakaura, Y.; Yamamoto, K. Injury and recovery of Escherichia coli cells in phosphate-buffered saline after high hydrostatic pressure treatment. Food Sci. Technol. Res. 2019, 25, 479–484. [Google Scholar] [CrossRef]
- Prabhakar, A.; Capece, M.C.; Petrov, A.; Choi, J.; Puglisi, J.D. Post-termination ribosome intermediate acts as the gateway to ribosome recycling. Cell Rep. 2017, 20, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Basu, S.; Inouye, M. Dissection of 16S rRNA methyltransferase (KsgA) function in Escherichia coli. J. Bacteriol. 2008, 189, 8510–8518. [Google Scholar] [CrossRef] [Green Version]
- Nord, S.; Bhatt, M.J.; Tükenmez, H.; Farabaugh, P.J.; Wikström, P.M. Mutation of ribosomal protein S5 suppress a defect in late-30S ribosomal subunit biogenesis caused by lack of the RbfA biogenesis factor. RNA 2015, 21, 1454–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksimova, E.M.; Korepanov, A.P.; Kravchenko, O.V.; Baymukhametov, T.N.; Myasnikov, A.G.; Vassilenko, K.S.; Afonina, Z.A.; Stolboushkina, E.A. RbfA is involved in two important stages of 30S subunit assembly: Formation of the central pseudoknot and docking of helix 44 to the decoding center. Int. J. Mol. Sci. 2021, 22, 6140. [Google Scholar] [CrossRef]
- Sharma, I.M.; Woodson, S.A. RbfA and IF3 couple ribosome biogenesis and translation initiation to increase stress tolerance. Nucleic Acids Res. 2020, 48, 359–372. [Google Scholar] [CrossRef]
- Kaczanowska, M.; Ryde’n-Aulin, M. Ribosome biogenesis and the translation process in Escherichia coli. Microbiol. Mol. Biol. Rev. 2007, 71, 477–494. [Google Scholar] [CrossRef] [Green Version]
- Bubunenko, M.; Court, D.; Refaii, A.; Saxena, S.; Korepanov, A.; Friedman, D.; Gottesman, M.E.; Alix, J.H. Nus transcription elongation factors and RNase III modulate small ribosome subunit biogenesis in Escherichia coli. Mol. Microbiol. 2012, 87, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Illigmann, A.; Thoma, Y.; Pan, S.; Reinhardt, L.; Brötz-Oesterhelt, H. Contribution of the Clp protease to bacterial survival and mitochondrial homoeostasis. Microb. Physiol. 2021, 31, 260–279. [Google Scholar] [CrossRef]
- Darsonval, M.; Julliat, F.; Msadek, T.; Alexandre, H.; Granvalet, C. CtsR, the master regulator of stress-response in Oenococcus oeni, is a heat sensor interacting with ClpL1. Front. Microbiol. 2018, 9, 3135. [Google Scholar] [CrossRef] [PubMed]
- Karatzas, K.A.G.; Wouters, J.A.; Gahan, C.G.M.; Hill, C.; Abee, T.; Bennik, M.H.J. The CtsR regulator of Listeria monocytogenes contains a variant glycine repeat region that affects piezotolerance, stress resistance, motility and virulence. Mol. Microbiol. 2003, 49, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Chastanet, A.; Derre, I.; Nair, S.; Msadek, T. ClpB, a novel member of the Listeria monocytogenes CtsR regulon, is involved in virulence but not in general stress tolerance. J. Bacteriol. 2004, 186, 1165–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hörmann, S.; Scheyhing, C.; Behr, J.; Pavlovic, M.; Ehrmann, M.; Vogel, R.F. Comparative proteome approach to characterize the high-pressure stress response of Lactobacillus sanfranciscensis DSM 20451T. Proteomics 2006, 6, 1878–1885. [Google Scholar] [CrossRef]
- Gaillot, O.; Bregenholt, S.; Jaubert, F.; DISanto, J.P.; Berche, P. Stress-induced ClpP serine protease of Listeria monocytogenes is essential for induction of listeriolysin O-dependent protective immunity. Infect. Immun. 2001, 69, 4938–4943. [Google Scholar] [CrossRef] [Green Version]
- Cesinger, M.R.; Thomason, M.K.; Edrozo, M.B.; Halsey, C.R.; Reniere, M.L. Listeria monocytogenes SpxA1 is a global regulator required to activate genes encoding catalase and heme biosynthesis enzymes for aerobic growth. Mol. Microbiol. 2020, 114, 230–243. [Google Scholar] [CrossRef] [Green Version]
- Gaballa, A.; Guariglia-Oropeza, V.; Wiedmann, M.; Boor, K.J. Cross talk between SigB and PrfA in Listeria monocytogenes facilitates transitions between extra- and intracellular environments. Microbiol. Mol. Biol. Rev. 2019, 83, e00034-19. [Google Scholar] [CrossRef]
- Tiensuu, T.; Guerreiro, D.N.; Oliveira, A.H.; O’Byrne, C.; Johansson, J. Flick of a switch: Regulatory mechanisms allowing Listeria monocytogenes to transition from a saprophyte to a killer. Microbiology 2019, 165, 819–833. [Google Scholar] [CrossRef]
- Uhlig, F.; Hyland, N.P. Making sense of quorum sensing at the intestinal mucosal interface. Cells 2022, 11, 1734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ku, X.; Zhang, X.; Zhang, Y.; Chen, G.; Chen, F.; Zeng, W.; Li, J.; Zhu, L.Z.; He, Q. The AI-2/luxS quorum sensing system affects the growth characteristics, biofilm formation, and virulence of Haemophilus parasuis. Front. Cell Infect. Microbiol. 2019, 9, 62. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Characterization | Upregulation | Downregulation | |
|---|---|---|---|
| Metabolism (M) | |||
| Global and overview maps | 01100 Metabolic pathways | tal12, adk, tpiA12, pdxS2, luxS, tarI2, dapF1 | thrB, hemE, hemA, deoC, Alr2, murE2, accA2, gatC, atpC2, gcvH2, aroD2, dltC2 |
| 01110 Biosynthesis of secondary metabolites | adk, tal12, tpiA12, dapF1 | thrB, hemE, accA2, gcvH2, hemA, aroD2 | |
| 01120 Microbial metabolism in diverse environments | adk, tal12, tpiA12, dapF1 | thrB, accA2, hemA | |
| 01200 Carbon metabolism | tal12, tpiA12 | accA2, gcvH2 | |
| 01230 Biosynthesis of amino acids | tal12, tpiA12, luxS2, dapF1 | thrB, aroD2 | |
| Carbohydrate metabolism | 00010 Glycolysis/Gluconeogenesis | tpiA12 | |
| 00030 Pentose phosphate pathway | tal12 | deoC | |
| 00040 Pentose and glucuronate interconversions | tarI2 | ||
| 00051 Fructose and mannose metabolism | tpiA12 | ||
| 00620 Pyruvate metabolism | accA2 | ||
| 00630 Glyoxylate and dicarboxylate metabolism | gcvH2 | ||
| 00640 Propanoate metabolism | accA2 | ||
| 00562 Inositol phosphate metabolism | tpiA12 | ||
| Energy metabolism | 00190 Oxidative phosphorylation | atpC2 | |
| ko03029 Mitochondrial biogenesis | infC, rpmF2, rpsP, dnaJ | gatC | |
| 00195 Photosynthesis | atpC2 | ||
| 00710 Carbon fixation in photosynthetic organisms | tpiA12 | ||
| 00720 Carbon fixation pathways in prokaryotes | accA2 | ||
| Lipid metabolism | 00061 Fatty acid biosynthesis | accA2 | |
| Nucleotide metabolism | 00230 Purine metabolism | adk, rpoZ2 | |
| Amino acid metabolism | 00260 Glycine, serine and threonine metabolism | thrB, gcvH2 | |
| 00270 Cysteine and methionine metabolism | luxS | ||
| 00300 Lysine biosynthesis | dapF1 | murE2 | |
| 00400 Phenylalanine, tyrosine and tryptophan biosynthesis | aroD2 | ||
| ko01002 Peptidases and inhibitors | clpP | lexA | |
| Glycan biosynthesis | 00473 D-alanine metabolism | Alr2, dltC2 | |
| 00550 ko01011 Peptidoglycan biosynthesis and degradation proteins | Alr2, murE2, dltC2 | ||
| Metabolism of cofactors and vitamins | 00730 Thiamine metabolism | adk | |
| 00750 Vitamin B6 metabolism | pdxS2 | ||
| 00860 Porphyrin and chlorophyll metabolism | hemE, hemA | ||
| Genetic Information Processing (GIP) | |||
| Transcription | 03020 RNA polymerase | rpoZ2 | |
| ko03021 Transcription machinery | rpoZ2 | nusB | |
| Transcription regulation: stress response | cspLA2, cspLB | ||
| ko03019 Messenger RNA biogenesis | cshA | ||
| Translation | 03010 Ribosome | rpsE, rpsG, rpsI, rpsJ rpsK, rpsL, rpsM, rpsO, rpsP, rpsS, rpsT, rpsU, rplB, rplC2, rplF, rplI, rplK, rplM, rplN, rplO, rplP, rplQ, rplR, rplS2, rplT, rplU, rplV, rplX, rpmA, rpmD, rpmE2, rpmF2 | rpmG11 |
| 00970 Aminoacyl-tRNA biosynthesis | pheS1 | thrS2, gatC | |
| ko03009 Ribosome biogenesis | cshA | rimM2, nusB, rbfA1 | |
| ko03016 Transfer RNA biogenesis | pheS1 | thrS2, queA2, gatC, rnz1 | |
| ko03012 Translation factors | infC, frr2 | efp | |
| Folding, sorting and degradation | 03013 RNA transport 03018 RNA degradation | cshA | rnz1 |
| ko03110 Chaperones and folding catalysts | dnaJ | ||
| Replication and repair | 03410 Base excision repair | mutM | |
| 03420 Nucleotide excision repair | uvrC2 | ||
| ko03400 DNA repair and recombination proteins | rpoZ2 | lexA, uvrC2, mutM | |
| Environmental Information Processing (EIP) | |||
| Membrane transport | 02010 ABC transporters | metN22, ecfA1 | |
| Signal transduction | 02020 Two-component system | dltC2 | |
| Cellular Processes (CP) | |||
| Cell growth and death | 04112 Cell cycle—Caulobacter | clpP | |
| Cellular community—prokaryotes | 02024 Quorum sensing | luxS | |
| 05111 Biofilm formation—Vibrio cholerae 02026 Biofilm formation—Escherichia coli | luxS | ||
| Regulation of cell septum | ko04812 Cytoskeleton proteins | spoVG12 | |
| Exosome | ko04147 Regulation of Exosome (2) | adk, tpiA12 | |
| Organismal Systems (OS) | |||
| Aging | |||
| 04212 Longevity regulating pathway—worm | clpP | ||
| Human Diseases (HD) | |||
| Infectious disease: bacterial | 05150 Staphylococcus aureus infection | dltC2 | |
| Drug resistance: antimicrobial | 01502 Vancomycin resistance | Alr2 | |
| 01503 Cationic antimicrobial peptide (CAMP) resistance | dltC2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-A.; Chen, G.-W.; Ku, H.-H.; Huang, T.-C.; Chang, H.-Y.; Wei, C.-I.; Tsai, Y.-H.; Chen, T.-Y. Differential Proteomic Analysis of Listeria monocytogenes during High-Pressure Processing. Biology 2022, 11, 1152. https://doi.org/10.3390/biology11081152
Chen Y-A, Chen G-W, Ku H-H, Huang T-C, Chang H-Y, Wei C-I, Tsai Y-H, Chen T-Y. Differential Proteomic Analysis of Listeria monocytogenes during High-Pressure Processing. Biology. 2022; 11(8):1152. https://doi.org/10.3390/biology11081152
Chicago/Turabian StyleChen, Yi-An, Guan-Wen Chen, Hao-Hsiang Ku, Tsui-Chin Huang, Hsin-Yi Chang, Cheng-I Wei, Yung-Hsiang Tsai, and Tai-Yuan Chen. 2022. "Differential Proteomic Analysis of Listeria monocytogenes during High-Pressure Processing" Biology 11, no. 8: 1152. https://doi.org/10.3390/biology11081152
APA StyleChen, Y.-A., Chen, G.-W., Ku, H.-H., Huang, T.-C., Chang, H.-Y., Wei, C.-I., Tsai, Y.-H., & Chen, T.-Y. (2022). Differential Proteomic Analysis of Listeria monocytogenes during High-Pressure Processing. Biology, 11(8), 1152. https://doi.org/10.3390/biology11081152