
PhIP-Seq Reveals Autoantibodies for Ubiquitously Expressed Antigens in Viral Myocarditis

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Peptides and Proteins
2.3. Infection Studies
2.3.1. CVB Infections
2.3.2. Influenza a Infection
2.4. Immunization Procedures
2.5. Phage ImmunoPrecipitation Sequencing (PhIP-Seq)
2.5.1. VirScan, Human, and Mouse Peptide Library Design and Cloning
2.5.2. Mouse Serum Antibody Profiling
2.6. Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Statistics
3. Results
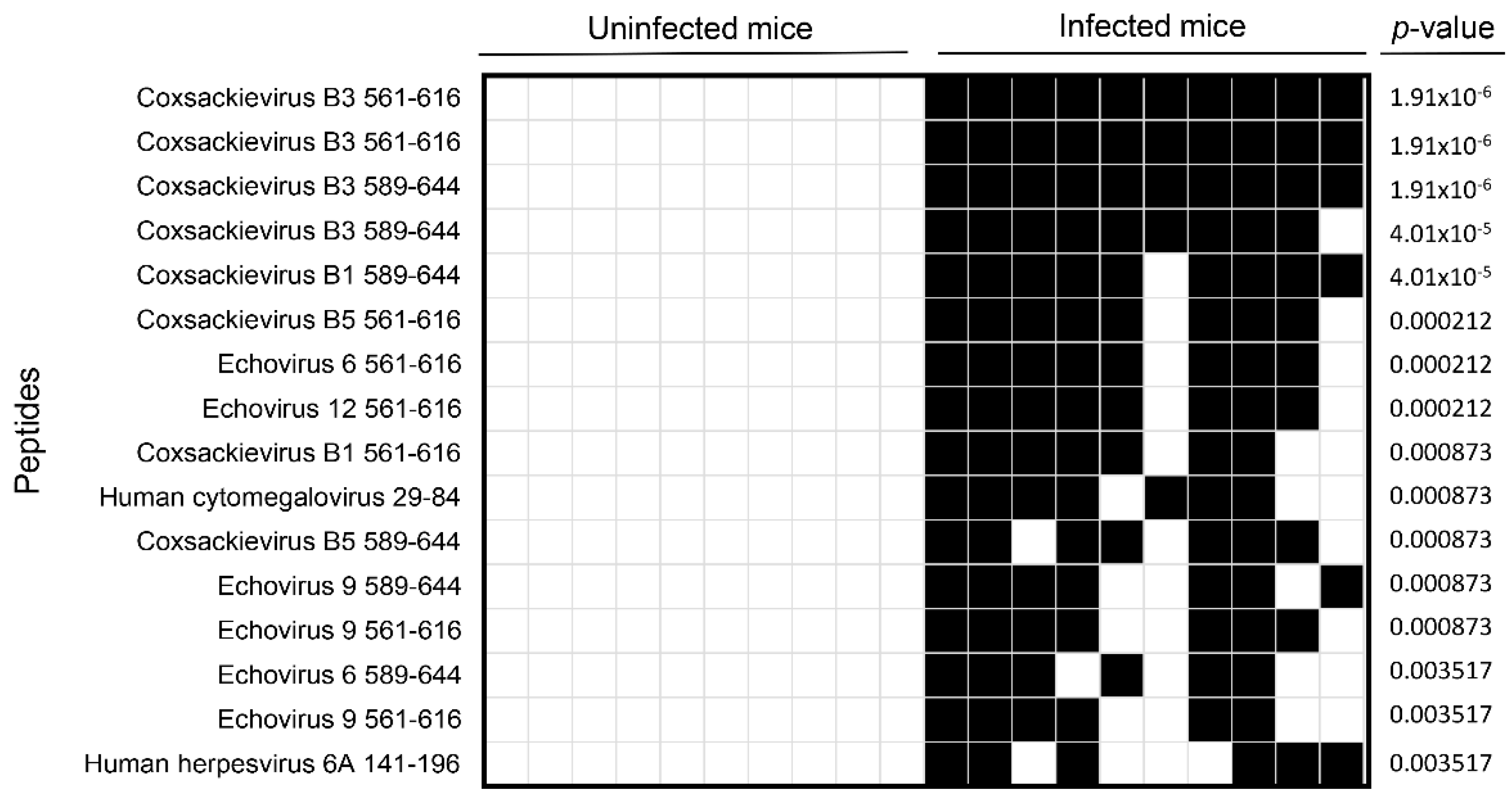
3.1. VirScan Screening of Sera from CVB3-Infected Mice Revealed Antibody Reactivity to CVB3 with a High Degree of Specificity and Sensitivity
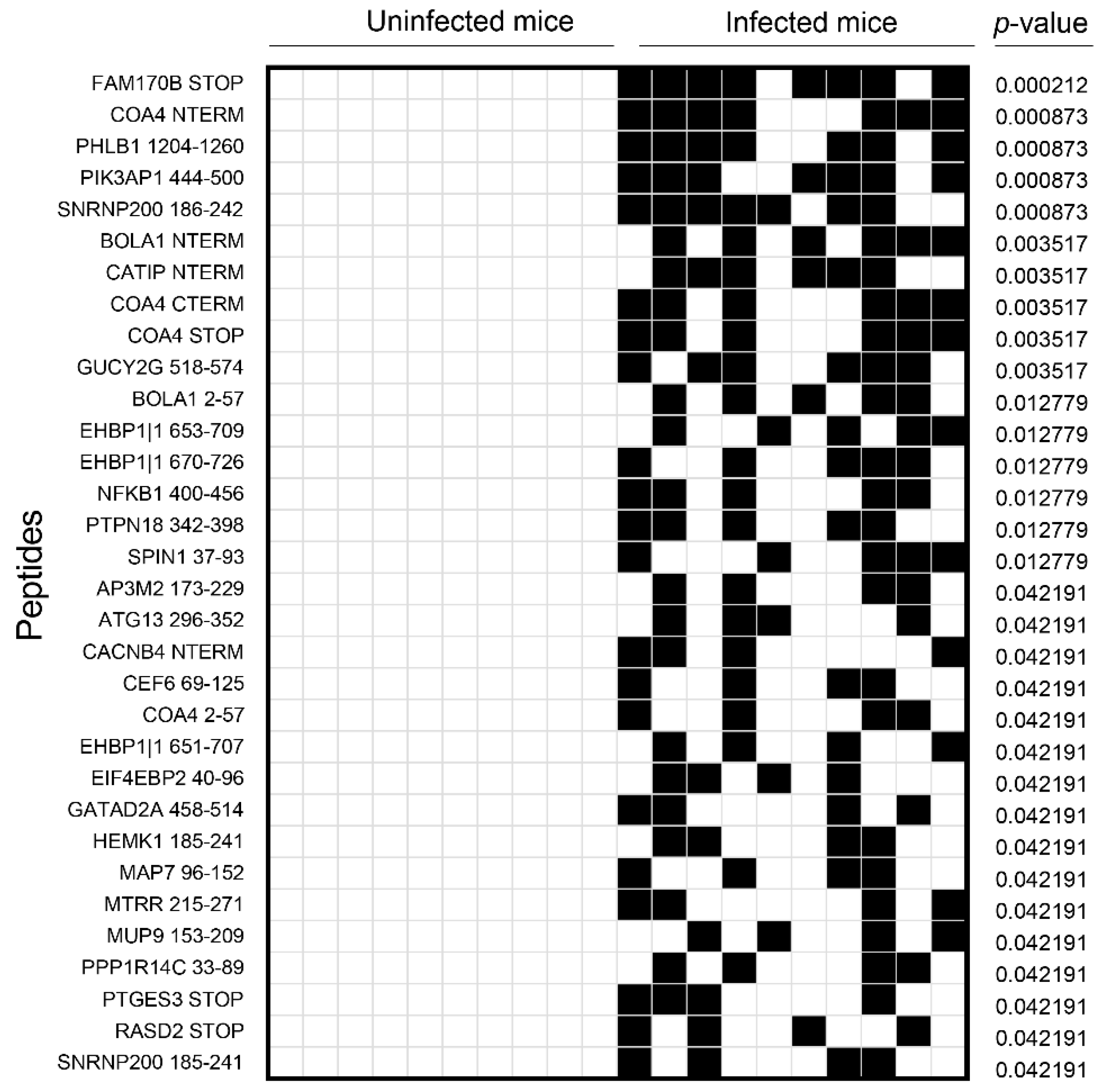
3.2. Analysis of the Mouse Peptide Library Revealed Antibodies to Novel Proteins That Were Previously Unknown in CVB3 Myocarditis
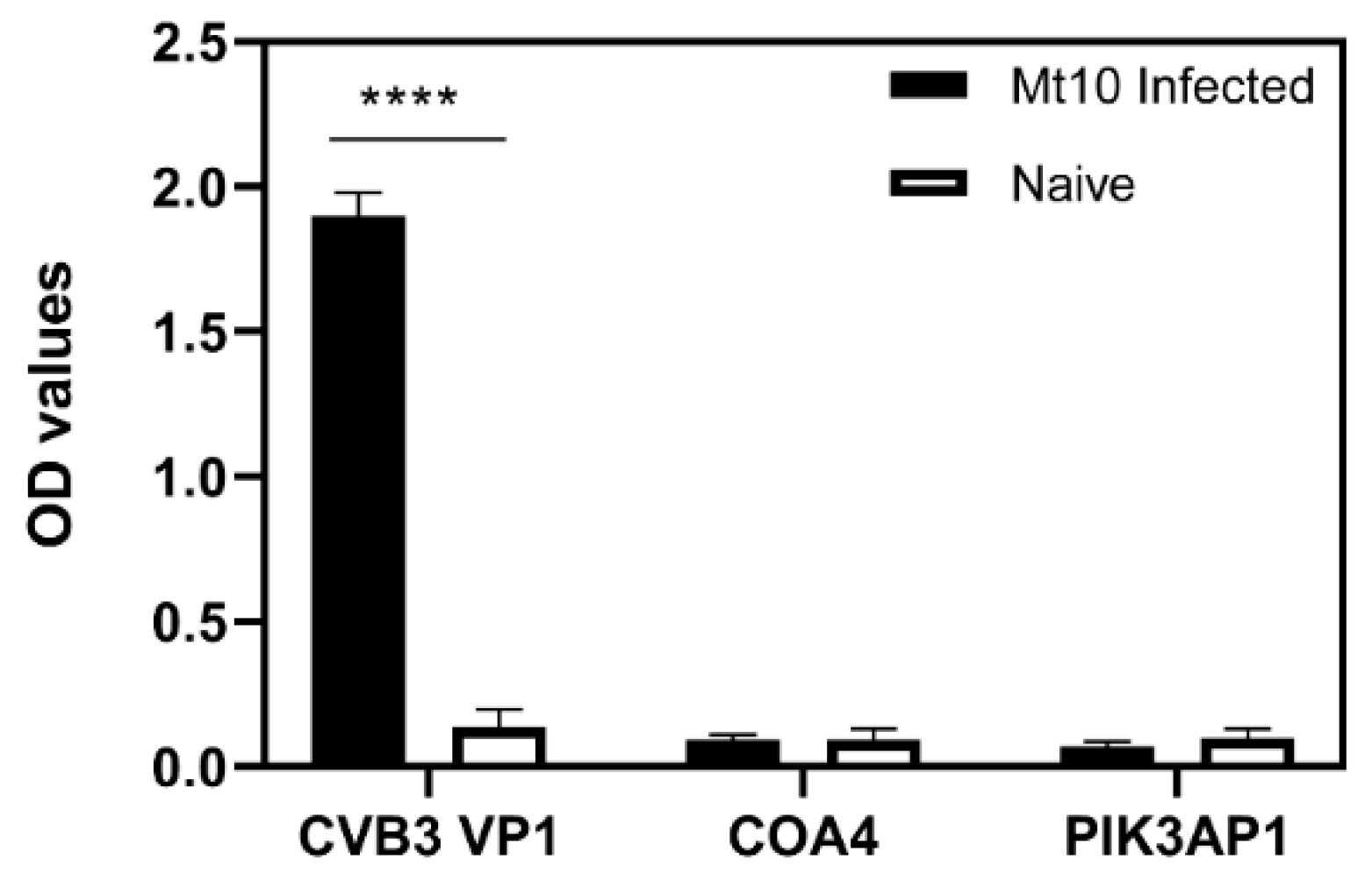
3.3. ELISA Analysis of Serum Samples from CVB3-Infected Animals Validated the Results Obtained with PhIP-Seq
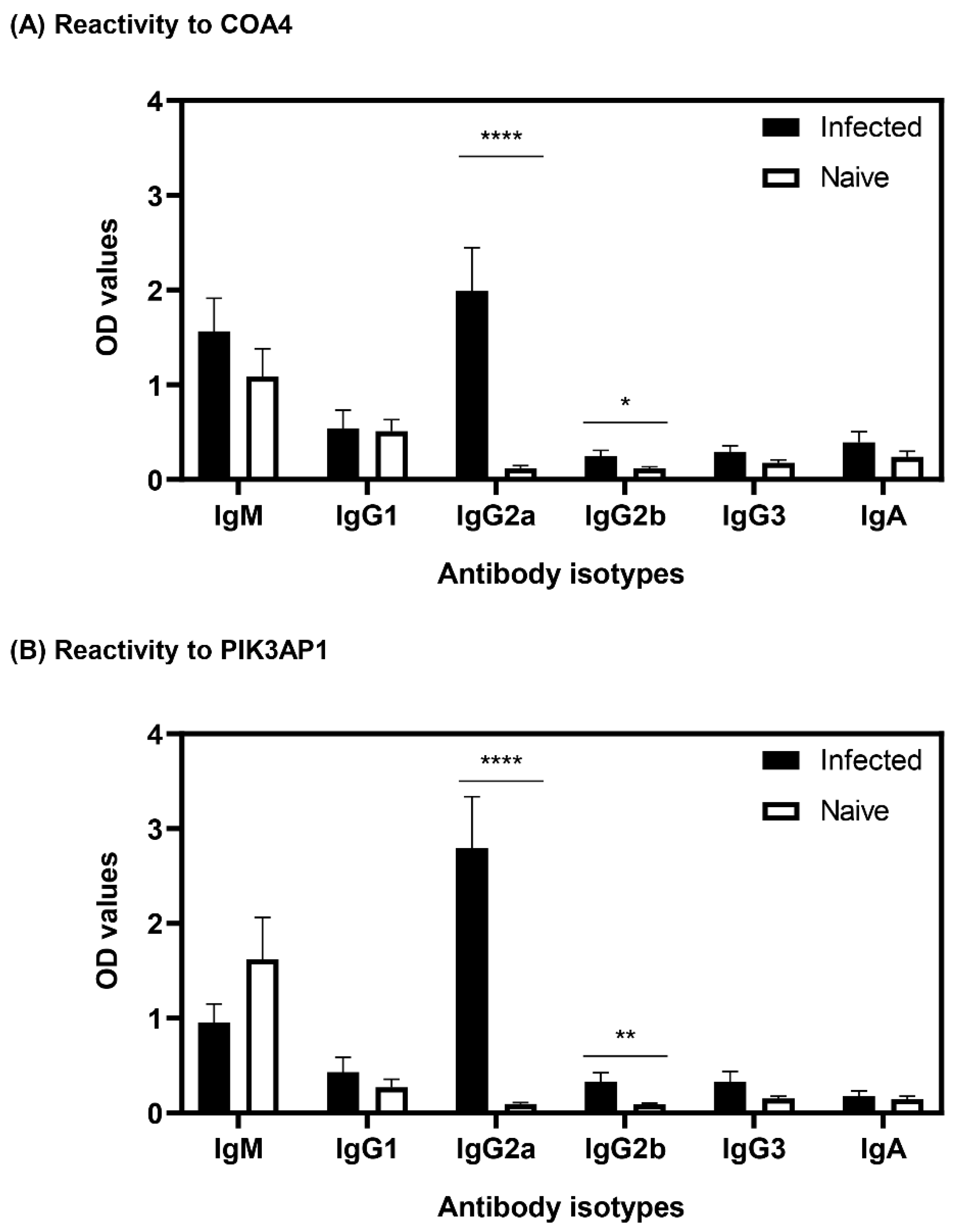
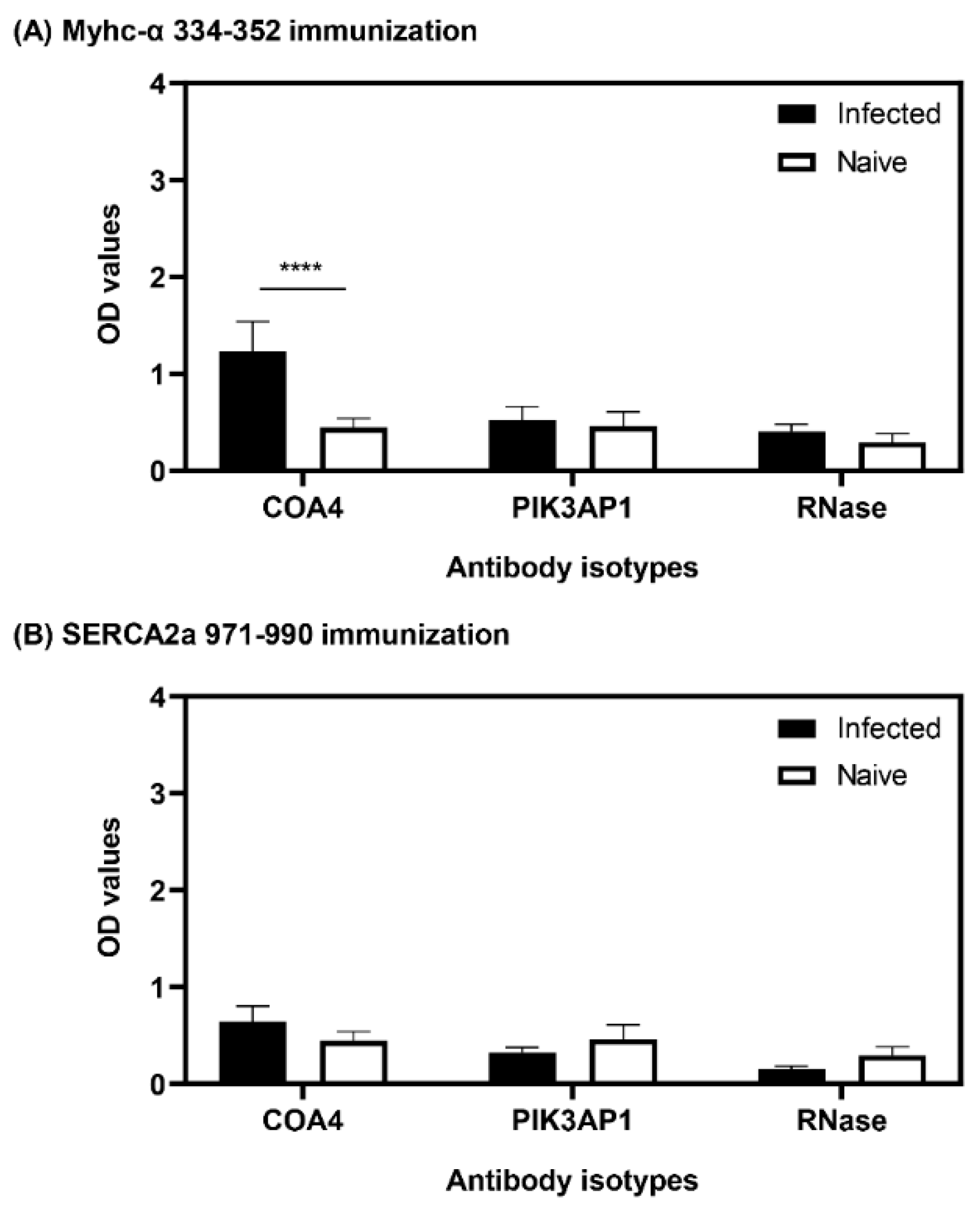
3.4. Antibodies to COA4 and PIK3AP1 Detected in CVB3-Infected Animals Were Predominantly of the IgG2a Isotype
3.5. Sera from CVB4-Infected Animals Showed Antibodies to COA4 and PIK3AP1 with Profiles Similar to Those of CVB3 Infection
3.6. Mice Infected with the Influenza Virus Did Not Reveal Detection of COA4 or PIK3AP1 Antibodies
3.7. Virulent CVB3 Is Necessary to Induce COA4- or PIK3AP1-Reactive Antibodies
3.8. The Presence of COA4-Reactive Antibodies Was Revealed in Animals Immunized with Myhc-α 334-352
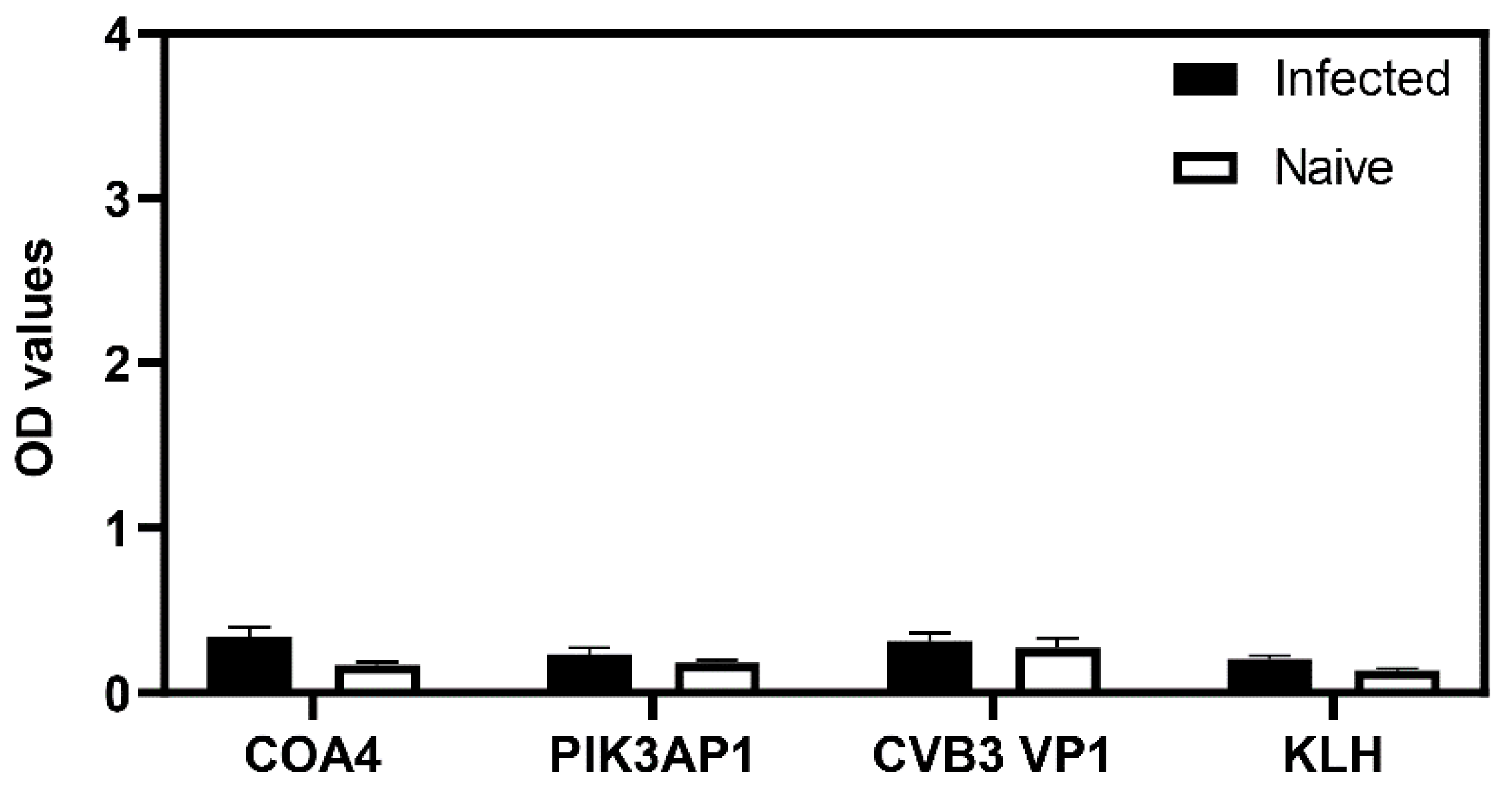
3.9. The Generation of COA4-Reactive Antibodies Did Not Involve Cross-Reactivity to CVB Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lasrado, N.; Reddy, J. An overview of the immune mechanisms of viral myocarditis. Rev. Med. Virol. 2020, 30, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lasrado, N.; Yalaka, B.; Reddy, J. Triggers of Inflammatory Heart Disease. Front. Cell Dev. Biol. 2020, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2020, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Pollack, A.; Kontorovich, A.R.; Fuster, V.; Dec, G.W. Viral myocarditis—Diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 2015, 12, 670–680. [Google Scholar] [CrossRef]
- Trachtenberg, B.H.; Hare, J.M. Inflammatory Cardiomyopathic Syndromes. Circ. Res. 2017, 121, 803–818. [Google Scholar] [CrossRef]
- Cihakova, D.; Rose, N.R. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv. Immunol. 2008, 99, 95–114. [Google Scholar]
- Molina, K.M.; Garcia, X.; Denfield, S.W.; Fan, Y.; Morrow, W.R.; Towbin, J.A.; Frazier, E.A.; Nelson, D.P. Parvovirus B19 Myocarditis Causes Significant Morbidity and Mortality in Children. Pediatr. Cardiol. 2013, 34, 390–397. [Google Scholar] [CrossRef]
- Das, B.B.; Reddy, S.; Eliassen, E.; Krueger, G.R. Human herpesvirus 6-induced inflammatory cardiomyopathy in immunocompetent children. Ann. Pediatr. Cardiol. 2017, 10, 259–268. [Google Scholar] [CrossRef]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, P.; Khanji, M.Y.; Cooper, L.T.; Chahal, C.A.A. Recognizing COVID-19–related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 2020, 17, 1463–1471. [Google Scholar] [CrossRef]
- Neumann, D.A.; Rose, N.R.; Ansari, A.A.; Herskowitz, A. Induction of multiple heart autoantibodies in mice with coxsackievirus B3- and cardiac myosin-induced autoimmune myocarditis. J. Immunol. 1994, 152, 343–350. [Google Scholar]
- Błyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Lodge, P.A.; Herzum, M.; Olszewski, J.; Huber, S.A. Coxsackievirus B-3 myocarditis. Acute and chronic forms of the disease caused by different immunopathogenic mechanisms. Am. J. Pathol. 1987, 128, 455–463. [Google Scholar] [PubMed]
- Fairweather, D.; Rose, N.R. Coxsackievirus-induced myocarditis in mice: A model of autoimmune disease for studying immunotoxicity. Methods 2007, 41, 118–122. [Google Scholar] [CrossRef]
- Caforio, A.L.; Mahon, N.J.; Tona, F.; McKenna, W.J. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: Pathogenetic and clinical significance. Eur. J. Heart Fail. 2002, 4, 411–417. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular Mimicry, Bystander Activation, or Viral Persistence: Infections and Autoimmune Disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Tona, F.; Bottaro, S.; Vinci, A.; Dequal, G.; Daliento, L.; Thiene, G.; Iliceto, S. Clinical implications of anti-heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity 2008, 41, 35–45. [Google Scholar] [CrossRef]
- Warraich, R.S.; Dunn, M.J.; Yacoub, M.H. Subclass Specificity of Autoantibodies against Myosin in Patients with Idiopathic Dilated Cardiomyopathy: Pro-inflammatory Antibodies in DCM Patients. Biochem. Biophys. Res. Commun. 1999, 259, 255–261. [Google Scholar] [CrossRef]
- Magnusson, Y.; Marullo, S.; Hoyer, S.; Waagstein, F.; Andersson, B.; Vahlne, A.; Guillet, J.G.; Strosberg, A.D.; Hjalmarson, A.; Hoebeke, J. Mapping of a functional autoimmune epitope on the beta 1-adrenergic receptor in patients with idiopathic dilated cardiomyopathy. J. Clin. Investig. 1990, 86, 1658–1663. [Google Scholar] [CrossRef]
- Landsberger, M.; Staudt, A.; Choudhury, S.; Trimpert, C.; Herda, L.R.; Klingel, K.; Kandolf, R.; Schultheiss, H.-P.; Kroemer, H.K.; Völker, U.; et al. Potential role of antibodies against cardiac Kv channel-interacting protein 2 in dilated cardiomyopathy. Am. Heart J. 2008, 156, 92–99.e2. [Google Scholar] [CrossRef]
- Lasrado, N.; Arumugam, R.; Rasquinha, M.T.; Sur, M.; Steffen, D.; Reddy, J. Mt10-CVB3 Vaccine Virus Protects against CVB4 Infection by Inducing Cross-Reactive, Antigen-Specific Immune Responses. Microorganisms 2021, 9, 2323. [Google Scholar] [CrossRef]
- Lasrado, N.; Gangaplara, A.; Massilamany, C.; Arumugam, R.; Shelbourn, A.; Rasquinha, M.T.; Basavalingappa, R.H.; Delhon, G.; Xiang, S.-H.; Pattnaik, A.K.; et al. Attenuated strain of CVB3 with a mutation in the CAR-interacting region protects against both myocarditis and pancreatitis. Sci. Rep. 2021, 11, 12432. [Google Scholar] [CrossRef]
- Xu, G.J.; Kula, T.; Xu, Q.; Li, M.Z.; Vernon, S.D.; Ndung’U, T.; Ruxrungtham, K.; Sanchez, J.; Brander, C.; Chung, R.T.; et al. Comprehensive serological profiling of human populations using a synthetic human virome. Science 2015, 348, aaa0698. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.J.; Shah, A.A.; Li, M.Z.; Xu, Q.; Rosen, A.; Casciola-Rosen, L.; Elledge, S.J. Systematic autoantigen analysis identifies a distinct subtype of scleroderma with coincident cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E7526–E7534. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Wansley, D.L.; Sie, B.M.; Noon, M.S.; Baer, A.N.; Laserson, U.; Larman, H.B. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc. 2018, 13, 1958–1978. [Google Scholar] [CrossRef]
- Larman, H.B.; Zhao, Z.; Laserson, U.; Li, M.Z.; Ciccia, A.; Gakidis, M.A.M.; Church, G.; Kesari, S.; LeProust, E.M.; Solimini, N.L.; et al. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011, 29, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Monaco, D.R.; Kottapalli, S.V.; Breitwieser, F.P.; Anderson, D.E.; Wijaya, L.; Tan, K.; Ni Chia, W.; Kammers, K.; Caturegli, P.; Waugh, K.; et al. Deconvoluting virome-wide antibody epitope reactivity profiles. eBioMedicine 2022, 75, 103747. [Google Scholar] [CrossRef] [PubMed]
- Massilamany, C.; Gangaplara, A.; Reddy, J. Intricacies of cardiac damage in coxsackievirus B3 infection: Implications for therapy. Int. J. Cardiol. 2014, 177, 330–339. [Google Scholar] [CrossRef]
- Batzoglou, S.; Pachter, L.; Mesirov, J.P.; Berger, B.; Lander, E.S. Human and Mouse Gene Structure: Comparative Analysis and Application to Exon Prediction. Genome Res. 2000, 10, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Makałowski, W.; Zhang, J.; Boguski, M.S. Comparative analysis of 1196 orthologous mouse and human full-length mRNA and protein sequences. Genome Res. 1996, 6, 846–857. [Google Scholar] [CrossRef]
- Chu, T.; Ni, M.; Chen, C.; Akilesh, S.; Hamerman, J.A. Cutting Edge: BCAP Promotes Lupus-like Disease and TLR-Mediated Type I IFN Induction in Plasmacytoid Dendritic Cells. J. Immunol. 2019, 202, 2529–2534. [Google Scholar] [CrossRef]
- Bossie, A.; Vitetta, E.S. IFN-gamma enhances secretion of IgG2a from IgG2a-committed LPS-stimulated murine B cells: Implications for the role of IFN-gamma in class switching. Cell. Immunol. 1991, 135, 95–104. [Google Scholar] [CrossRef]
- Duarte, J.H. Functional switching. Nat. Immunol. 2016, 17, S12. [Google Scholar] [CrossRef]
- Lasrado, N.; Gangaplara, A.; Arumugam, R.; Massilamany, C.; Pokal, S.; Zhou, Y.; Xiang, S.-H.; Steffen, D.; Reddy, J. Identification of Immunogenic Epitopes That Permit the Detection of Antigen-Specific T Cell Responses in Multiple Serotypes of Group B Coxsackievirus Infections. Viruses 2020, 12, 347. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Frisancho-Kiss, S.; Gatewood, S.; Njoku, D.; Steele, R.; Barrett, M.; Rose, N.R. Mast Cells and Innate Cytokines are Associated with Susceptibility to Autoimmune Heart Disease Following Coxsackievirus B3 Infection. Autoimmunity 2004, 37, 131–145. [Google Scholar] [CrossRef]
- Fairweather, D.; Stafford, K.A.; Sung, Y.K. Update on coxsackievirus B3 myocarditis. Curr. Opin. Rheumatol. 2012, 24, 401–407. [Google Scholar] [CrossRef]
- Huber, S.; Ramsingh, A.I. Coxsackievirus-induced pancreatitis. Viral Immunol. 2004, 17, 358–369. [Google Scholar] [CrossRef]
- Bullard, B.L.; Corder, B.N.; DeBeauchamp, J.; Rubrum, A.; Korber, B.; Webby, R.J.; Weaver, E.A. Epigraph hemagglutinin vaccine induces broad cross-reactive immunity against swine H3 influenza virus. Nat. Commun. 2021, 12, 1203. [Google Scholar] [CrossRef]
- Corder, B.N.; Bullard, B.L.; DeBeauchamp, J.L.; Ilyushina, N.A.; Webby, R.J.; Weaver, E.A. Influenza H1 Mosaic Hemagglutinin Vaccine Induces Broad Immunity and Protection in Mice. Vaccines 2019, 7, 195. [Google Scholar] [CrossRef]
- Donermeyer, D.L.; Beisel, K.W.; Allen, P.M.; Smith, S.C. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J. Exp. Med. 1995, 182, 1291–1300. [Google Scholar] [CrossRef]
- Krishnan, B.; Massilamany, C.; Basavalingappa, R.H.; Gangaplara, A.; Rajasekaran, R.A.; Afzal, M.Z.; Khalilzad-Sharghi, V.; Zhou, Y.; Riethoven, J.-J.; Nandi, S.S.; et al. Epitope Mapping of SERCA2a Identifies an Antigenic Determinant That Induces Mainly Atrial Myocarditis in A/J Mice. J. Immunol. 2018, 200, 523–537. [Google Scholar] [CrossRef]
- Arumugam, R.; Yalaka, B.; Massilamany, C.; Ali, M.S.H.; Lasrado, N.; Jayaraja, S.; Riethoven, J.-J.; Sun, X.; Reddy, J. An evidence for surface expression of an immunogenic epitope of sarcoplasmic/endoplasmic reticulum calcium-ATPase2a on antigen-presenting cells from naive mice in the mediation of autoimmune myocarditis. Immunobiology 2020, 225, 151896. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, J.; Sreekumar, A.; Varambally, S.; Shen, R.; Giacherio, D.; Mehra, R.; Montie, J.E.; Pienta, K.J.; Sanda, M.G.; et al. Autoantibody Signatures in Prostate Cancer. N. Engl. J. Med. 2005, 353, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Eisenbarth, G.S. Accepting clocks that tell time poorly: Fluid-phase versus standard ELISA autoantibody assays. Clin. Immunol. 2007, 125, 120–126. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mandel-Brehm, C.; Dubey, D.; Kryzer, T.J.; O’Donovan, B.D.; Tran, B.; Vazquez, S.E.; Sample, H.A.; Zorn, K.C.; Khan, L.M.; Bledsoe, I.O.; et al. Kelch-like Protein 11 Antibodies in Seminoma-Associated Paraneoplastic Encephalitis. N. Engl. J. Med. 2019, 381, 47–54. [Google Scholar] [CrossRef]
- Robinson, W.H.; Digennaro, C.; Hueber, W.; Haab, B.B.; Kamachi, M.; Dean, E.J.; Fournel, S.; Fong, D.; Genovese, M.C.; De Vegvar, H.E.N.; et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med. 2002, 8, 295–301. [Google Scholar] [CrossRef]
- Deutscher, S. Phage Display to Detect and Identify Autoantibodies in Disease. N. Engl. J. Med. 2019, 381, 89–91. [Google Scholar] [CrossRef]
- Tiu, C.K.; Zhu, F.; Wang, L.F.; de Alwis, R. Phage ImmunoPrecipitation Sequencing (PhIP-Seq): The Promise of High Throughput Serology. Pathogens 2022, 11, 568. [Google Scholar] [CrossRef]
- Angkeow, J.W.; Monaco, D.R.; Chen, A.; Venkataraman, T.; Jayaraman, S.; Valencia, C.; Sie, B.M.; Liechti, T.; Farhadi, P.N.; Funez-Depagnier, G.; et al. Phage display of environmental protein toxins and virulence factors reveals the prevalence, persistence, and genetics of antibody responses. Immunity 2022, 55, 1051–1066.e4. [Google Scholar] [CrossRef]
- Liu, J.; Tang, W.; Budhu, A.; Forgues, M.; Hernandez, M.O.; Candia, J.; Kim, Y.; Bowman, E.D.; Ambs, S.; Zhao, Y.; et al. A Viral Exposure Signature Defines Early Onset of Hepatocellular Carcinoma. Cell 2020, 182, 317–328.e10. [Google Scholar] [CrossRef]
- Chen, G.; Shrock, E.L.; Li, M.Z.; Spergel, J.M.; Nadeau, K.C.; Pongracic, J.A.; Umetsu, D.T.; Rachid, R.; MacGinnitie, A.J.; Phipatanakul, W.; et al. High-resolution epitope mapping by AllerScan reveals relationships between IgE and IgG repertoires during peanut oral immunotherapy. Cell Rep. Med. 2021, 2, 100410. [Google Scholar] [CrossRef]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell 2020, 183, 982–995.e14. [Google Scholar] [CrossRef] [PubMed]
- Larman, H.B.; Laserson, U.; Querol, L.; Verhaeghen, K.; Solimini, N.L.; Xu, G.J.; Klarenbeek, P.L.; Church, G.M.; Hafler, D.A.; Plenge, R.M.; et al. PhIP-Seq characterization of autoantibodies from patients with multiple sclerosis, type 1 diabetes and rheumatoid arthritis. J. Autoimmun. 2013, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, S.E.; Ferre, E.M.N.; Scheel, D.W.; Sunshine, S.; Miao, B.; Mandel-Brehm, C.; Quandt, Z.; Chan, A.Y.; Cheng, M.; German, M.; et al. Identification of novel, clinically correlated autoantigens in the monogenic autoimmune syndrome APS1 by proteome-wide PhIP-Seq. eLife 2020, 9, e55053. [Google Scholar] [CrossRef]
- O’Donovan, B.; Mandel-Brehm, C.; Vazquez, S.E.; Liu, J.; Parent, A.V.; Anderson, M.S.; Kassimatis, T.; Zekeridou, A.; Hauser, S.L.; Pittock, S.J.; et al. High-resolution epitope mapping of anti-Hu and anti-Yo autoimmunity by programmable phage display. Brain Commun. 2020, 2, fcaa059. [Google Scholar] [CrossRef]
- Larman, H.B.; Salajegheh, M.; Bs, R.N.; Lam, T.; Sauld, J.; Steen, H.; Kong, S.W.; Pinkus, J.L.; Amato, A.A.; Elledge, S.J.; et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann. Neurol. 2013, 73, 408–418. [Google Scholar] [CrossRef]
- Basavalingappa, R.H.; Arumugam, R.; Lasrado, N.; Yalaka, B.; Massilamany, C.; Gangaplara, A.; Riethoven, J.-J.; Xiang, S.-H.; Steffen, D.; Reddy, J. Viral myocarditis involves the generation of autoreactive T cells with multiple antigen specificities that localize in lymphoid and non-lymphoid organs in the mouse model of CVB3 infection. Mol. Immunol. 2020, 124, 218–228. [Google Scholar] [CrossRef]
- Gangaplara, A.; Massilamany, C.; Brown, D.M.; Delhon, G.; Pattnaik, A.K.; Chapman, N.; Rose, N.; Steffen, D.; Reddy, J. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-α-reactive CD4 T cells in A/J mice. Clin. Immunol. 2012, 144, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Yoshikawa, T.; Baba, A.; Anzai, T.; Nakamura, I.; Wainai, Y.; Takahashi, T.; Ogawa, S. Autoimmunity against the second extracellular loop of beta(1)-adrenergic receptors induces beta-adrenergic receptor desensitization and myocardial hypertrophy in vivo. Circ. Res. 2001, 88, 578–586. [Google Scholar] [CrossRef]
- Maisch, B.; Trostel-Soeder, R.; Stechemesser, E.; Berg, P.A.; Kochsiek, K. Diagnostic relevance of humoral and cell-mediated immune reactions in patients with acute viral myocarditis. Clin. Exp. Immunol. 1982, 48, 533–545. [Google Scholar]
- Katzmann, J.L.; Schlattmann, P.; Rigopoulos, A.G.; Noutsias, E.; Bigalke, B.; Pauschinger, M.; Tschope, C.; Sedding, D.; Schulze, P.C.; Noutsias, M. Meta-analysis on the immunohistological detection of inflammatory cardiomyopathy in endomyocardial biopsies. Heart Fail. Rev. 2020, 25, 277–294. [Google Scholar] [CrossRef]
- Basavalingappa, R.H.; Massilamany, C.; Krishnan, B.; Gangaplara, A.; Kang, G.; Khalilzad-Sharghi, V.; Han, Z.; Othman, S.; Li, Q.; Riethoven, J.-J.; et al. Identification of an Epitope from Adenine Nucleotide Translocator 1 That Induces Inflammation in Heart in A/J Mice. Am. J. Pathol. 2016, 186, 3160–3175. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Steinman, L. Human peptidome display. Nat. Biotechnol. 2011, 29, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Purcell, A.W.; Rossjohn, J. Post-translationally modified T cell epitopes: Immune recognition and immunotherapy. Klin. Wochenschr. 2009, 87, 1045–1051. [Google Scholar] [CrossRef]
- Irving, M.B.; Pan, O.; Scott, J.K. Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr. Opin. Chem. Biol. 2001, 5, 314–324. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Bolte, H.D. Immunological analysis of auto-antibodies against the adenine nucleotide translocator in dilated cardiomyopathy. J. Mol. Cell. Cardiol. 1985, 17, 603–617. [Google Scholar] [CrossRef]
- Ansari, A.A.; Neckelmann, N.; Villinger, F.; Leung, P.; Danner, D.J.; Brar, S.S.; Zhao, S.; Gravanis, M.B.; Mayne, A.; Gershwin, M.E. Epitope mapping of the branched chain alpha-ketoacid dehydrogenase dihydrolipoyl transacylase (BCKD-E2) protein that reacts with sera from patients with idiopathic dilated cardiomyopathy. J. Immunol. 1994, 153, 4754–4765. [Google Scholar]
- Pohlner, K.; Portig, I.; Pankuweit, S.; Lottspeich, F.; Maisch, B. Identification of mitochondrial antigens recognized by antibodies in sera of patients with idiopathic dilated cardiomyopathy by two-dimensional gel electrophoresis and protein sequencing. Am. J. Cardiol. 1997, 80, 1040–1045. [Google Scholar] [CrossRef]
- Otto, A.; Stähle, I.; Klein, R.; Berg, P.A.; Pankuweit, S.; Brandsch, R. Anti-mitochondrial antibodies in patients with dilated cardiomyopathy (anti-M7) are directed against flavoenzymes with covalently bound FAD. Clin. Exp. Immunol. 1998, 111, 541–547. [Google Scholar] [CrossRef]
- Becker, Y.; Loignon, R.-C.; Julien, A.-S.; Marcoux, G.; Allaeys, I.; Lévesque, T.; Rollet-Labelle, E.; Benk-Fortin, H.; Cloutier, N.; Melki, I.; et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci. Rep. 2019, 9, 4530. [Google Scholar] [CrossRef]
- Tanaka, A. Anti-mitochondrial autoantibodies-milestone or byway to primary biliary cholangitis? Ann. Transl. Med. 2017, 5, 50. [Google Scholar] [CrossRef]
- Lauritsen, K.; Diederichsen, H. Arthritis in Patients with Antimitochondrial Antibodies. Scand. J. Rheumatol. 1983, 12, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Helft, J.; Manicassamy, B.; Guermonprez, P.; Hashimoto, D.; Silvin, A.; Agudo, J.; Brown, B.D.; Schmolke, M.; Miller, J.C.; Leboeuf, M.; et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Investig. 2012, 122, 4037–4047. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Martinez, E.; Planes, R.; Anselmi, G.; Reynolds, M.; Menezes, S.; Adiko, A.C.; Saveanu, L.; Guermonprez, P. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front. Immunol. 2015, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Troutman, T.D.; Hu, W.; Fulenchek, S.; Yamazaki, T.; Kurosaki, T.; Bazan, J.F.; Pasare, C. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc. Natl. Acad. Sci. USA 2012, 109, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Takeda, K.; Gotoh, K.; Takeshima, H.; Akira, S.; Kurosaki, T. Essential Immunoregulatory Role for BCAP in B Cell Development and Function. J. Exp. Med. 2002, 195, 535–545. [Google Scholar] [CrossRef]
- Mackay, I.R.; Leskovsek, N.V.; Rose, N.R. Cell damage and autoimmunity: A critical appraisal. J. Autoimmun. 2008, 30, 5–11. [Google Scholar] [CrossRef]
- Zwaka, T.P.; Manolov, D.; Ozdemir, C.; Marx, N.; Kaya, Z.; Kochs, M.; Hoher, M.; Hombach, V.; Torzewski, J. Complement and dilated cardiomyopathy: A role of sublytic terminal complement complex-induced tumor necrosis factor-alpha synthesis in cardiac myocytes. Am. J. Pathol. 2002, 161, 449–457. [Google Scholar] [CrossRef]
- Nakayama, T.; Sugano, Y.; Yokokawa, T.; Nagai, T.; Matsuyama, T.-A.; Ohta-Ogo, K.; Ikeda, Y.; Ishibashi-Ueda, H.; Nakatani, T.; Ohte, N.; et al. Clinical impact of the presence of macrophages in endomyocardial biopsies of patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 490–498. [Google Scholar] [CrossRef]
- Jane-wit, D.; Altuntas, C.Z.; Johnson, J.M.; Yong, S.; Wickley, P.J.; Clark, P.; Wang, Q.; Popovic, Z.B.; Penn, M.S.; Damron, D.S.; et al. Beta 1-adrenergic receptor autoantibodies mediate dilated cardiomyopathy by agonistically inducing cardiomyocyte apoptosis. Circulation 2007, 116, 399–410. [Google Scholar] [CrossRef]
- Teubner, A.; Tillmann, H.L.; Schuppan, D.; Gericke, G.; Manns, M.P.; Stolzel, U. Prevalence of circulating autoantibodies in healthy individuals. Med. Klin. 2002, 97, 645–649. [Google Scholar] [CrossRef]
- Lacroix-Desmazes, S.; Kaveri, S.V.; Mouthon, L.; Ayouba, A.; Malanchère, E.; Coutinho, A.; Kazatchkine, M.D. Self-reactive antibodies (natural autoantibodies) in healthy individuals. J. Immunol. Methods 1998, 216, 117–137. [Google Scholar] [CrossRef]
- Pashnina, I.A.; Krivolapova, I.M.; Fedotkina, T.V.; Ryabkova, V.A.; Chereshneva, M.V.; Churilov, L.P.; Chereshnev, V.A. Antinuclear Autoantibodies in Health: Autoimmunity Is Not a Synonym of Autoimmune Disease. Antibodies 2021, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Fritzler, M.J.; Martinez-Prat, L.; Choi, M.Y.; Mahler, M. The Utilization of Autoantibodies in Approaches to Precision Health. Front. Immunol. 2018, 9, 2682. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasquinha, M.T.; Lasrado, N.; Petro-Turnquist, E.; Weaver, E.; Venkataraman, T.; Anderson, D.; Laserson, U.; Larman, H.B.; Reddy, J. PhIP-Seq Reveals Autoantibodies for Ubiquitously Expressed Antigens in Viral Myocarditis. Biology 2022, 11, 1055. https://doi.org/10.3390/biology11071055
Rasquinha MT, Lasrado N, Petro-Turnquist E, Weaver E, Venkataraman T, Anderson D, Laserson U, Larman HB, Reddy J. PhIP-Seq Reveals Autoantibodies for Ubiquitously Expressed Antigens in Viral Myocarditis. Biology. 2022; 11(7):1055. https://doi.org/10.3390/biology11071055
Chicago/Turabian StyleRasquinha, Mahima T., Ninaad Lasrado, Erika Petro-Turnquist, Eric Weaver, Thiagarajan Venkataraman, Daniel Anderson, Uri Laserson, H. Benjamin Larman, and Jay Reddy. 2022. "PhIP-Seq Reveals Autoantibodies for Ubiquitously Expressed Antigens in Viral Myocarditis" Biology 11, no. 7: 1055. https://doi.org/10.3390/biology11071055
APA StyleRasquinha, M. T., Lasrado, N., Petro-Turnquist, E., Weaver, E., Venkataraman, T., Anderson, D., Laserson, U., Larman, H. B., & Reddy, J. (2022). PhIP-Seq Reveals Autoantibodies for Ubiquitously Expressed Antigens in Viral Myocarditis. Biology, 11(7), 1055. https://doi.org/10.3390/biology11071055