
Epithelial-Mesenchymal Plasticity Induced by Discontinuous Exposure to TGFβ1 Promotes Tumour Growth

 , , , ,
, , , ,  , ,
, ,  ,
,  , , add
Show full author list
, , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Extraction and Quantification
2.3. Immunocytochemistry
2.4. Whole-Transcriptome Sequencing
2.5. BrdU Assay
2.6. Focus Formation Assay
2.7. Wound Healing Assay
2.8. In Vivo Tumourigenesis Assay, H&E Staining and KI67 Immunohistochemistry
2.9. First Passage Mammosphere Formation Assay
3. Results
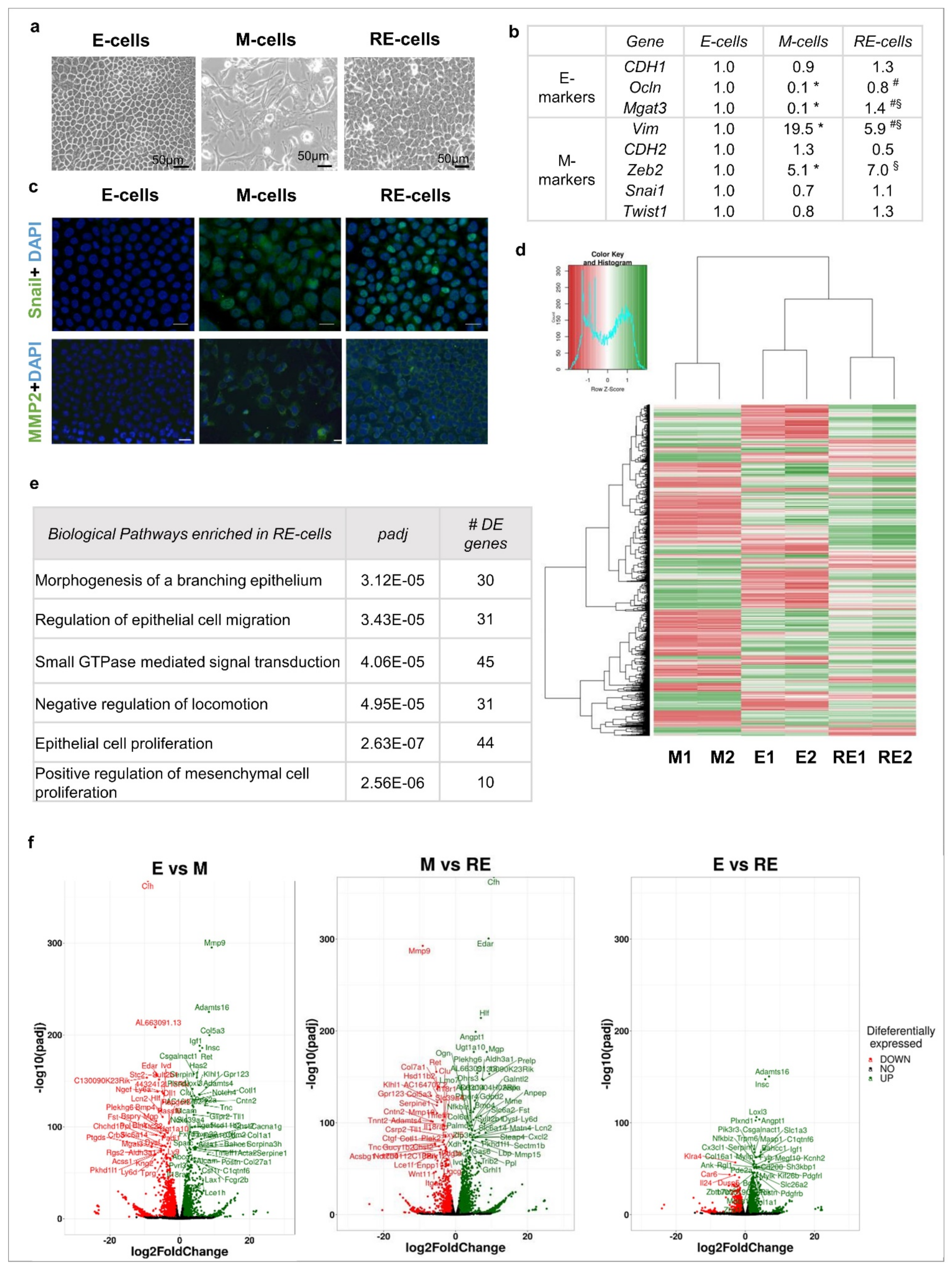
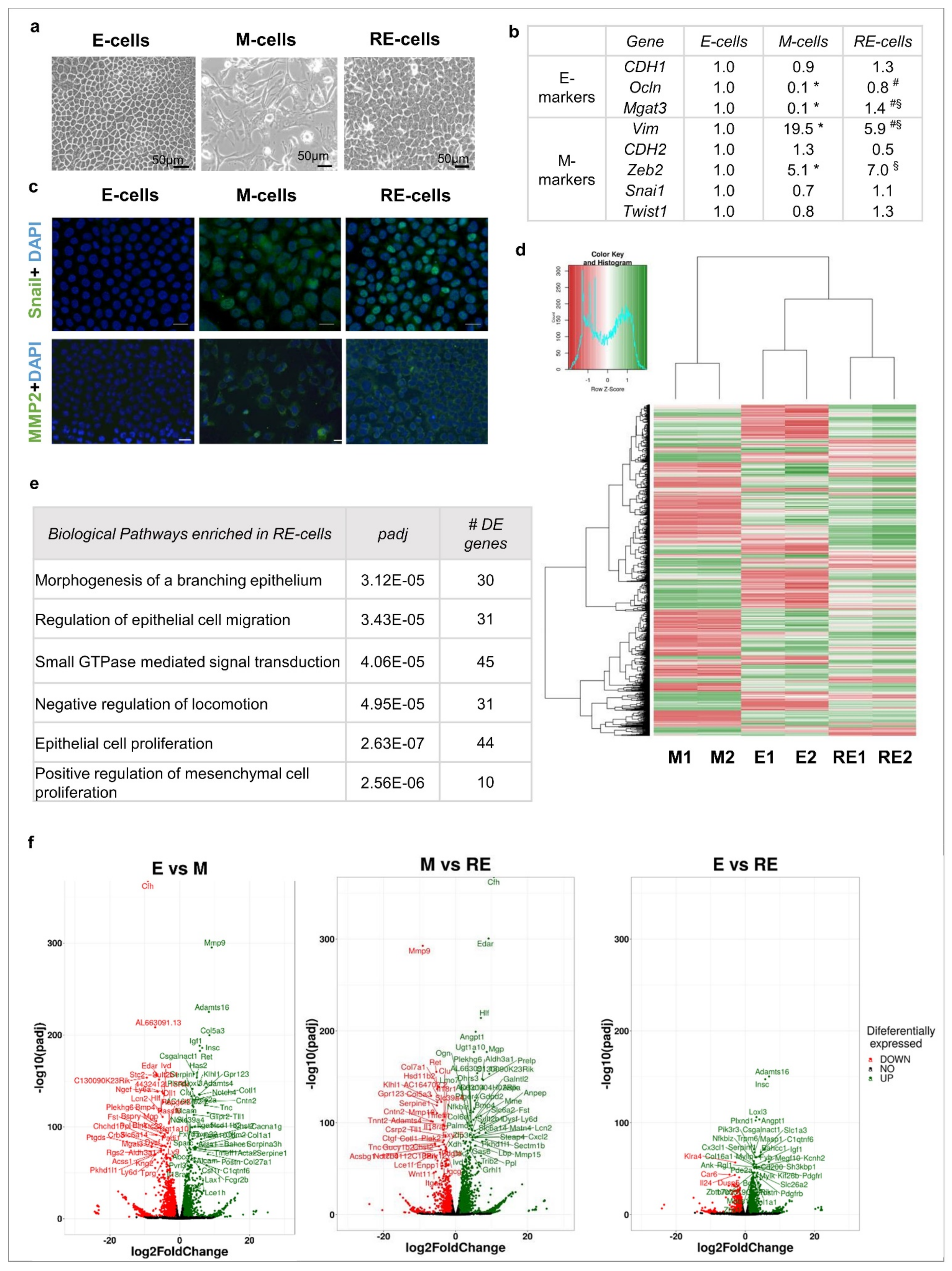
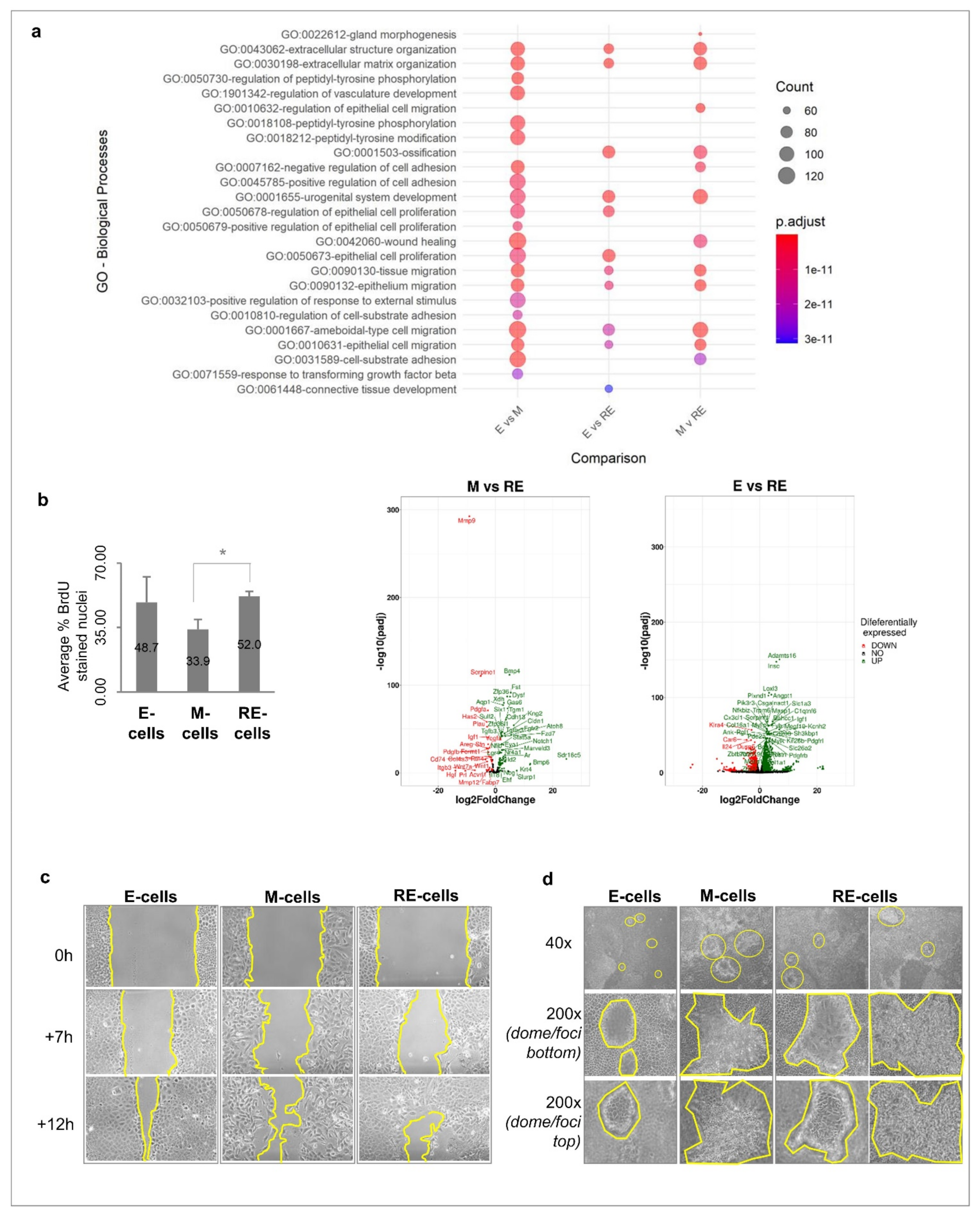
3.1. Phenotypic EMT/MET vs. Molecular EMT/MET
3.2. Phenotypic MET Is Not Supported by Complete Molecular Reversion Back to Epithelia
3.3. Phenotypic MET Generates E-like, M-like and Novel Cellular Subpopulations
3.4. Cellular Heterogeneity, Generated after Phenotypic MET In Vitro, Creates Functional Heterogeneity
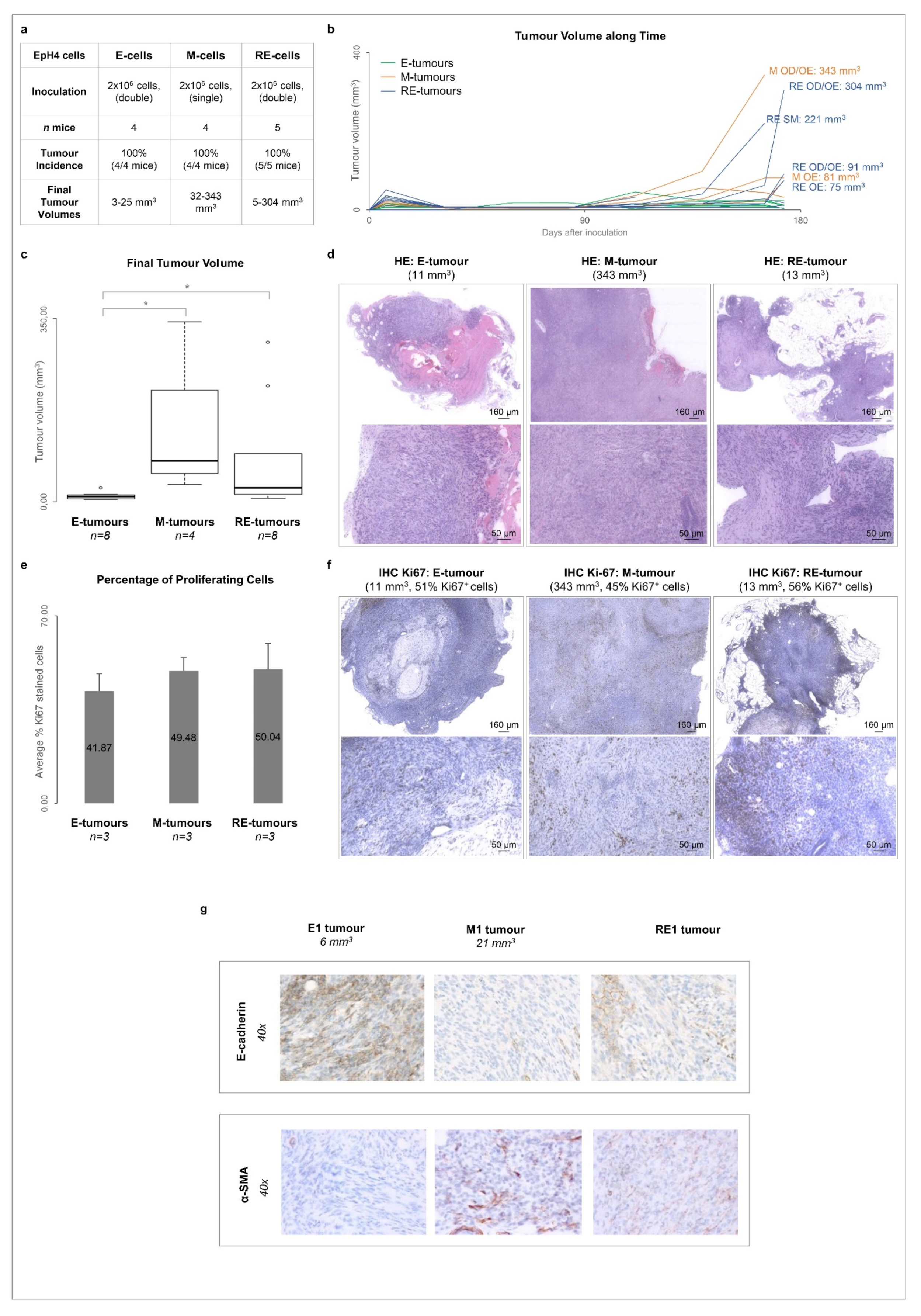
3.5. Cellular Heterogeneity, Generated after Phenotypic MET In Vitro, Is Maintained in Tumours Growing In Vivo
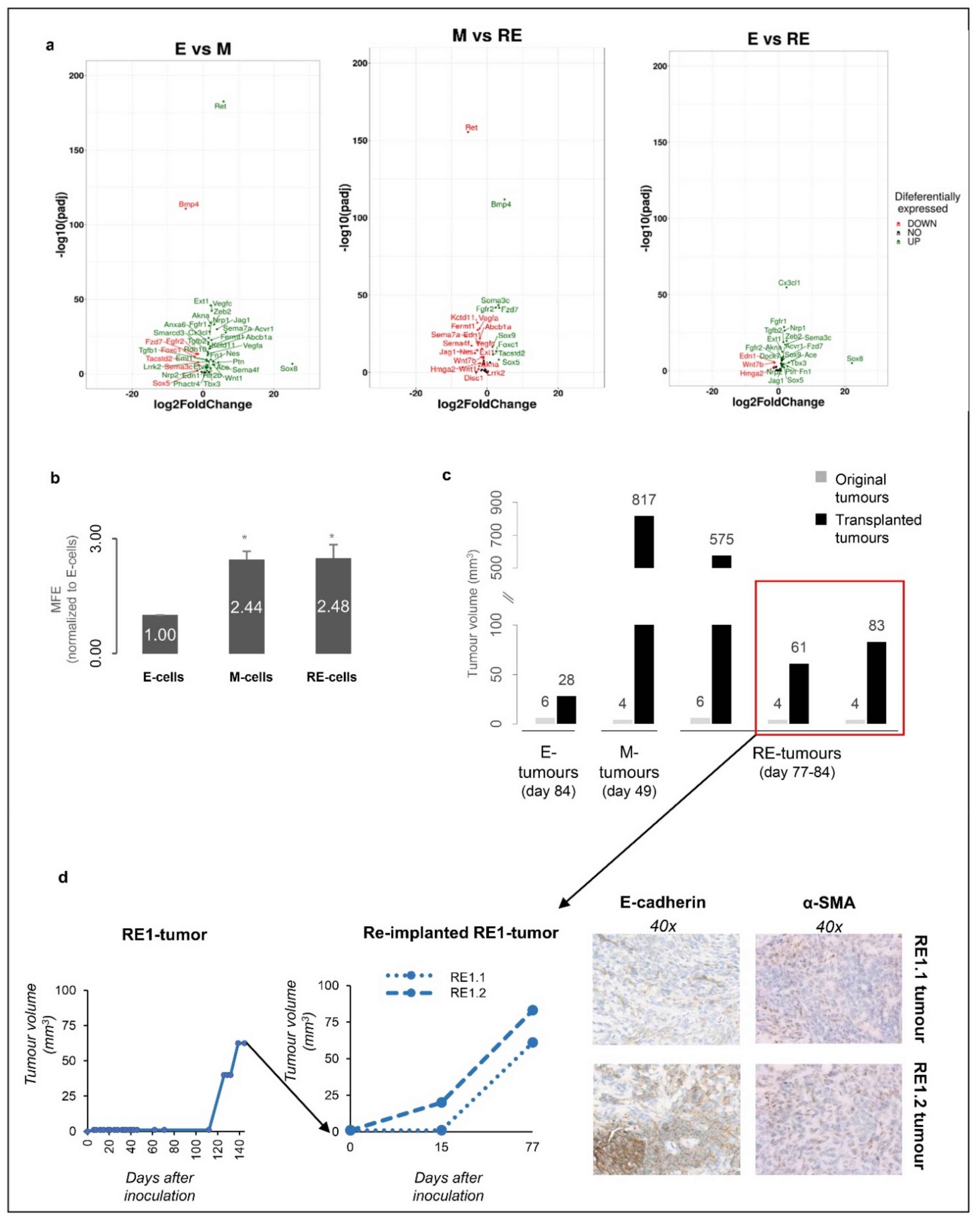
3.6. RE Cells Mimic M Cells in Self-Renewal Capacity In Vitro and Fast Growth of Tumour Transplants In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial—Mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A. Epithelial-Mesenchymal Transitions in development and disease: Old views and new perspectives. Int. J. Dev. Biol. 2009, 53, 1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta—Rev. Cancer 2009, 1796, 75–90. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.-P.; Chua, K.; Jing Sim, W.; Huang, R. La transition épithéliomésenchymateuse au cours du développement dans la fibrose et dans la progression tumorale. Bull. Cancer 2010, 97, 1285–1295. [Google Scholar] [CrossRef]
- Gunasinghe, N.P.A.D.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Richardson, F.; Young, G.D.; Sennello, R.; Wolf, J.; Argast, G.M.; Mercado, P.; Davies, A.; Epstein, D.M.; Wacker, B. The evaluation of E-cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small cell lung cancer treated with erlotinib as second- or third-line therapy. Anticancer Res. 2012, 32, 537–552. [Google Scholar]
- Van Denderen, B.J.W.; Thompson, E.W. Cancer: The to and fro of tumour spread. Nature 2013, 493, 487–488. [Google Scholar] [CrossRef]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2016, 10, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Tu, G.; Liu, Z.; Liu, M. Cancer-associated fibroblasts: A multifaceted driver of breast cancer progression. Cancer Lett. 2015, 361, 155–163. [Google Scholar] [CrossRef]
- Kim, H.Y.; Jackson, T.R.; Davidson, L.A. On the role of mechanics in driving mesenchymal-to-epithelial transitions. Semin. Cell Dev. Biol. 2017, 67, 113–122. [Google Scholar] [CrossRef]
- Di Donato, M.; Cernera, G.; Migliaccio, A.; Castoria, G. Nerve Growth Factor Induces Proliferation and Aggressiveness In Prostate Cancer Cells. Cancers 2019, 11, 784. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Scheel, C.; Eaton, E.N.; Li, S.H.-J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.-J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and Autocrine Signals Induce and Maintain Mesenchymal and Stem Cell States in the Breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Bruna, A.; Greenwood, W.; Le Quesne, J.; Teschendorff, A.; Miranda-Saavedra, D.; Rueda, O.M.; Sandoval, J.L.; Vidakovic, A.T.; Saadi, A.; Pharoah, P.; et al. TGFβ induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat. Commun. 2012, 3, 1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.; Pereira, P.M.; Varanda, A.S.; Carvalho, J.; Azevedo, M.; Mateus, D.D.; Mendes, N.; Oliveira, P.; Trindade, F.; Pinto, M.T.; et al. Codon misreading tRNAs promote tumor growth in mice. RNA Biol. 2018, 15, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, L.G.; Wu, X.; Guan, J.-L. Wound-healing assay. Methods Mol. Biol. 2005, 294, 23–29. [Google Scholar] [CrossRef]
- Vieira, A.F.; Ricardo, S.; Ablett, M.P.; Dionísio, M.R.; Mendes, N.; Albergaria, A.; Farnie, G.; Gerhard, R.; Cameselle-Teijeiro, J.F.; Seruca, R.; et al. P-Cadherin Is Coexpressed with CD44 and CD49f and Mediates Stem Cell Properties in Basal-like Breast Cancer. Stem Cells 2012, 30, 854–864. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.S.; Oliveira, P.; Cabral, J.; Carvalho, S.; Huntsman, D.; Gärtner, F.; Seruca, R.; Reis, C.A.; Oliveira, C. Loss and recovery of Mgat3 and GnT-III mediated E-cadherin N-glycosylation is a mechanism involved in epithelial-Mesenchymal-Epithelial transitions. PLoS ONE 2012, 7, e33191. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.E.; Binas, B.; Chapman, R.S.; Kurian, K.M.; Clarkson, R.W.E.; Clark, A.J.; Lane, E.B.; Watson, C.J. A novel cell culture model for studying differentiation and apoptosis in the mouse mammary gland. Breast Cancer Res. 2000, 2, 222–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakiri, L.; Macho-Maschler, S.; Custic, I.; Niemiec, J.; Guío-Carrión, A.; Hasenfuss, S.C.; Eger, A.; Müller, M.; Beug, H.; Wagner, E.F. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2015, 22, 336–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.S.; Baek, S.H. Mouse models for breast cancer metastasis. Biochem. Biophys. Res. Commun. 2010, 394, 443–447. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H.M. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Wei, H.; Lu, T. Targeting microenvironment in cancer therapeutics. Oncotarget 2016, 7, 52575–52583. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.-Y.; Liu, X.-Z.; Liang, C.-M. Inflammatory microenvironment contributes to epithelial-mesenchymal transition in gastric cancer. World J. Gastroenterol. 2016, 22, 6619–6628. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.M.; Panzilius, E.; Bartsch, H.S.; Irmler, M.; Beckers, J.; Kari, V.; Linnemann, J.R.; Dragoi, D.; Hirschi, B.; Kloos, U.J.; et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient twist1 activation. Cell Rep. 2015, 10, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Grosse-Wilde, A.; Fouquier d’Hérouël, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.-H.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 Cooperatively Determine the Mammary Stem Cell State. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Ibaragi, S.; Shima, K.; Hu, M.G.; Katsurano, M.; Sasaki, A.; Hu, G.F. Epithelial-mesenchymal transition induced by growth suppressor p12 CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008, 68, 10377–10386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, M.; Ferreira, M.; Oliveira, P.; Mendes, N.; André, A.; Vieira, A.F.; Nunes, J.B.; Carvalho, J.; Rocha, S.; Azevedo, M.; et al. Epithelial-Mesenchymal Plasticity Induced by Discontinuous Exposure to TGFβ1 Promotes Tumour Growth. Biology 2022, 11, 1046. https://doi.org/10.3390/biology11071046
Santos M, Ferreira M, Oliveira P, Mendes N, André A, Vieira AF, Nunes JB, Carvalho J, Rocha S, Azevedo M, et al. Epithelial-Mesenchymal Plasticity Induced by Discontinuous Exposure to TGFβ1 Promotes Tumour Growth. Biology. 2022; 11(7):1046. https://doi.org/10.3390/biology11071046
Chicago/Turabian StyleSantos, Mafalda, Marta Ferreira, Patrícia Oliveira, Nuno Mendes, Ana André, André F. Vieira, Joana B. Nunes, Joana Carvalho, Sara Rocha, Mafalda Azevedo, and et al. 2022. "Epithelial-Mesenchymal Plasticity Induced by Discontinuous Exposure to TGFβ1 Promotes Tumour Growth" Biology 11, no. 7: 1046. https://doi.org/10.3390/biology11071046
APA StyleSantos, M., Ferreira, M., Oliveira, P., Mendes, N., André, A., Vieira, A. F., Nunes, J. B., Carvalho, J., Rocha, S., Azevedo, M., Ferreira, D., Reis, I., Vinagre, J., Paredes, J., Heravi-Moussavi, A., Lima, J., Máximo, V., Burleigh, A., Roskelley, C., ... Oliveira, C. (2022). Epithelial-Mesenchymal Plasticity Induced by Discontinuous Exposure to TGFβ1 Promotes Tumour Growth. Biology, 11(7), 1046. https://doi.org/10.3390/biology11071046