
GJB2 Is a Major Cause of Non-Syndromic Hearing Impairment in Senegal

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approvals
2.2. Study Population
2.3. Mutation Screening of GJB2 and GJB6
2.4. Bioinformatic and Statistical Analyses
3. Results
3.1. Socio-Demographic Data
3.2. Audiological Patterns
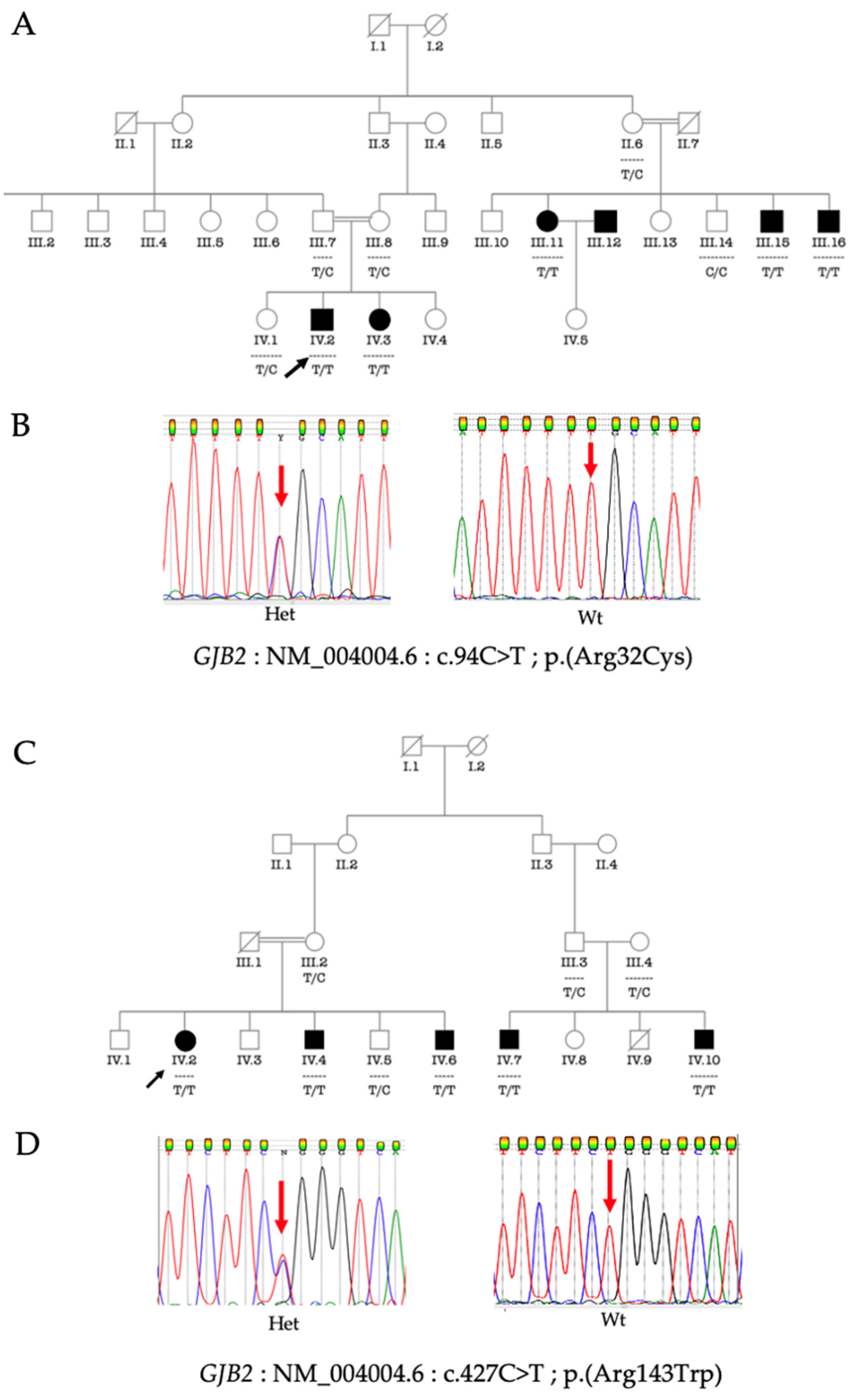
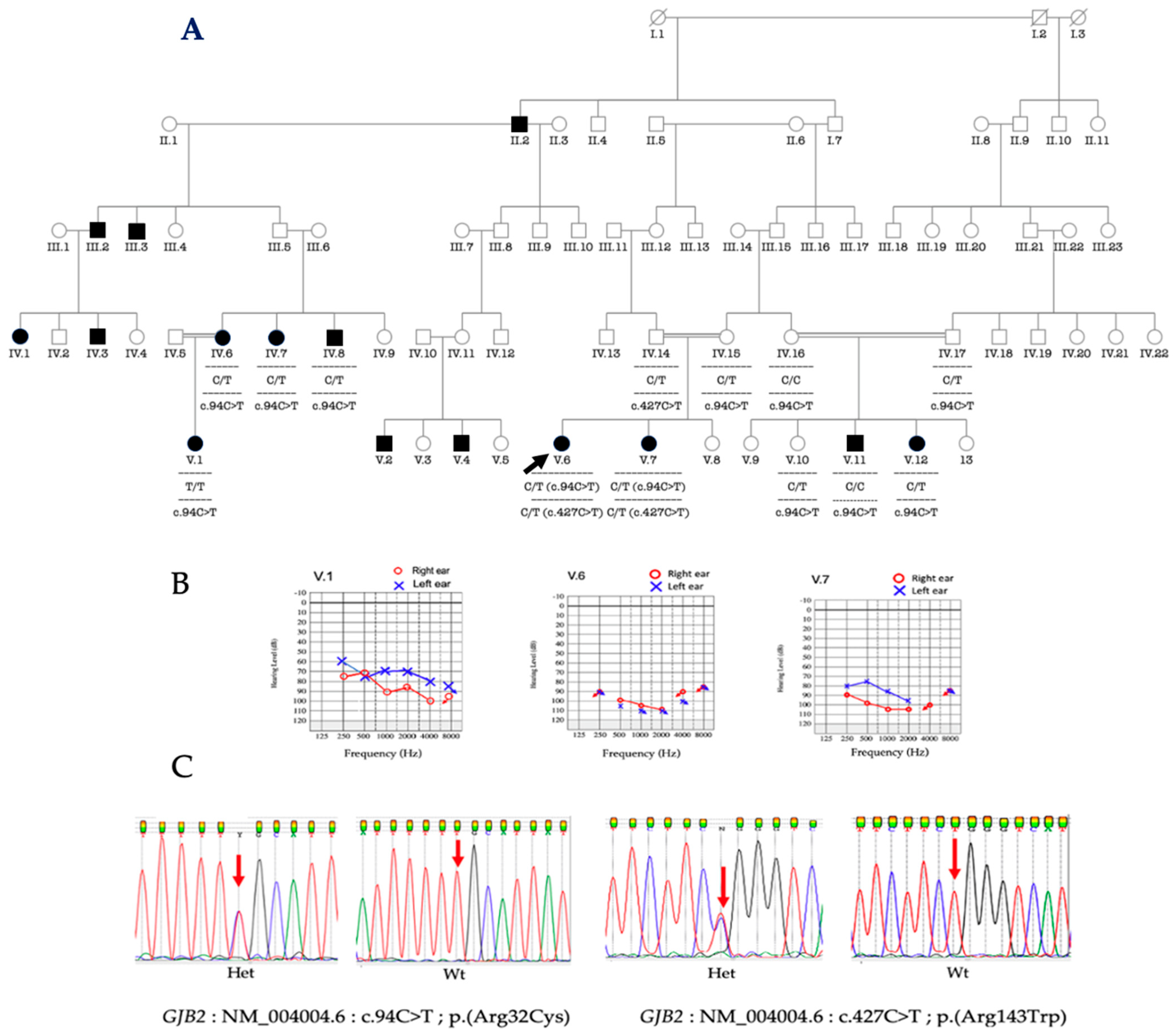
3.3. Molecular Analysis of GJB2 and GJB6
3.4. Phenotype-Genotype Correlation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2013 DALYs and HALE Collaborators; Murray, C.J.L.; Barber, R.M.; Foreman, K.J.; Ozgoren, A.A.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Abraham, J.P.; Abubakar, I.; et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: Quantifying the epidemiological transition. Lancet 2015, 386, 2145–2191. [Google Scholar] [CrossRef]
- WHO. The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Wolff, R.; Hommerich, J.; Riemsma, R.; Antes, G.; Lange, S.; Kleijnen, J. Hearing screening in newborns: Systematic review of accuracy, effectiveness, and effects of interventions after screening. Arch. Dis. Child. 2009, 95, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Olusanya, B.O.; Neumann, K.J.; Saunders, J.E. The global burden of disabling hearing impairment: A call to action. Bull. World Health Organ. 2014, 92, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Korver, A.M.H.; Smith, R.J.H.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.K.; Lustig, L.R.; Usami, S.; Boudewyns, A.N. Congenital hearing loss. Nat. Rev. Dis. Prim 2017, 3, 16094. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary Hearing Loss and Deafness Overview. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1434/ (accessed on 28 March 2022).
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. 2018. Available online: http://hereditaryhearingloss.org (accessed on 17 December 2021).
- Guilford, P.; Ben Arab, S.; Blanchard, S.; Levilliers, J.; Weissenbach, J.; Belkahia, A.; Petit, C. A non–syndromic form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat. Genet. 1994, 6, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Braun, K.V.N.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef]
- Adhikary, B.; Ghosh, S.; Paul, S.; Bankura, B.; Pattanayak, A.K.; Biswas, S.; Maity, B.; Das, M. Spectrum and frequency of GJB2, GJB6 and SLC26A4 gene mutations among nonsyndromic hearing loss patients in eastern part of India. Gene 2015, 573, 239–245. [Google Scholar] [CrossRef]
- Adadey, S.M.; Manyisa, N.; Mnika, K.; De Kock, C.; Nembaware, V.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. GJB2 and GJB6 Mutations in Non-Syndromic Childhood Hearing Impairment in Ghana. Front. Genet. 2019, 10, 841. [Google Scholar] [CrossRef]
- Lasisi, A.O.; Bademci, G.; Foster, J.; Blanton, S.; Tekin, M. Common genes for non-syndromic deafness are uncommon in sub-Saharan Africa: A report from Nigeria. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1870–1873. [Google Scholar] [CrossRef][Green Version]
- Gasmelseed, N.M.A.; Schmidt, M.; Magzoub, M.M.A.; Macharia, M.; Elmustafa, O.M.; Ototo, B.; Winkler, E.; Ruge, G.; Horstmann, R.D.; Meyer, C.G. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum. Mutat. 2004, 23, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Kabahuma, R.I.; Ouyang, X.; Du, L.L.; Yan, D.; Hutchin, T.; Ramsay, M.; Penn, C.; Liu, X.-Z. Absence of GJB2 gene mutations, the GJB6 deletion (GJB6-D13S1830) and four common mitochondrial mutations in nonsyndromic genetic hearing loss in a South African population. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 611–617. [Google Scholar] [CrossRef][Green Version]
- Tingang Wonkam, E.; Chimusa, E.; Noubiap, J.J.; Adadey, S.M.F.; Fokouo, J.V.; Wonkam, A. GJB2 and GJB6 Mutations in Hereditary Recessive Non-Syndromic Hearing Impairment in Cameroon. Genes 2019, 10, 844. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, I.; Moreno-Pelayo, M.A.; Del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Moreno, F.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: A multicenter study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Sc, M.; Ibis Menéndez, M.D.; Felipe Moreno, P.D. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef]
- Zhao, H.-B.; Kikuchi, T.; Ngezahayo, A.; White, T.W. Gap Junctions and Cochlear Homeostasis. J. Membr. Biol. 2006, 209, 177–186. [Google Scholar] [CrossRef]
- Del Castillo, F.J.; Rodríguez-Ballesteros, M.; Alvarez, A.; Hutchin, T.; Leonardi, E.; De Oliveira, C.A.; Azaiez, H.; Brownstein, Z.; Avenarius, M.R.; Marlin, S.; et al. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef]
- A Bliznets, E.; Makienko, O.N.; Okuneva, E.G.; Markova, T.G.; Poliakov, A.V. New recurrent extended deletion, including GJB2 and GJB6 genes, results in isolated sensorineural hearing impairment with autosomal recessive type of inheritance. Genetika 2014, 50, 474–480. [Google Scholar]
- Moisan, S.; Le Nabec, A.; Quillévéré, A.; Le Maréchal, C.; Férec, C. Characterization of GJB2 cis-regulatory elements in the DFNB1 locus. Qual. Life Res. 2019, 138, 1275–1286. [Google Scholar] [CrossRef]
- Yalcouyé, A.; Traoré, O.; Taméga, A.; Maïga, A.B.; Kané, F.; Oluwole, O.G.; Guinto, C.O.; Kéita, M.; Timbo, S.K.; DeKock, C.; et al. Etiologies of Childhood Hearing Impairment in Schools for the Deaf in Mali. Front. Pediatr. 2021, 9, 726726. [Google Scholar] [CrossRef]
- ISO 8253-1:2010; Acoustics—Audiometric Test Methods—Part 1: Basic Pure Tone Air and Bone Conduction Audiometry. International Organization for Standardization: Geneva, Switzerland, 2010.
- Olusanya, B.O.; Davis, A.C.; Hoffman, H.J. Hearing loss grades and the International classification of functioning, disability and health. Bull. World Health Organ. 2019, 97, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Lebeko, K.; Nziale, J.J.N.; Dandara, C.; Makubalo, N.; Wonkam, A. In Search of Genetic Markers for Nonsyndromic Deafness in Africa: A Study in Cameroonians and Black South Africans with the GJB6 and GJA1 Candidate Genes. OMICS A J. Integr. Biol. 2014, 18, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Homo Sapiens Gap Junction Protein Beta 2 (GJB2), mRNA. 2021 [Cited 29 September 2021]. Available online: http://www.ncbi.nlm.nih.gov/nuccore/NM_004004.6 (accessed on 1 March 2022).
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M.; et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Grosse, S.D.; Mason, C.A.; Gaffney, M.; Thomson, V.; White, K.R. What Contribution Did Economic Evidence Make to the Adoption of Universal Newborn Hearing Screening Policies in the United States? Int. J. Neonatal Screen. 2018, 4, 25. [Google Scholar] [CrossRef]
- Masson, E. Évolution De L’âge Du Diagnostic Des Surdités Congénitales. EM-Consulte. [Cited 12 February 2022]. Available online: https://www.em-consulte.com/article/287793/evolution-de-lage-du-diagnostic-des-surdites-conge (accessed on 30 March 2022).
- Engelman, D. The Status of Neonatal Hearing Screening in Sub-Saharan Africa: A Systematic Review. Ph.D. Thesis, City University of New York, New York, NY, USA, 2014. [Google Scholar]
- Moctar, E.C.M.; Riahi, Z.; El Hachmi, H.; Veten, F.; Meiloud, G.; Bonnet, C.; Abdelhak, S.; Errami, M.; Houmeida, A. Etiology and associated GJB2 mutations in Mauritanian children with non-syndromic hearing loss. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 3693–3698. [Google Scholar] [CrossRef]
- Zheng, J.; Ying, Z.; Cai, Z.; Sun, D.; He, Z.; Gao, Y.; Zhang, T.; Zhu, Y.; Chen, Y.; Guan, M.-X. GJB2 Mutation Spectrum and Genotype-Phenotype Correlation in 1067 Han Chinese Subjects with Non-Syndromic Hearing Loss. PLoS ONE 2015, 10, e0128691. [Google Scholar] [CrossRef]
- Kasakura-Kimura, N.; Masuda, M.; Mutai, H.; Masuda, S.; Morimoto, N.; Ogahara, N.; Misawa, H.; Sakamoto, H.; Saito, K.; Matsunaga, T. WFS1andGJB2mutations in patients with bilateral low-frequency sensorineural hearing loss. Laryngoscope 2017, 127, E324–E329. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, A.R.; Kim, N.K.D.; Lee, C.; Kim, M.Y.; Jeon, E.H.; Park, W.Y.; Choi, B.Y. Unraveling of Enigmatic Hearing-Impaired GJB2 Single Heterozygotes by Massive Parallel Sequencing: DFNB1 or Not? Medicine 2016, 95, e3029. [Google Scholar] [CrossRef] [PubMed]
- Adadey, S.M.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. Screening for GJB2-R143W-Associated Hearing Impairment: Implications for Health Policy and Practice in Ghana. Public Health Genom. 2020, 23, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Adadey, S.M.; Wonkam, E.T.; Aboagye, E.T.; Quansah, D.; Asante-Poku, A.; Quaye, O.; Amedofu, G.K.; Awandare, G.A.; Wonkam, A. Enhancing Genetic Medicine: Rapid and Cost-Effective Molecular Diagnosis for a GJB2 Founder Mutation for Hearing Impairment in Ghana. Genes 2020, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Fareed, M.; Afzal, M. Genetics of consanguinity and inbreeding in health and disease. Ann. Hum. Biol. 2016, 44, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Aboagye, E.T.; Adadey, S.M.; Esoh, K.; Jonas, M.; De Kock, C.; Amenga-Etego, L.; Awandare, G.A.; Wonkam, A. Age Estimate of GJB2-p.(Arg143Trp) Founder Variant in Hearing Impairment in Ghana, Suggests Multiple Independent Origins across Populations. Biology 2022, 11, 476. [Google Scholar] [CrossRef] [PubMed]
- Fofana, D.M. Senegal, the African Slave Trade, and the Door of No Return: Giving Witness to Gorée Island. Humanities 2020, 9, 57. [Google Scholar] [CrossRef]
- Cryns, K.; Orzan, E.; Murgia, A.; Huygen, P.L.M.; Moreno, F.; Del Castillo, I.; Chamberlin, G.P.; Azaiez, H.; Prasad, S.; Cucci, R.A.; et al. A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J. Med. Genet. 2004, 41, 147–154. [Google Scholar] [CrossRef]
- Wang, X.; Huang, L.; Zhao, X.; Wang, X.; Cheng, X.; Du, Y.; Liu, D. Children with GJB2 gene mutations have various audiological phenotypes. Biosci. Trends 2018, 12, 419–425. [Google Scholar] [CrossRef]
- Carranza, C.; Menendez, I.; Herrera, M.; Castellanos, P.; Amado, C.; Maldonado, F.; Rosales, L.; Escobar, N.; Guerra, M.; Alvarez, D.; et al. A Mayan founder mutation is a common cause of deafness in Guatemala. Clin. Genet. 2015, 89, 461–465. [Google Scholar] [CrossRef]
- Nadeau, J.H. Modifier genes in mice and humans. Nat. Rev. Genet. 2001, 2, 165–174. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Degree of HI | Number of Patients(n) | Mean Age at Medical Diagnosis |
|---|---|---|
| Moderate (41–60 dB) | 8 (6.42%) | 8.37 ± 3.81 [5–14 years] |
| Severe (61–80 dB) | 14 (11.29%) | 4.25 ± 3.77 [1.5–13 years] |
| Profound (≥81 dB) | 107 (82.26%) | 2.33 ± 1.14 [1–6 years] |
| Genotypes | Multiplex Families | |
|---|---|---|
| n * | % (n/N) | |
| [c.94C>T]; [c.94C>T] | 11 | 25 |
| [c.427C>T]; [c.427C>T] | 2 | 4.54 |
| [c.427C>T]; [c.94C>T] | 1 | 2.27 |
| [c.427C>T]; [c.132G>A] | 1 | 2.27 |
| Total | 15 | 34.09 |
| Allele Frequency (n/N) | Allele Frequency from Ensembl | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variants | rs Number | Allele | Cases | Controls | p-Value (Cases vs. Controls) | Global | Africa | America | East Asia | Europe |
| c.94C>T | rs371024165 | C | 0.78 (86/106) | 0.99 (294/296) | <0.0001 | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| T | 0.22 (23/106) | 0.01 (2/296) | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | |||
| c.427C>T | rs80338948 | C | 0.94 (100/106) | 0.98 (292/296) | 0.024 | 0.9998 | 1.0000 | 1.0000 | 0.9990 | 1.0000 |
| T | 0.06 (6/106) | 0.02 (4/296) | 0.0002 | 0.0000 | 0.0000 | 0.0010 | 0.0000 | |||
| c.132G>A | rs104894407 | G | 0.99 (105/106) | 0.996 (295/296) | 0.458 | 0.9998 | 1.0000 | 1.0000 | 1.0000 | 1.0000 |
| A | 0.01 (1/106) | 0.004 (1/296) | 0.0002 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | |||
| Genotypes | Degree of HI | ||
|---|---|---|---|
| Moderate (41–60 dB) | Severe (61–80 dB) | Profound (≥81 dB) | |
| [c.94C>T]; [c.94C>T] | 4 | 3 | 9 |
| [c.427C>T]; [c.427C>T] | 0 | 0 | 7 |
| [c.427C>T]; [c.94C>T] | 0 | 0 | 2 |
| [c.427C>T]; [c.132G>A] | 0 | 0 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dia, Y.; Adadey, S.M.; Diop, J.P.D.; Aboagye, E.T.; Ba, S.A.; De Kock, C.; Ly, C.A.T.; Oluwale, O.G.; Sène, A.R.G.; Sarr, P.D.; et al. GJB2 Is a Major Cause of Non-Syndromic Hearing Impairment in Senegal. Biology 2022, 11, 795. https://doi.org/10.3390/biology11050795
Dia Y, Adadey SM, Diop JPD, Aboagye ET, Ba SA, De Kock C, Ly CAT, Oluwale OG, Sène ARG, Sarr PD, et al. GJB2 Is a Major Cause of Non-Syndromic Hearing Impairment in Senegal. Biology. 2022; 11(5):795. https://doi.org/10.3390/biology11050795
Chicago/Turabian StyleDia, Yacouba, Samuel Mawuli Adadey, Jean Pascal Demba Diop, Elvis Twumasi Aboagye, Seydi Abdoul Ba, Carmen De Kock, Cheikh Ahmed Tidjane Ly, Oluwafemi Gabriel Oluwale, Andrea Regina Gnilane Sène, Pierre Diaga Sarr, and et al. 2022. "GJB2 Is a Major Cause of Non-Syndromic Hearing Impairment in Senegal" Biology 11, no. 5: 795. https://doi.org/10.3390/biology11050795
APA StyleDia, Y., Adadey, S. M., Diop, J. P. D., Aboagye, E. T., Ba, S. A., De Kock, C., Ly, C. A. T., Oluwale, O. G., Sène, A. R. G., Sarr, P. D., Diallo, B. K., Diallo, R. N., & Wonkam, A. (2022). GJB2 Is a Major Cause of Non-Syndromic Hearing Impairment in Senegal. Biology, 11(5), 795. https://doi.org/10.3390/biology11050795