
Biology of Cardiac Troponins: Emphasis on Metabolism


Abstract
Simple Summary
Abstract
1. Introduction
2. Metabolic Pathway of cTns
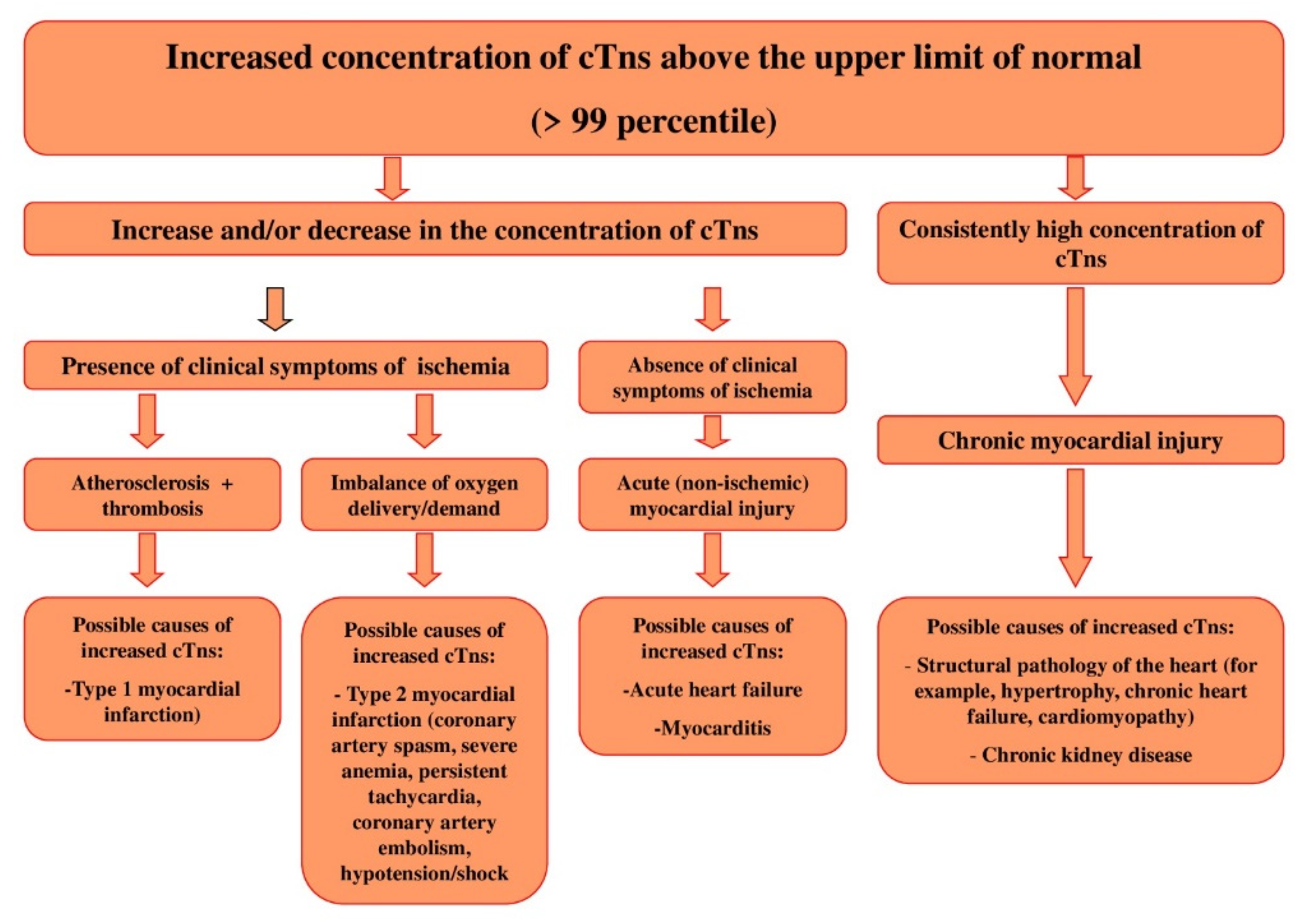
2.1. Release of cTns from MCs: Mechanisms and Diagnostic Value
2.2. Release of cTns as a Result of the Processes of Regeneration and Renewal of MCs
2.3. Release of cTns as a Result of Apoptosis of MCs
2.4. Release of cTns as a Result of the Formation of Membrane Vesicles on the Surface of MCs
2.5. Intracellular Proteolytic Degradation of cTns Molecules into Small Fragments and the Release of the Latter through the Intact Membrane of MCs
2.6. Release of cTns as a Result of Increased Membrane Permeability of MCs
2.7. Release of cTns as a Result of Small-Scale (Subclinical) Necrosis of Cardiomyocytes
2.8. Release of cTns from Non-Cardiac Cells
2.9. Circulation of cTns in Blood Plasma: Influencing Factors and Diagnostic Value
- (1)
- Study of the fundamental specific mechanisms of proteolytic degradation of cTns in the bloodstream, both under normal conditions and under the conditions of simulated concomitant pathologies. This requires a targeted and thorough study of the potential effect of individual serum proteolytic enzymes (for example, the specific thrombin-mediated degradation of cTnT).
- (2)
- Search for specific fragments of cTns, which are released at the earliest possible time after the onset of myocardial ischemia and the creation of antibodies to them, which will increase the sensitivity and specificity of troponin immunoassays.
- (3)
- Search for specific fragments of cTns, which have a small molecular weight and are able to pass through the glomerular and blood-salivary barriers. The creation of antibodies to these fragments will make it possible to develop specific highly sensitive test systems for the analysis of non-invasive biological fluids (urine and oral fluid) and for the introduction of new methods of non-invasive diagnostics and monitoring of cardiovascular pathologies, including MI, into routine clinical practice.
- (4)
- Study and identification of potentially possible specific mechanisms of proteolytic cleavage of cTns under the action of other (non-ischemic) factors. This will allow the development of specific troponin immunoassays to identify those fragments that, for example, will increase exclusively with the stretching of the myocardium or exclusively with an increase in the activity of the adrenergic nervous system and an increase in β-AR stimulation, etc. Thus, it will be possible to carry out a more specific diagnosis of non-ischemic myocardial damage in some physiological and pathological conditions not associated with ischemia of the cardiac muscle tissue.
2.10. Removal of cTns from the Bloodstream: Mechanisms and Diagnostic Value
2.11. Circadian Rhythms of cTns: Possible Mechanisms of Formation and Diagnostic Role
3. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takeda, S. Crystal structure of troponin and the molecular mechanism of muscle regulation. J. Electron. Microsc. Tokyo 2005, 54 (Suppl. 1), i35–i41. [Google Scholar] [CrossRef] [PubMed]
- Chaulin, A.M. Main analytical characteristics of laboratory methods for the determination of cardiac troponins: A review from the historical and modern points of view. Orv. Hetil. 2022, 163, 12–20. (In Hungarian) [Google Scholar] [CrossRef] [PubMed]
- Katrukha, I.A. Human cardiac troponin complex. Structure and functions. Biochem. Mosc. 2013, 78, 1447–1465. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar] [CrossRef]
- Chaulin, A. Clinical and Diagnostic Value of Highly Sensitive Cardiac Troponins in Arterial Hypertension. Vasc. Health Risk Manag. 2021, 17, 431–443. [Google Scholar] [CrossRef]
- Chaulin, A.M. Cardiac troponins: Current information on the main analytical characteristics of determination methods and new diagnostic possibilities. Medwave 2021, 21, e8498. [Google Scholar] [CrossRef]
- Wei, B.; Jin, J.P. Troponin T isoforms and posttranscriptional modifications: Evolution, regulation and function. Arch. Biochem. Biophys. 2011, 505, 144–154. [Google Scholar] [CrossRef]
- Jin, J.P. Evolution, Regulation, and Function of N-terminal Variable Region of Troponin T: Modulation of Muscle Contractility and Beyond. Int. Rev. Cell Mol. Biol. 2016, 321, 1–28. [Google Scholar] [CrossRef]
- Chaulin, A. Cardiac Troponins: Contemporary Biological Data and New Methods of Determination. Vasc. Health Risk Manag. 2021, 17, 299–316. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhang, F.; Zhang, C.; Zheng, L.R.; Yang, J. The Biomarkers for Acute Myocardial Infarction and Heart Failure. Biomed. Res. Int. 2020, 2020, 2018035. [Google Scholar] [CrossRef]
- Smith, J.N.; Negrelli, J.M.; Manek, M.B.; Hawes, E.M.; Viera, A.J. Diagnosis and management of acute coronary syndrome: An evidence-based update. J. Am. Board Fam. Med. 2015, 28, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.A. Acute coronary syndrome: Optimising management through risk assessment. Clin. Med. 2013, 13, 602–606. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Grigorieva, Y.u.V.; Pavlova, T.V.; Duplyakov, D.V. Diagnostic significance of complete blood count in cardiovascular patients; Samara State Medical University. Russ. J. Cardiol. 2020, 25, 3923. [Google Scholar] [CrossRef]
- Makki, N.; Brennan, T.M.; Girotra, S. Acute coronary syndrome. J. Intensive Care Med. 2015, 30, 186–200. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; Writing Group on the Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction; Thygesen, K.; Alpert, J.S.; White, H.D.; et al. Third universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2012, 60, 1581–1598, Erratum in Circulation 2018, 138, e652. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D.; Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). Circulation 2018, 138, e618–e651, Erratum in Circulation 2018, 138, e652. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakov, D.V. MicroRNAs in atrial fibrillation: Pathophysiological aspects and potential biomarkers. Int. J. Biomed. 2020, 10, 198–205. [Google Scholar] [CrossRef]
- Eckner, D.; Pauschinger, M.; Ademaj, F.; Martinovic, K. Klinische Bedeutung der 4. Universellen Definition des Myokardinfarkts [Clinical implications of the fourth universal definition of myocardial infarction]. Herz 2020, 45, 520–527. (In German) [Google Scholar] [CrossRef]
- Apple, F.S.; Sandoval, Y.; Jaffe, A.S.; Ordonez-Llanos, J.; IFCC Task Force on Clinical Applications of Cardiac Bio-Markers. Cardiac Troponin Assays: Guide to Understanding Analytical Characteristics and Their Impact on Clinical Care. Clin. Chem. 2017, 63, 73–81. [Google Scholar] [CrossRef]
- Aakre, K.M.; Saeed, N.; Wu, A.H.B.; Kavsak, P.A. Analytical performance of cardiac troponin assays—Current status and future needs. Clin. Chim. Acta 2020, 509, 149–155. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakov, D.V. Arrhythmogenic effects of doxorubicin. Complex Issues Cardiovasc. Dis. 2020, 9, 69–80. (In Russian) [Google Scholar] [CrossRef]
- Chaulin, A.M.; Abashina, O.E.; Duplyakov, D.V. Pathophysiological mechanisms of cardiotoxicity in chemotherapeutic agents. Russ. Open Med. J. 2020, 9, e0305. [Google Scholar] [CrossRef]
- Chuang, A.M.; Nguyen, M.T.; Kung, W.M.; Lehman, S.; Chew, D.P. High-sensitivity troponin in chronic kidney disease: Considerations in myocardial infarction and beyond. Rev. Cardiovasc. Med. 2020, 21, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Chaulin, A.M.; Duplyakov, D.V. Increased natriuretic peptides not associated with heart failure. Russ. J. Cardiol. 2020, 25, 4140. [Google Scholar] [CrossRef]
- Stavroulakis, G.A.; George, K.P. Exercise-induced release of troponin. Clin. Cardiol. 2020, 43, 872–881. [Google Scholar] [CrossRef]
- Chaulin, A.M. Elevation Mechanisms and Diagnostic Consideration of Cardiac Troponins under Conditions Not Associated with Myocardial Infarction. Part 1. Life 2021, 11, 914. [Google Scholar] [CrossRef]
- Chaulin, A.M. Elevation Mechanisms and Diagnostic Consideration of Cardiac Troponins under Conditions Not Associated with Myocardial Infarction. Part 2. Life 2021, 11, 1175. [Google Scholar] [CrossRef]
- Chaulin, A.M. False-Positive Causes in Serum Cardiac Troponin Levels. J. Clin. Med. Res. 2022, 14, 80–87. [Google Scholar] [CrossRef]
- Lindner, G.; Pfortmueller, C.A.; Braun, C.T.; Exadaktylos, A.K. Non-acute myocardial infarction-related causes of elevated high-sensitive troponin T in the emergency room: A cross-sectional analysis. Intern. Emerg. Med. 2014, 9, 335–339. [Google Scholar] [CrossRef]
- Wu, W.; Li, D.X.; Wang, Q.; Xu, Y.; Cui, Y.J. Relationship between high-sensitivity cardiac troponin T and the prognosis of elderly inpatients with non-acute coronary syndromes. Clin. Interv. Aging 2018, 13, 1091–1098. [Google Scholar] [CrossRef]
- Askin, L.; Tanriverdi, O.; Turkmen, S. Clinical importance of high- sensitivity troponin T in patients without coronary artery disease. North. Clin. Istanb. 2020, 7, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.J.; Wu, Q.Y.; Deng, Y.; Li, X.; Wei, X.D.; Tang, C.J.; Jia, J.F. Association Between High-Sensitivity Troponin T on Admission and Organ Dysfunction During Hospitalization in Patients Aged 80 Years and Older with Hip Fracture: A Single-Centered Prospective Cohort Study. Clin. Interv. Aging 2021, 16, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2021, 42, 1289–1367, Erratum in Eur. Heart J. 2021, 42, 1908; Erratum in Eur. Heart J. 2021, 42, 1925; Erratum in Eur. Heart J. 2021, 42, 2298. [Google Scholar] [CrossRef] [PubMed]
- Odsæter, I.H.; Grenne, B.; Hov, G.G.; Laugsand, L.E.; Wiseth, R.; Mikkelsen, G. Establishing the 99th percentile of a novel assay for high-sensitivity troponin I in a healthy blood donor population. Clin. Chem. Lab. Med. 2020, 58, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, K.; Ijaz, A.; Salahuddin, M.; Alam, A. Determination of high sensitive cardiac troponin I 99th percentile upper reference limits in a healthy Pakistani population. Pak. J. Med. Sci. 2020, 36, 1303–1307. [Google Scholar] [CrossRef]
- Koerbin, G.; Tate, J.; Potter, J.M.; Cavanaugh, J.; Glasgow, N.; Hickman, P.E. Characterisation of a highly sensitive troponin I assay and its application to a cardio-healthy population. Clin. Chem. Lab. Med. 2012, 50, 871–878. [Google Scholar] [CrossRef]
- Abe, N.; Tomita, K.; Teshima, M.; Kuwabara, M.; Sugawa, S.; Hinata, N.; Matsuura, M.; Fujiwara, M.; Takaya, K.; Hiyoshi, T.; et al. Distribution of cardiac troponin I in the Japanese general population and factors influencing its concentrations. J. Clin. Lab. Anal. 2018, 32, e22294. [Google Scholar] [CrossRef]
- Chen, J.Y.; Lee, S.Y.; Li, Y.H.; Lin, C.Y.; Shieh, M.D.; Ciou, D.S. Urine High-Sensitivity Troponin I Predict Incident Cardiovascular Events in Patients with Diabetes Mellitus. J. Clin. Med. 2020, 9, 3917. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Karslyan, L.S.; Bazyuk, E.V.; Nurbaltaeva, D.A.; Duplyakov, D.V. Clinical and Diagnostic Value of Cardiac Markers in Human Biological Fluids. Kardiologiia 2019, 59, 66–75. (In Russian) [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakova, P.D.; Bikbaeva, G.R.; Tukhbatova, A.A.; Grigorieva, E.V.; Duplyakov, D.V. Concentration of high-sensitivity cardiac troponin I in the oral fluid in patients with acute myocardial infarction: A pilot study. Russ. J. Cardiol. 2020, 25, 3814. [Google Scholar] [CrossRef]
- Mirzaii-Dizgah, I.; Riahi, E. Salivary high-sensitivity cardiac troponin T levels in patients with acute myocardial infarction. Oral Dis. 2013, 19, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Osuna, A.; Gaze, D.; Grau-Agramunt, M.; Morris, T.; Telha, C.; Bartolome, A.; Bishop, J.J.; Monsalve, L.; Livingston, R.; Estis, J.; et al. Ultrasensitive quantification of cardiac troponin I by a Single Molecule Counting method: Analytical validation and biological features. Clin. Chim. Acta 2018, 486, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Giannitsis, E.; Mueller-Hennessen, M.; Zeller, T.; Schuebler, A.; Aurich, M.; Biener, M.; Vafaie, M.; Stoyanov, K.M.; Ochs, M.; Riffel, J.; et al. Gender-specific reference values for high-sensitivity cardiac troponin T and I in well-phenotyped healthy individuals and validity of high-sensitivity assay designation. Clin. Biochem. 2020, 78, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Rocco, E.; La Rosa, G.; Liuzzo, G.; Biasucci, L.M. High-sensitivity cardiac troponin assays and acute coronary syndrome: A matter of sex? J. Cardiovasc. Med. 2019, 20, 504–509. [Google Scholar] [CrossRef]
- Romiti, G.F.; Cangemi, R.; Toriello, F.; Ruscio, E.; Sciomer, S.; Moscucci, F.; Vincenti, M.; Crescioli, C.; Proietti, M.; Basili, S.; et al. Sex-Specific Cut-Offs for High-Sensitivity Cardiac Troponin: Is Less More? Cardiovasc. Ther. 2019, 2019, 9546931. [Google Scholar] [CrossRef]
- Monneret, D.; Gellerstedt, M.; Bonnefont-Rousselot, D. Determination of age- and sex-specific 99th percentiles for high-sensitive troponin T from patients: An analytical imprecision- and partitioning-based approach. Clin. Chem. Lab. Med. 2018, 56, 818–829. [Google Scholar] [CrossRef]
- Bohn, M.K.; Higgins, V.; Kavsak, P.; Hoffman, B.; Adeli, K. High-Sensitivity Generation 5 Cardiac Troponin T Sex- and Age-Specific 99th Percentiles in the CALIPER Cohort of Healthy Children and Adolescents. Clin. Chem. 2019, 65, 589–591. [Google Scholar] [CrossRef]
- Boeddinghaus, J.; Nestelberger, T.; Twerenbold, R.; Neumann, J.T.; Lindahl, B.; Giannitsis, E.; Sörensen, N.A.; Badertscher, P.; Jann, J.E.; Wussler, D.; et al. Impact of age on the performance of the ESC 0/1h-algorithms for early diagnosis of myocardial infarction. Eur. Heart J. 2018, 39, 3780–3794. [Google Scholar] [CrossRef]
- Gore, M.O.; Seliger, S.L.; Defilippi, C.R.; Nambi, V.; Christenson, R.H.; Hashim, I.A.; Hoogeveen, R.C.; Ayers, C.R.; Sun, W.; McGuire, D.K.; et al. Age- and sex-dependent upper reference limits for the high-sensitivity cardiac troponin T assay. J. Am. Coll. Cardiol. 2014, 63, 1441–1448. [Google Scholar] [CrossRef]
- Fournier, S.; Iten, L.; Marques-Vidal, P.; Boulat, O.; Bardy, D.; Beggah, A.; Calderara, R.; Morawiec, B.; Lauriers, N.; Monney, P.; et al. Circadian rhythm of blood cardiac troponin T concentration. Clin. Res. Cardiol. 2017, 106, 1026–1032. [Google Scholar] [CrossRef]
- Klinkenberg, L.J.; van Dijk, J.W.; Tan, F.E.; van Loon, L.J.; van Dieijen-Visser, M.P.; Meex, S.J. Circulating cardiac troponin T exhibits a diurnal rhythm. J. Am. Coll. Cardiol. 2014, 63, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Chaulin, A.M.; Duplyakov, D.V. High-sensitivity cardiac troponins: Circadian rhythms. Cardiovasc. Ther. Prev. 2021, 20, 2639. (In Russian) [Google Scholar] [CrossRef]
- Chaulin, A.M.; Abashina, O.E.; Duplyakov, D.V. High-sensitivity cardiac troponins: Detection and central analytical characteristics. Cardiovasc. Ther. Prev. 2021, 20, 2590. (In Russian) [Google Scholar] [CrossRef]
- Eggers, K.M.; Lindahl, B. Impact of Sex on Cardiac Troponin Concentrations-A Critical Appraisal. Clin. Chem. 2017, 63, 1457–1464. [Google Scholar] [CrossRef]
- Sedighi, S.M.; Prud’Homme, P.; Ghachem, A.; Lepage, S.; Nguyen, M.; Fulop, T.; Khalil, A. Increased level of high-sensitivity cardiac Troponin T in a geriatric population is determined by comorbidities compared to age. Int. J. Cardiol. Heart Vasc. 2019, 22, 187–191. [Google Scholar] [CrossRef]
- Hickman, P.E.; Abhayaratna, W.P.; Potter, J.M.; Koerbin, G. Age-related differences in hs-cTnI concentration in healthy adults. Clin. Biochem. 2019, 69, 26–29. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakova, P.D.; Duplyakov, D.V. Circadian rhythms of cardiac troponins: Mechanisms and clinical significance. Russ. J. Cardiol. 2020, 25, 4061. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakov, D.V. On the potential effect of circadian rhythms of cardiac troponins on the diagnosis of acute myocardial infarction. Signa Vitae. 2021, 17, 79–84. [Google Scholar] [CrossRef]
- Vogiatzis, I. Circadian rhythm of cardiac troponins. Does it really exist? Int. J. Cardiol. 2018, 270, 72–73. [Google Scholar] [CrossRef]
- Zaninotto, M.; Padoan, A.; Mion, M.M.; Marinova, M.; Plebani, M. Short-term biological variation and diurnal rhythm of cardiac troponin I (Access hs-TnI) in healthy subjects. Clin. Chim. Acta 2020, 504, 163–167. [Google Scholar] [CrossRef]
- Wildi, K.; Singeisen, H.; Twerenbold, R.; Badertscher, P.; Wussler, D.; Klinkenberg, L.J.J.; Meex, S.J.R.; Nestelberger, T.; Boeddinghaus, J.; Miró, Ò.; et al. Circadian rhythm of cardiac troponin I and its clinical impact on the diagnostic accuracy for acute myocardial infarction. Int. J. Cardiol. 2018, 270, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.R.; Bunk, D.M.; Christenson, R.H.; Katrukha, A.; Noble, J.E.; Porter, R.A.; Schimmel, H.; Wang, L.; Panteghini, M.; IFCC Working Group on Standardization of Troponin I. Standardisation of cardiac troponin I measurement: Past and present. Pathology 2010, 42, 402–408. [Google Scholar] [CrossRef]
- Panteghini, M.; Bunk, D.M.; Christenson, R.H.; Katrukha, A.; Porter, R.A.; Schimmel, H.; Wang, L.; Tate, J.R.; IFCC Working Group on Standardization of Troponin I. Standardization of troponin I measurements: An update. Clin. Chem. Lab. Med. 2008, 46, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Jarolim, P. High sensitivity cardiac troponin assays in the clinical laboratories. Clin. Chem. Lab. Med. 2015, 53, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Katrukha, I.A.; Kogan, A.E.; Vylegzhanina, A.V.; Kharitonov, A.V.; Tamm, N.N.; Filatov, V.L.; Bereznikova, A.V.; Koshkina, E.V.; Katrukha, A.G. Full-Size Cardiac Troponin I and Its Proteolytic Fragments in Blood of Patients with Acute Myocardial Infarction: Antibody Selection for Assay Development. Clin. Chem. 2018, 64, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Streng, A.S.; de Boer, D.; van der Velden, J.; van Dieijen-Visser, M.P.; Wodzig, W.K. Posttranslational modifications of cardiac troponin T: An overview. J. Mol. Cell Cardiol. 2013, 63, 47–56. [Google Scholar] [CrossRef]
- Mirzaii-Dizgah, I.; Riahi, E. Salivary troponin I as an indicator of myocardial infarction. Indian J. Med. Res. 2013, 138, 861–865. [Google Scholar]
- Bahbah, E.I.; Noehammer, C.; Pulverer, W.; Jung, M.; Weinhaeusel, A. Salivary biomarkers in cardiovascular disease: An insight into the current evidence. FEBS J. 2021, 288, 6392–6405. [Google Scholar] [CrossRef]
- Abdul Rehman, S.; Khurshid, Z.; Hussain Niazi, F.; Naseem, M.; Al Waddani, H.; Sahibzada, H.A.; Sannam Khan, R. Role of Salivary Biomarkers in Detection of Cardiovascular Diseases (CVD). Proteomes 2017, 5, 21. [Google Scholar] [CrossRef]
- Klichowska-Palonka, M.; Załęska-Chromińska, K.; Bachanek, T. Possibility of using saliva as a diagnostic test material in cardiovascular diseases. Wiad. Lek. 2015, 68 Pt 2, 354–357. (In Polish) [Google Scholar]
- Pervan, P.; Svaguša, T.; Prkačin, I.; Savuk, A.; Bakos, M.; Perkov, S. Urine high sensitive Troponin I measuring in patients with hypertension. Signa Vitae 2017, 13, 62–64. [Google Scholar] [CrossRef]
- Mishra, V.; Patil, R.; Khanna, V.; Tripathi, A.; Singh, V.; Pandey, S.; Chaurasia, A. Evaluation of Salivary Cardiac Troponin-I as Potential Marker for Detection of Acute Myocardial Infarction. J. Clin. Diagn. Res. 2018, 12, 44–47. [Google Scholar] [CrossRef]
- Chaulin, A.M. Phosphorylation and Fragmentation of the Cardiac Troponin T: Mechanisms, Role in Pathophysiology and Laboratory Diagnosis. Int. J. Biomed. 2021, 11, 250–259. [Google Scholar] [CrossRef]
- Ziebig, R.; Lun, A.; Hocher, B.; Priem, F.; Altermann, C.; Asmus, G.; Kern, H.; Krause, R.; Lorenz, B.; Möbes, R.; et al. Renal elimination of troponin T and troponin I. Clin. Chem. 2003, 49, 1191–1193. [Google Scholar] [CrossRef]
- Ellis, K.; Dreisbach, A.W.; Lertora, J.L. Plasma elimination of cardiac troponin I in end-stage renal disease. South Med. J. 2001, 94, 993–996. [Google Scholar] [CrossRef]
- Hernández-Romero, D.; Valverde-Vázquez, M.D.R.; Hernández Del Rincón, J.P.; Noguera-Velasco, J.A.; Pérez-Cárceles, M.D.; Osuna, E. Diagnostic Application of Postmortem Cardiac Troponin I Pericardial Fluid/Serum Ratio in Sudden Cardiac Death. Diagnostics 2021, 11, 614. [Google Scholar] [CrossRef]
- Zhu, B.L.; Ishikawa, T.; Michiue, T.; Li, D.R.; Zhao, D.; Bessho, Y.; Kamikodai, Y.; Tsuda, K.; Okazaki, S.; Maeda, H. Postmortem cardiac troponin I and creatine kinase MB levels in the blood and pericardial fluid as markers of myocardial damage in medicolegal autopsy. Leg. Med. 2007, 9, 241–250. [Google Scholar] [CrossRef]
- González-Herrera, L.; Valenzuela, A.; Ramos, V.; Blázquez, A.; Villanueva, E. Cardiac troponin T determination by a highly sensitive assay in postmortem serum and pericardial fluid. Forensic Sci. Med. Pathol. 2016, 12, 181–188. [Google Scholar] [CrossRef]
- Maeda, H.; Michiue, T.; Zhu, B.L.; Ishikawa, T.; Quan, L. Analysis of cardiac troponins and creatine kinase MB in cerebrospinal fluid in medicolegal autopsy cases. Leg. Med. 2009, 11 (Suppl. 1), S266–S268. [Google Scholar] [CrossRef]
- Wang, Q.; Michiue, T.; Ishikawa, T.; Zhu, B.L.; Maeda, H. Combined analyses of creatine kinase MB, cardiac troponin I and myoglobin in pericardial and cerebrospinal fluids to investigate myocardial and skeletal muscle injury in medicolegal autopsy cases. Leg. Med. 2011, 13, 226–232. [Google Scholar] [CrossRef]
- Chen, J.H.; Inamori-Kawamoto, O.; Michiue, T.; Ikeda, S.; Ishikawa, T.; Maeda, H. Cardiac biomarkers in blood, and pericardial and cerebrospinal fluids of forensic autopsy cases: A reassessment with special regard to postmortem interval. Leg. Med. 2015, 17, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Sessa, F.; Esposito, M.; Messina, G.; Di Mizio, G.; Di Nunno, N.; Salerno, M. Sudden Death in Adults: A Practical Flow Chart for Pathologist Guidance. Healthcare 2021, 9, 870. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, V.; Loukovaara, M. Amniotic fluid cardiac troponin T in pathological pregnancies with evidence of chronic fetal hypoxia. Croat. Med. J. 2005, 46, 801–807. [Google Scholar] [PubMed]
- Yoshida, M.; Matsuda, H.; Yoshinaga, Y.; Asai, K.; Kawashima, A.; Sei, K.; Horii, M.; Nakanishi, A.; Soyama, H.; Furuya, K. Analysis about the influence on the fetus infected with parvovirus B19 using amniotic erythropoietin and troponin-T. Arch. Gynecol. Obstet. 2013, 288, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Blohm, M.E.; Arndt, F.; Fröschle, G.M.; Langenbach, N.; Sandig, J.; Vettorazzi, E.; Mir, T.S.; Hecher, K.; Weil, J.; Kozlik-Feldmann, R.; et al. Cardiovascular Biomarkers in Amniotic Fluid, Umbilical Arterial Blood, Umbilical Venous Blood, and Maternal Blood at Delivery, and Their Reference Values for Full-Term, Singleton, Cesarean Deliveries. Front. Pediatr. 2019, 7, 271. [Google Scholar] [CrossRef] [PubMed]
- Van Mieghem, T.; Doné, E.; Gucciardo, L.; Klaritsch, P.; Allegaert, K.; Van Bree, R.; Lewi, L.; Deprest, J. Amniotic fluid markers of fetal cardiac dysfunction in twin-to-twin transfusion syndrome. Am. J. Obstet. Gynecol. 2010, 202, 48.e1–48.e7. [Google Scholar] [CrossRef] [PubMed]
- Chaulin, A.M.; Duplyakov, D.V. Cardiac troponins: Current data on the diagnostic value and analytical characteristics of new determination methods. Cor. Vasa. 2021, 63, 486–493. [Google Scholar] [CrossRef]
- Kavsak, P.A. Should detectable cardiac troponin concentrations in a healthy population be the only criterion for classifying high-sensitivity cardiac troponin assays? Clin. Biochem. 2018, 56, 1–3. [Google Scholar] [CrossRef]
- Ji, M.; Moon, H.W.; Hur, M.; Yun, Y.M. Determination of high-sensitivity cardiac troponin I 99th percentile upper reference limits in a healthy Korean population. Clin. Biochem. 2016, 49, 756–761. [Google Scholar] [CrossRef]
- Tjora, S.; Hall, T.S.; Larstorp, A.C.; Hallen, J.; Atar, D. Increases in Circulating Cardiac Troponin Are Not Always Associated with Myocardial Cell Death. Clin. Lab. 2018, 64, 1961–1962. [Google Scholar] [CrossRef]
- Jaffe, A.S.; Wu, A.H. Troponin release--reversible or irreversible injury? Should we care? Clin. Chem. 2012, 58, 148–150, Erratum in Clin. Chem. 2012, 58, 796. [Google Scholar] [CrossRef] [PubMed]
- Mair, J.; Lindahl, B.; Hammarsten, O.; Müller, C.; Giannitsis, E.; Huber, K.; Möckel, M.; Plebani, M.; Thygesen, K.; Jaffe, A.S. How is cardiac troponin released from injured myocardium? Eur. Heart J. Acute Cardiovasc. Care 2018, 7, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Hammarsten, O.; Mair, J.; Möckel, M.; Lindahl, B.; Jaffe, A.S. Possible mechanisms behind cardiac troponin elevations. Biomarkers 2018, 23, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Chaulin, A.M.; Duplyakov, D.V. Mechanisms of increase and diagnostic role of highly sensitive troponins in arterial hypertension. Ann Cardiol. Angeiol. 2021; in press. (In French). [Google Scholar] [CrossRef]
- Gumprecht, J.; Domek, M.; Lip, G.Y.H.; Shantsila, A. Invited review: Hypertension and atrial fibrillation: Epidemiology, pathophysiology, and implications for management. J. Hum. Hypertens. 2019, 33, 824–836. [Google Scholar] [CrossRef]
- Liao, X.D.; Wang, X.H.; Jin, H.J.; Chen, L.Y.; Chen, Q. Mechanical stretch induces mitochondria-dependent apoptosis in neonatal rat cardiomyocytes and G2/M accumulation in cardiac fibroblasts. Cell Res. 2004, 14, 16–26. [Google Scholar] [CrossRef]
- Cheng, W.P.; Wang, B.W.; Lo, H.M.; Shyu, K.G. Mechanical Stretch Induces Apoptosis Regulator TRB3 in Cultured Cardiomyocytes and Volume-Overloaded Heart. PLoS ONE 2015, 10, e0123235. [Google Scholar] [CrossRef]
- Jiang, S.; Huo, D.; Wang, X.; Zhao, H.; Tan, J.; Zeng, Q.; O’Rourke, S.T.; Sun, C. β-adrenergic Receptor-stimulated Cardiac Myocyte Apoptosis: Role of Cytochrome P450 ω-hydroxylase. J. Cardiovasc. Pharmacol. 2017, 70, 94–101. [Google Scholar] [CrossRef]
- Communal, C.; Colucci, W.S. The control of cardiomyocyte apoptosis via the beta-adrenergic signaling pathways. Arch. Mal. Coeur Vaiss. 2005, 98, 236–241. [Google Scholar]
- Dalal, S.; Foster, C.R.; Das, B.C.; Singh, M.; Singh, K. Β-adrenergic receptor stimulation induces endoplasmic reticulum stress in adult cardiac myocytes: Role in apoptosis. Mol. Cell Biochem. 2012, 364, 59–70. [Google Scholar] [CrossRef]
- Chen, Q.M.; Tu, V.C. Apoptosis and heart failure: Mechanisms and therapeutic implications. Am. J. Cardiovasc. Drugs. 2002, 2, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Kunapuli, S.; Rosanio, S.; Schwarz, E.R. “How do cardiomyocytes die?” apoptosis and autophagic cell death in cardiac myocytes. J. Card. Fail. 2006, 12, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Ricchiuti, V.; Apple, F.S. RNA expression of cardiac troponin T isoforms in diseased human skeletal muscle. Clin. Chem. 1999, 45, 2129–2135, Erratum in Clin. Chem 2000, 46, 437. [Google Scholar] [CrossRef]
- Ricchiuti, V.; Voss, E.M.; Ney, A.; Odland, M.; Anderson, P.A.; Apple, F.S. Cardiac troponin T isoforms expressed in renal diseased skeletal muscle will not cause false-positive results by the second generation cardiac troponin T assay by Boehringer Mannheim. Clin. Chem. 1998, 44, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Frisén, J.; Bernard, S.; Druid, H.; Jovinge, S. Cardiomyocyte renewal in humans. Circ. Res. 2012, 110, e17–e18, author reply e19–e21. [Google Scholar] [CrossRef]
- White, H.D. Pathobiology of troponin elevations: Do elevations occur with myocardial ischemia as well as necrosis? J. Am. Coll. Cardiol. 2011, 57, 2406–2408, Erratum in J. Am. Coll. Cardiol. 2011, 58, 2356. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef]
- Lázár, E.; Sadek, H.A.; Bergmann, O. Cardiomyocyte renewal in the human heart: Insights from the fall-out. Eur. Heart J. 2017, 38, 2333–2342. [Google Scholar] [CrossRef]
- Foglia, M.J.; Poss, K.D. Building and re-building the heart by cardiomyocyte proliferation. Development 2016, 143, 729–740. [Google Scholar] [CrossRef]
- Docshin, P.M.; Karpov, A.A.; Eyvazova, S.D.; Puzanov, M.V.; Kostareva, A.A.; Galagudza, M.; Malashicheva, A.B. Activation of Cardiac Stem Cells in Myocardial Infarction. Cell Tissue Biol. 2018, 12, 175–182. [Google Scholar] [CrossRef]
- Waring, C.D.; Vicinanza, C.; Papalamprou, A.; Smith, A.J.; Purushothaman, S.; Goldspink, D.F.; Nadal-Ginard, B.; Torella, D.; Ellison, G.M. The adult heart responds to increased workload with physiologic hypertrophy, cardiac stem cell activation, and new myocyte formation. Eur. Heart J. 2014, 35, 2722–2731. [Google Scholar] [CrossRef] [PubMed]
- Rovira, M.; Borràs, D.M.; Marques, I.J.; Puig, C.; Planas, J.V. Physiological Responses to Swimming-Induced Exercise in the Adult Zebrafish Regenerating Heart. Front. Physiol. 2018, 9, 1362. [Google Scholar] [CrossRef] [PubMed]
- Schüttler, D.; Clauss, S.; Weckbach, L.T.; Brunner, S. Molecular Mechanisms of Cardiac Remodeling and Regeneration in Physical Exercise. Cells 2019, 8, 1128. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- Isomi, M.; Sadahiro, T.; Ieda, M. Progress and Challenge of Cardiac Regeneration to Treat Heart Failure. J. Cardiol. 2019, 73, 97–101. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Zhang, M.; Zhang, Y. Programmed necrosis in cardiomyocytes: Mitochondria, death receptors and beyond. Br. J. Pharmacol. 2019, 176, 4319–4339. [Google Scholar] [CrossRef]
- Lee, Y.; Gustafsson, A.B. Role of apoptosis in cardiovascular disease. Apoptosis 2009, 14, 536–548. [Google Scholar] [CrossRef]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of apoptosis by TUNEL assay. Methods Mol. Biol. 2012, 887, 41–47. [Google Scholar] [CrossRef]
- Zorc-Pleskovic, R.; Alibegović, A.; Zorc, M.; Milutinović, A.; Radovanović, N.; Petrović, D. Apoptosis of cardiomyocytes in myocarditis. Folia Biol. 2006, 52, 6–9. [Google Scholar]
- Zhang, Q.; Yu, N.; Yu, B.T. MicroRNA-298 regulates apoptosis of cardiomyocytes after myocardial infarction. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Weil, B.R.; Young, R.F.; Shen, X.; Suzuki, G.; Qu, J.; Malhotra, S.; Canty, J.M., Jr. Brief Myocardial Ischemia Produces Cardiac Troponin I Release and Focal Myocyte Apoptosis in the Absence of Pathological Infarction in Swine. JACC Basic Transl. Sci. 2017, 2, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Li, B.; Kajstura, J.; Li, P.; Wolin, M.S.; Sonnenblick, E.H.; Hintze, T.H.; Olivetti, G.; Anversa, P. Stretch-induced programmed myocyte cell death. J. Clin. Investig. 1995, 96, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Gherasim, L. Troponins in Heart Failure—A Perpetual Challenge. Maedica 2019, 14, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Aengevaeren, V.L.; Baggish, A.L.; Chung, E.H.; George, K.; Kleiven, Ø.; Mingels, A.M.A.; Ørn, S.; Shave, R.E.; Thompson, P.D.; Eijsvogels, T.M.H. Exercise-Induced Cardiac Troponin Elevations: From Underlying Mechanisms to Clinical Relevance. Circulation 2021, 144, 1955–1972. [Google Scholar] [CrossRef]
- Park, K.C.; Gaze, D.C.; Collinson, P.O.; Marber, M.S. Cardiac troponins: From myocardial infarction to chronic disease. Cardiovasc. Res. 2017, 113, 1708–1718. [Google Scholar] [CrossRef]
- Weil, B.R.; Suzuki, G.; Young, R.F.; Iyer, V.; Canty, J.M., Jr. Troponin Release and Reversible Left Ventricular Dysfunction after Transient Pressure Overload. J. Am. Coll. Cardiol. 2018, 71, 2906–2916. [Google Scholar] [CrossRef]
- Felker, G.M.; Fudim, M. Unraveling the Mystery of Troponin Elevation in Heart Failure. J. Am. Coll. Cardiol. 2018, 71, 2917–2918. [Google Scholar] [CrossRef]
- Sanchez, O.; Planquette, B.; Wermert, D.; Marié, E.; Meyer, G. Embolies pulmonaires graves [Massive pulmonary embolism]. Presse Med. 2008, 37, 1439–1446. (In French) [Google Scholar] [CrossRef]
- El-Menyar, A.; Sathian, B.; Al-Thani, H. Elevated serum cardiac troponin and mortality in acute pulmonary embolism: Systematic review and meta-analysis. Respir. Med. 2019, 157, 26–35. [Google Scholar] [CrossRef]
- Daquarti, G.; March Vecchio, N.; Mitrione, C.S.; Furmento, J.; Ametrano, M.C.; Dominguez Pace, M.P.; Costabel, J.P. High-sensitivity troponin and right ventricular function in acute pulmonary embolism. Am. J. Emerg. Med. 2016, 34, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Communal, C.; Sawyer, D.B.; Colucci, W.S. Adrenergic regulation of myocardial apoptosis. Cardiovasc. Res. 2000, 45, 713–719. [Google Scholar] [CrossRef]
- Colucci, W.S.; Sawyer, D.B.; Singh, K.; Communal, C. Adrenergic overload and apoptosis in heart failure: Implications for therapy. J. Card. Fail. 2000, 6 (Suppl. 1), 1–7. [Google Scholar] [PubMed]
- Xiao, R.P.; Tomhave, E.D.; Wang, D.J.; Ji, X.; Boluyt, M.O.; Cheng, H.; Lakatta, E.G.; Koch, W.J. Age-associated reductions in cardiac beta1- and beta2-adrenergic responses without changes in inhibitory G proteins or receptor kinases. J. Clin. Investig. 1998, 101, 1273–1282. [Google Scholar] [CrossRef]
- Mougenot, N.; Mika, D.; Czibik, G.; Marcos, E.; Abid, S.; Houssaini, A.; Vallin, B.; Guellich, A.; Mehel, H.; Sawaki, D.; et al. Cardiac adenylyl cyclase overexpression precipitates and aggravates age-related myocardial dysfunction. Cardiovasc. Res. 2019, 115, 1778–1790. [Google Scholar] [CrossRef]
- de Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling during Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef]
- Schwartz, P.; Piper, H.M.; Spahr, R.; Spieckermann, P.G. Ultrastructure of cultured adult myocardial cells during anoxia and reoxygenation. Am. J. Pathol. 1984, 115, 349–361. [Google Scholar]
- Siegmund, B.; Koop, A.; Klietz, T.; Schwartz, P.; Piper, H.M. Sarcolemmal integrity and metabolic competence of cardiomyocytes under anoxia-reoxygenation. Am. J. Physiol. 1990, 258 Pt 2, H285–H291. [Google Scholar] [CrossRef]
- Piper, H.M.; Schwartz, P.; Spahr, R.; Hütter, J.F.; Spieckermann, P.G. Absence of reoxygenation damage in isolated heart cells after anoxic injury. Pflug. Arch. 1984, 401, 71–76. [Google Scholar] [CrossRef]
- Chaulin, A.M. Updated information about methods of identification and diagnostic opportunities of cardiac troponins. Riv. Ital. Della Med. Lab. 2021, 17, 154–164. [Google Scholar] [CrossRef]
- Aakre, K.M.; Omland, T. Physical activity, exercise and cardiac troponins: Clinical implications. Prog. Cardiovasc. Dis. 2019, 62, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Sheyin, O.; Davies, O.; Duan, W.; Perez, X. The prognostic significance of troponin elevation in patients with sepsis: A meta-analysis. Heart Lung 2015, 44, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Gibler, W.B.; Gibler, C.D.; Weinshenker, E.; Abbottsmith, C.; Hedges, J.R.; Barsan, W.G.; Sperling, M.; Chen, I.W.; Embry, S.; Kereiakes, D. Myoglobin as an early indicator of acute myocardial infarction. Ann. Emerg. Med. 1987, 16, 851–856. [Google Scholar] [CrossRef]
- Bhayana, V.; Henderson, A.R. Biochemical markers of myocardial damage. Clin. Biochem. 1995, 28, 1–29. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, Y.; Zhang, L.; Xu, W.; Zhou, X. Diagnostic and prognostic value of biomarkers in acute myocardial infarction. Postgrad. Med. J. 2019, 95, 210–216. [Google Scholar] [CrossRef]
- McDonough, J.L.; Arrell, D.K.; Van Eyk, J.E. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ. Res. 1999, 84, 9–20. [Google Scholar] [CrossRef]
- Feng, J.; Schaus, B.J.; Fallavollita, J.A.; Lee, T.C.; Canty, J.M., Jr. Preload induces troponin I degradation independently of myocardial ischemia. Circulation 2001, 103, 2035–2037. [Google Scholar] [CrossRef]
- Gao, C.Q.; Sawicki, G.; Suarez-Pinzon, W.L.; Csont, T.; Wozniak, M.; Ferdinandy, P.; Schulz, R. Matrix metalloproteinase-2 mediates cytokine-induced myocardial contractile dysfunction. Cardiovasc. Res. 2003, 57, 426–433. [Google Scholar] [CrossRef]
- Lin, N.N.; Cheng, C.C.; Lee, Y.F.; Fu, Y.C.; Chen, J.S.; Ho, S.P.; Chiu, Y.T. Early activation of myocardial matrix metalloproteinases and degradation of cardiac troponin I after experimental subarachnoid hemorrhage. J. Surg. Res. 2013, 179, e41–e48. [Google Scholar] [CrossRef]
- Parente, J.M.; Blascke de Mello, M.M.; Silva, P.H.L.D.; Omoto, A.C.M.; Pernomian, L.; Oliveira, I.S.; Mahmud, Z.; Fazan, R., Jr.; Arantes, E.C.; Schulz, R.; et al. MMP inhibition attenuates hypertensive eccentric cardiac hypertrophy and dysfunction by preserving troponin I and dystrophin. Biochem. Pharmacol. 2021, 193, 114744. [Google Scholar] [CrossRef]
- Streng, A.S.; de Boer, D.; van Doorn, W.P.; Kocken, J.M.; Bekers, O.; Wodzig, W.K. Cardiac troponin T degradation in serum is catalysed by human thrombin. Biochem. Biophys. Res. Commun. 2016, 481, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Katrukha, I.A.; Kogan, A.E.; Vylegzhanina, A.V.; Serebryakova, M.V.; Koshkina, E.V.; Bereznikova, A.V.; Katrukha, A.G. Thrombin-Mediated Degradation of Human Cardiac Troponin T. Clin. Chem. 2017, 63, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S. Cardiac Troponins: Molecules of Many Surprises. Clin. Chem. 2017, 63, 1059–1060. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Date, T.; Ikegami, M.; Hongo, K.; Fujisaki, M.; Katoh, D.; Yoshino, T.; Anzawa, R.; Nagoshi, T.; Yamashita, S.; et al. An immunohistochemical analysis of tissue thrombin expression in the human atria. PLoS ONE 2013, 8, e65817. [Google Scholar] [CrossRef]
- Ito, K.; Hongo, K.; Date, T.; Ikegami, M.; Hano, H.; Owada, M.; Morimoto, S.; Kashiwagi, Y.; Katoh, D.; Yoshino, T.; et al. Tissue thrombin is associated with the pathogenesis of dilated cardiomyopathy. Int. J. Cardiol. 2017, 228, 821–827. [Google Scholar] [CrossRef]
- Matsukura, U.; Okitani, A.; Nishimuro, T.; Kato, H. Mode of degradation of myofibrillar proteins by an endogenous protease, cathepsin L. Biochim. Biophys. Acta 1981, 662, 41–47. [Google Scholar] [CrossRef]
- Peng, K.; Liu, H.; Yan, B.; Meng, X.W.; Song, S.Y.; Ji, F.H.; Xia, Z. Inhibition of cathepsin S attenuates myocardial ischemia/reperfusion injury by suppressing inflammation and apoptosis. J. Cell Physiol. 2021, 236, 1309–1320. [Google Scholar] [CrossRef]
- Hickman, P.E.; Potter, J.M.; Aroney, C.; Koerbin, G.; Southcott, E.; Wu, A.H.; Roberts, M.S. Cardiac troponin may be released by ischemia alone, without necrosis. Clin. Chim. Acta 2010, 411, 318–323. [Google Scholar] [CrossRef]
- Hessel, M.H.; Atsma, D.E.; van der Valk, E.J.; Bax, W.H.; Schalij, M.J.; van der Laarse, A. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Pflug. Arch. 2008, 455, 979–986. [Google Scholar] [CrossRef]
- Ross, R.S.; Borg, T.K. Integrins and the myocardium. Circ. Res. 2001, 88, 1112–1119. [Google Scholar] [CrossRef]
- Khabbaz, K.R.; Feng, J.; Boodhwani, M.; Clements, R.T.; Bianchi, C.; Sellke, F.W. Nonischemic myocardial acidosis adversely affects microvascular and myocardial function and triggers apoptosis during cardioplegia. J. Thorac. Cardiovasc. Surg. 2008, 135, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Thatte, H.S.; Rhee, J.H.; Zagarins, S.E.; Treanor, P.R.; Birjiniuk, V.; Crittenden, M.D.; Khuri, S.F. Acidosis-induced apoptosis in human and porcine heart. Ann. Thorac. Surg. 2004, 77, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Frazier, D.P.; Thompson, J.W.; Haliko, S.; Li, H.; Wasserlauf, B.J.; Spiga, M.G.; Bishopric, N.H.; Webster, K.A. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J. Exp. Biol. 2004, 207 Pt 18, 3189–3200. [Google Scholar] [CrossRef]
- Wasfy, M.M.; Hutter, A.M.; Weiner, R.B. Sudden Cardiac Death in Athletes. Methodist Debakey Cardiovasc. J. 2016, 12, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Schlesinger, S.; Hamer, M.; Norat, T.; Riboli, E. Physical activity and the risk of sudden cardiac death: A systematic review and meta-analysis of prospective studies. BMC Cardiovasc. Disord. 2020, 20, 318. [Google Scholar] [CrossRef] [PubMed]
- DeFroda, S.F.; McDonald, C.; Myers, C.; Cruz, A.I.; Owens, B.D.; Daniels, A.H. Sudden Cardiac Death in the Adolescent Athlete: History, Diagnosis, and Prevention. Am. J. Med. 2019, 132, 1374–1380. [Google Scholar] [CrossRef]
- Sollazzo, F.; Palmieri, V.; Gervasi, S.F.; Cuccaro, F.; Modica, G.; Narducci, M.L.; Pelargonio, G.; Zeppilli, P.; Bianco, M. Sudden Cardiac Death in Athletes in Italy during 2019: Internet-Based Epidemiological Research. Medicina 2021, 57, 61. [Google Scholar] [CrossRef]
- Klinkenberg, L.J.; Luyten, P.; van der Linden, N.; Urgel, K.; Snijders, D.P.; Knackstedt, C.; Dennert, R.; Kietselaer, B.L.; Mingels, A.M.; Cardinaels, E.P.; et al. Cardiac Troponin T and I Release After a 30-km Run. Am. J. Cardiol. 2016, 118, 281–287. [Google Scholar] [CrossRef]
- Martínez-Navarro, I.; Sánchez-Gómez, J.; Sanmiguel, D.; Collado, E.; Hernando, B.; Panizo, N.; Hernando, C. Immediate and 24-h post-marathon cardiac troponin T is associated with relative exercise intensity. Eur. J. Appl. Physiol. 2020, 120, 1723–1731. [Google Scholar] [CrossRef]
- Marshall, L.; Lee, K.K.; Stewart, S.D.; Wild, A.; Fujisawa, T.; Ferry, A.V.; Stables, C.L.; Lithgow, H.; Chapman, A.R.; Anand, A.; et al. Effect of Exercise Intensity and Duration on Cardiac Troponin Release. Circulation 2020, 141, 83–85. [Google Scholar] [CrossRef]
- O’Hanlon, R.; Wilson, M.; Wage, R.; Smith, G.; Alpendurada, F.D.; Wong, J.; Dahl, A.; Oxborough, D.; Godfrey, R.; Sharma, S.; et al. Troponin release following endurance exercise: Is inflammation the cause? A cardiovascular magnetic resonance study. J. Cardiovasc. Magn. Reson. 2010, 12, 38. [Google Scholar] [CrossRef] [PubMed]
- Lazzarino, A.I.; Hamer, M.; Gaze, D.; Collinson, P.; Steptoe, A. The association between cortisol response to mental stress and high-sensitivity cardiac troponin T plasma concentration in healthy adults. J. Am. Coll. Cardiol. 2013, 62, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Eggers, K.M. Mental stress and cardiac troponin: Keep calm and carry on? J. Am. Coll. Cardiol. 2013, 62, 1702–1703. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yamaji, M.; Tsutamoto, T.; Kawahara, C.; Nishiyama, K.; Yamamoto, T.; Fujii, M.; Horie, M. Serum cortisol as a useful predictor of cardiac events in patients with chronic heart failure: The impact of oxidative stress. Circ. Heart Fail. 2009, 2, 608–615. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iwaszczuk, P.; Łosiak, W.; Szczeklik, W.; Musiałek, P. Patient periprocedural stress in cardiovascular medicine: Friend or foe? Postępy Kardiol. Interwencyjnej 2021, 17, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Bakay, M.; Zhao, P.; Chen, J.; Hoffman, E.P. A web-accessible complete transcriptome of normal human and DMD muscle. Neuromuscul. Disord. 2002, 12 (Suppl. 1), S125–S141. [Google Scholar] [CrossRef]
- Messner, B.; Baum, H.; Fischer, P.; Quasthoff, S.; Neumeier, D. Expression of messenger RNA of the cardiac isoforms of troponin T and I in myopathic skeletal muscle. Am. J. Clin. Pathol. 2000, 114, 544–549, Erratum in Am. J. Clin. Pathol. 2000, 114, 986. [Google Scholar] [CrossRef]
- Rusakov, D.Y.; Yamshcikov, N.V.; Tulayeva, O.N.; Suvorova, L.A.; Metlenko, O.I. Histogenesis and pecularities of structural organization of the cardiac muscle tissue un the walls of human caval and pulmonary veins. Morphology 2015, 148, 38–42. (In Russian) [Google Scholar]
- Rusakov, D.Y.; Vologdina, N.N.; Tulayeva, O.N. The development of striated cardiac muscle tissue in the walls of the caval and pulmonary veins. J. Anat. Histopathol. 2015, 4, 105. (In Russian) [Google Scholar] [CrossRef]
- Bodor, G.S.; Porterfield, D.; Voss, E.M.; Smith, S.; Apple, F.S. Cardiac troponin-I is not expressed in fetal and healthy or diseased adult human skeletal muscle tissue. Clin. Chem. 1995, 41 Pt 1, 1710–1715. [Google Scholar] [CrossRef]
- Hammerer-Lercher, A.; Erlacher, P.; Bittner, R.; Korinthenberg, R.; Skladal, D.; Sorichter, S.; Sperl, W.; Puschendorf, B.; Mair, J. Clinical and experimental results on cardiac troponin expression in Duchenne muscular dystrophy. Clin. Chem. 2001, 47, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Schmid, J.; Liesinger, L.; Birner-Gruenberger, R.; Stojakovic, T.; Scharnagl, H.; Dieplinger, B.; Asslaber, M.; Radl, R.; Beer, M.; Polacin, M.; et al. Elevated Cardiac Troponin T in Patients With Skeletal Myopathies. J. Am. Coll. Cardiol. 2018, 71, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.A.; Greig, A.; Mark, T.M.; Malouf, N.N.; Oakeley, A.E.; Ungerleider, R.M.; Allen, P.D.; Kay, B.K. Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ. Res. 1995, 76, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Bates, K.J.; Hall, E.M.; Fahie-Wilson, M.N.; Kindler, H.; Bailey, C.; Lythall, D.; Lamb, E.J. Circulating immunoreactive cardiac troponin forms determined by gel filtration chromatography after acute myocardial infarction. Clin. Chem. 2010, 56, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, A.; Lee, J.K.; Nagaya, T.; Kamiya, K.; Yasui, K.; Horiba, M.; Miwa, K.; Uzzaman, M.; Maki, M.; Ueda, Y.; et al. Overexpression of calpastatin by gene transfer prevents troponin I degradation and ameliorates contractile dysfunction in rat hearts subjected to ischemia/reperfusion. J. Mol. Cell Cardiol. 2003, 35, 1277–1284. [Google Scholar] [CrossRef]
- Zahran, S.; Figueiredo, V.P.; Graham, M.M.; Schulz, R.; Hwang, P.M. Proteolytic Digestion of Serum Cardiac Troponin I as Marker of Ischemic Severity. J. Appl. Lab. Med. 2018, 3, 450–455. [Google Scholar] [CrossRef]
- Vylegzhanina, A.V.; Kogan, A.E.; Katrukha, I.A.; Koshkina, E.V.; Bereznikova, A.V.; Filatov, V.L.; Bloshchitsyna, M.N.; Bogomolova, A.P.; Katrukha, A.G. Full-Size and Partially Truncated Cardiac Troponin Complexes in the Blood of Patients with Acute Myocardial Infarction. Clin. Chem. 2019, 65, 882–892. [Google Scholar] [CrossRef]
- Katus, H.A.; Remppis, A.; Looser, S.; Hallermeier, K.; Scheffold, T.; Kubler, W. Enzyme linked immune assay of cardiac troponin T for the detection of acute myocardial infarction in patients. J. Mol. Cell Cardiol. 1989, 21, 1349–1353. [Google Scholar] [CrossRef]
- Labugger, R.; Organ, L.; Collier, C.; Atar, D.; Van Eyk, J.E. Extensive troponin I and T modification detected in serum from patients with acute myocardial infarction. Circulation 2000, 102, 1221–1226. [Google Scholar] [CrossRef]
- Gaze, D.C.; Collinson, P.O. Multiple molecular forms of circulating cardiac troponin: Analytical and clinical significance. Ann. Clin. Biochem. 2008, 45 Pt 4, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Katrukha, A.G.; Bereznikova, A.V.; Esakova, T.V.; Pettersson, K.; Lövgren, T.; Severina, M.E.; Pulkki, K.; Vuopio-Pulkki, L.M.; Gusev, N.B. Troponin I is released in bloodstream of patients with acute myocardial infarction not in free form but as complex. Clin. Chem. 1997, 43 Pt 1, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S.; Oakeley, A.E.; Allen, P.D.; Crimmins, D.L.; Ladenson, J.H.; Anderson, P.A. Troponin I phosphorylation in the normal and failing adult human heart. Circulation 1997, 96, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Notkins, A.L. Clearance of LDH-5 from the circulation of inbred mice correlates with binding to macrophages. Int. J. Exp. Pathol. 1994, 75, 165–168. [Google Scholar]
- Prabhudas, M.; Bowdish, D.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; Means, T.K.; Moestrup, S.K.; et al. Standardizing scavenger receptor nomenclature. J. Immunol. 2014, 192, 1997–2006. [Google Scholar] [CrossRef] [PubMed]
- De Zoysa, J.R. Cardiac troponins and renal disease. Nephrology 2004, 9, 83–88. [Google Scholar] [CrossRef]
- Dubin, R.F.; Li, Y.; He, J.; Jaar, B.G.; Kallem, R.; Lash, J.P.; Makos, G.; Rosas, S.E.; Soliman, E.Z.; Townsend, R.R.; et al. Predictors of high sensitivity cardiac troponin T in chronic kidney disease patients: A cross-sectional study in the chronic renal insufficiency cohort (CRIC). BMC Nephrol. 2013, 14, 229. [Google Scholar] [CrossRef]
- Di Lullo, L.; Barbera, V.; Santoboni, A.; Bellasi, A.; Cozzolino, M.; De Pascalis, A.; Rivera, R.; Balducci, A.; Russo, D.; Ronco, C. Malattia renale cronica e sindrome coronarica acuta: Il ruolo della troponina [Troponins and chronic kidney disease]. G. Ital. Nefrol. 2015, 32, gin/32.4.1. (In Italian) [Google Scholar]
- Han, X.; Zhang, S.; Chen, Z.; Adhikari, B.K.; Zhang, Y.; Zhang, J.; Sun, J.; Wang, Y. Cardiac biomarkers of heart failure in chronic kidney disease. Clin. Chim Acta. 2020, 510, 298–310. [Google Scholar] [CrossRef]
- Wilhelm, J.; Hettwer, S.; Schuermann, M.; Bagger, S.; Gerhardt, F.; Mundt, S.; Muschik, S.; Zimmermann, J.; Amoury, M.; Ebelt, H.; et al. Elevated troponin in septic patients in the emergency department: Frequency, causes, and prognostic implications. Clin. Res. Cardiol. 2014, 103, 561–567. [Google Scholar] [CrossRef]
- Røsjø, H.; Varpula, M.; Hagve, T.A.; Karlsson, S.; Ruokonen, E.; Pettilä, V.; Omland, T.; FINNSEPSIS Study Group. Circulating high sensitivity troponin T in severe sepsis and septic shock: Distribution, associated factors, and relation to outcome. Intensive Care Med. 2011, 37, 77–85. [Google Scholar] [CrossRef]
- Daly, M.; Long, B.; Koyfman, A.; Lentz, S. Identifying cardiogenic shock in the emergency department. Am. J. Emerg. Med. 2020, 38, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Muslimovic, A.; Fridén, V.; Tenstad, O.; Starnberg, K.; Nyström, S.; Wesén, E.; Esbjörner, E.K.; Granholm, K.; Lindahl, B.; Hammarsten, O. The Liver and Kidneys mediate clearance of cardiac troponin in the rat. Sci. Rep. 2020, 10, 6791. [Google Scholar] [CrossRef] [PubMed]
- Fridén, V.; Starnberg, K.; Muslimovic, A.; Ricksten, S.E.; Bjurman, C.; Forsgard, N.; Wickman, A.; Hammarsten, O. Clearance of cardiac troponin T with and without kidney function. Clin. Biochem. 2017, 50, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Kavsak, P.A.; Worster, A.; Shortt, C.; Ma, J.; Clayton, N.; Sherbino, J.; Hill, S.A.; McQueen, M.; Griffith, L.E.; Mehta, S.R.; et al. Performance of high-sensitivity cardiac troponin in the emergency department for myocardial infarction and a composite cardiac outcome across different estimated glomerular filtration rates. Clin. Chim. Acta 2018, 479, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Patke, A.; Young, M.W.; Axelrod, S. Molecular mechanisms and physiological importance of circadian rhythms. Nat. Rev. Mol. Cell Biol. 2020, 21, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Cribbet, M.R.; Logan, R.W.; Edwards, M.D.; Hanlon, E.; Bien Peek, C.; Stubblefield, J.J.; Vasudevan, S.; Ritchey, F.; Frank, E. Circadian rhythms and metabolism: From the brain to the gut and back again. Ann. N. Y. Acad. Sci. 2016, 1385, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Thosar, S.S.; Butler, M.P.; Shea, S.A. Role of the circadian system in cardiovascular disease. J. Clin. Investig. 2018, 128, 2157–2167. [Google Scholar] [CrossRef]
- Klinkenberg, L.J.J.; Wildi, K.; van der Linden, N.; Kouw, I.W.K.; Niens, M.; Twerenbold, R.; Gimenez, M.R.; Puelacher, C.; Neuhaus, J.D.; Hillinger, P.; et al. Diurnal rhythm of cardiac troponin: Consequences for the diagnosis of acute myocardial infarction. Clin. Chem. 2016, 62, 1602–1611. [Google Scholar] [CrossRef]
- van der Linden, N.; Cornelis, T.; Klinkenberg, L.J.J.; Kimenai, D.M.; Hilderink, J.M.; Litjens, E.J.R. Strong diurnal rhythm of troponin T, but not troponin I, in a patient with renal dysfunction. Int. J. Cardiol. 2016, 221, 287–288. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakov, D.V. Cardiac troponins in hypertension: Mechanisms of increase and diagnostic value. Arter. Gipertenz. Arter. Hypertens. 2021, 27, 390–401. (In Russian) [Google Scholar] [CrossRef]
- Tofler, G.H.; Brezinski, D.; Schafer, A.I.; Czeisler, C.A.; Rutherford, J.D.; Willich, S.N.; Gleason, R.E.; Williams, G.H.; Muller, J.E. Concurrent morning increase in platelet aggregability and the risk of myocardial infarction and sudden cardiac death. N. Engl. J. Med. 1987, 316, 1514–1518. [Google Scholar] [CrossRef] [PubMed]
- Chaulin, A.M.; Duplyakov, D.V. Comorbidity in chronic obstructive pulmonary disease and cardiovascular disease. Cardiovasc. Ther. Prev. 2021, 20, 2539. (In Russian) [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakov, D.V. Microrna: The role in the pathophysiology of atrial fibrillation and potential use as a biomarker. Bull. Sib. Med. 2021, 20, 203–212. [Google Scholar] [CrossRef]
- Chaulin, A.M.; Duplyakov, D.V. Environmental factors and cardiovascular diseases. Hyg. Sanit. 2021, 100, 223–228. [Google Scholar] [CrossRef]
- Panza, J.A.; Epstein, S.E.; Quyyumi, A.A. Circadian variation in vascular tone and its relation to alpha-sympathetic vasoconstrictor activity. N. Engl. J. Med. 1991, 325, 986–990. [Google Scholar] [CrossRef]
- Tsareva, Y.O.; Mayskova, E.A.; Fedotov, E.A.; Shvarts, Y.G. Circadian rhythms of thyroid hormones in patients with ischemic heart disease, arterial hypertension, and atrial fibrillation. Kardiologiia 2019, 59, 23–29. (In Russian) [Google Scholar] [CrossRef]
- Chaulin, A.M.; Grigorieva, J.V.; Suvorova, G.N.; Duplyakov, D.V. Experimental Modeling of Hypothyroidism: Principles, Methods, Several Advanced Research Directions in Cardiology. Russ. Open Med. J. 2021, 10, e0311. [Google Scholar] [CrossRef]
- Chaulin, A.M. Diagnostic value of highly sensitive cardiac troponins and mechanisms of their increase in serum and urine in arterial hypertension. Riv. Ital. Med. Lab. 2021, 17, 99–107. [Google Scholar] [CrossRef]
- Suárez-Barrientos, A.; López-Romero, P.; Vivas, D.; Castro-Ferreira, F.; Núñez-Gil, I.; Franco, E.; Ruiz-Mateos, B.; García-Rubira, J.C.; Fernández-Ortiz, A.; Macaya, C.; et al. Circadian variations of infarct size in acute myocardial infarction. Heart 2011, 97, 970–976. [Google Scholar] [CrossRef]
- Arroyo Úcar, E.; Dominguez-Rodriguez, A.; Abreu-Gonzalez, P. Influencia de la variabilidad diurna en el tamaño del infarto agudo de miocardio [Influence of diurnal variation in the size of acute myocardial infarction]. Med. Intensiva 2012, 36, 11–14. (In Spanish) [Google Scholar] [CrossRef]
- Seneviratna, A.; Lim, G.H.; Devi, A.; Carvalho, L.P.; Chua, T.; Koh, T.H.; Tan, H.C.; Foo, D.; Tong, K.L.; Ong, H.Y.; et al. Circadian Dependence of Infarct Size and Acute Heart Failure in ST Elevation Myocardial Infarction. PLoS ONE 2015, 10, e0128526. [Google Scholar] [CrossRef] [PubMed]
- Chaulin, A.M.; Duplyakov, D.V. Cardioprotective Strategies for Doxorubicin-induced Cardiotoxicity: Present and Future. Ration. Pharmacother. Cardiol. 2022, 18, 103–112. (In Russian) [Google Scholar] [CrossRef]
- Manfredini, R.; Boari, B.; Bressan, S.; Gallerani, M.; Salmi, R.; Portaluppi, F.; Mehta, R.H. Influence of circadian rhythm on mortality after myocardial infarction: Data from a prospective cohort of emergency calls. Am. J. Emerg. Med. 2004, 22, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Fournier, S.; Puricel, S.; Morawiec, B.; Eeckhout, E.; Mangiacapra, F.; Trana, C.; Tapponnier, M.; Iglesias, J.F.; Michiels, V.; Stauffer, J.C.; et al. Relationship between time of day and periprocedural myocardial infarction after elective angioplasty. Chronobiol. Int. 2014, 31, 206–213. [Google Scholar] [CrossRef]
- Chaulin, A.M. Cardiac Troponins Metabolism: From Biochemical Mechanisms to Clinical Practice (Literature Review). Int. J. Mol. Sci. 2021, 22, 10928. [Google Scholar] [CrossRef]
- Fournier, S.; Muller, O. Commentary “Recent advances in circadian rhythms in cardiovascular system”. Front. Pharmacol. 2015, 6, 132. [Google Scholar] [CrossRef][Green Version]



{kind=link}
{kind=link}
| One-Hour NSTEMI Diagnostic Algorithm | |||||
| Troponin Immunoassay, Company (Manufacturer) | Biomarker Concentration That Indicates an Extremely Low Probability of an NSTEMI Diagnosis, ng/L | Biomarker Concentration That Indicates a Low Probability of an NSTEMI Diagnosis, ng/L | Changes in Biomarker Concentration after 1 h at which a Diagnosis of NSTEMI Should be Excluded, ng/L | Biomarker Concentration That Indicates a High Probability of an NSTEMI Diagnosis, ng/L | Changes in Biomarker Concentration after 1 h at which a Diagnosis of NSTEMI Should be Confirmed, ng/L |
| hs-cTnT (Elecsys; Roche) | <5 | <12 | <3 | ≥52 | ≥5 |
| hs-cTnI (Architect; Abbott) | <4 | <5 | <2 | ≥64 | ≥6 |
| hs-cTnI (Centaur; Siemens) | <3 | <6 | <3 | ≥120 | ≥12 |
| hs-cTnI (Access; Beckman Coulter) | <4 | <5 | <4 | ≥50 | ≥15 |
| hs-cTn I (Clarity; Singulex) | <1 | <2 | <1 | ≥30 | ≥6 |
| hs-cTn I (Vitros; Clinical Diagnostics) | <1 | <2 | <1 | ≥40 | ≥4 |
| hs-cTnI (Pathfast; LSI Medience) | <3 | <4 | <3 | ≥90 | ≥20 |
| Two-Hour NSTEMI Diagnostic Algorithm | |||||
| Troponin immunoassay, company (manufacturer) | Biomarker concentration that indicates an extremely low probability of an NSTEMI diagnosis, ng/L | Biomarker concentration that indicates a low probability of an NSTEMI diagnosis, ng/L | Changes in biomarker concentration after 2 h at which a diagnosis of NSTEMI should be excluded, ng/L | Biomarker concentration that indicates a high probability of an NSTEMI diagnosis, ng/L | Changes in biomarker concentration after 2 h at which a diagnosis of NSTEMI should be confirmed, ng/L |
| hs-cTnT (Elecsys; Roche) | <5 | <14 | <4 | ≥52 | ≥10 |
| hs-cTnI (Architect; Abbott) | <4 | <6 | <2 | ≥64 | ≥15 |
| hs-cTnI (Centaur; Siemens) | <3 | <8 | <7 | ≥120 | ≥20 |
| hs-cTnI (Access; Beckman Coulter) | <4 | <5 | <5 | ≥50 | ≥20 |
| hs-cTn I (Clarity; Singulex) | <1 | to be determined | to be determined | ≥30 | to be determined |
| hs-cTn I (Vitros; Clinical Diagnostics) | <1 | to be determined | to be determined | ≥40 | to be determined |
| hs-cTn I (Pathfast; LSI Medience) | <3 | to be determined | to be determined | ≥90 | to be determined |
| Human Biological Fluids | Diagnostic Role | References |
|---|---|---|
| Blood (whole, serum, plasma) | It is the main biological fluid used to diagnose MI and assess the prognosis of patients suffering from non-ischemic cardiac (myocardites, Takotsubo syndrome, cardiomyopathies, etc.) and non-cardiac (sepsis, renal failure, neurogenic pathologies, etc.) pathologies that cause damage to MCs. | [15,16,17,23,24,25,26] |
| Urine | Molecules of cTns can be detected in this biological fluid via highly sensitive test systems. Increased cTns levels have a high prognostic value in diabetes mellitus and arterial hypertension. The method of obtaining this biological fluid is non-invasive, which has a number of advantages over the use of blood. It should be noted that the possibilities of examination of highly sensitive cTns in urine are still poorly studied and have not been finally validated. Further research is needed before the introduction of this method into clinical practice. | [38,71] |
| Oral fluid | The levels of cTns in oral fluid increase in MI and moderately correlate with serum troponin levels; therefore, further study of this area of non-invasive diagnostics is very promising. | [39,40,41,70,72] |
| Pericardial fluid and cerebrospinal fluid | Molecules of cTns are detected in pericardial fluid and cerebrospinal fluid via moderately sensitive and highly sensitive test systems and, according to some studies, may correlate with serum levels of cTns. Increased troponin levels in these biological fluids may reflect the degree of myocardial damage and may be used in forensic medicine to determine the cause of death. Thus, according to Hernández-Romero et al., the concentration of troponin I in the pericardial fluid and the ratio of pericardial and serum levels of troponin I are associated with the cause of death. Highest cTnI ratio values were shown for AMI deaths, followed by asphyctic, traumatic and deaths by other natural causes [76]. However, due to the relative paucity of such studies, further investigation of these possibilities is necessary. | [76,77,78,79,80,81,82] |
| Amniotic fluid | cTns molecules can be detected in amniotic fluid via moderately sensitive and highly sensitive immunoassays. Increased cTns levels may indicate chronic fetal hypoxia, abnormal development of the cardiovascular system and fetal myocardial injury, and an increased risk of fetal death during the intrauterine growth period. However, it is worth noting that such studies are few in number. Further research is needed to clarify the diagnostic capabilities of amniotic fluid. | [83,84,85,86] |
| Mechanism | Diagnostic Value | References |
|---|---|---|
| Myocardial cell necrosis | This is the main proven mechanism underlying the increase in cTns in MI. Cardiomyocyte necrosis will result in the release of all molecules (biomarkers) from the cell into the bloodstream. | [14,15,16] |
| Release of cTns as a result of the processes of regeneration and renewal of MCs | The renewal of MCs gradually occurring throughout life, hypothetically, may be associated with normal (less than the upper limit of the 99th percentile) concentrations of cTns in the bloodstream. | [105,106,107,108,109,110,111,112,113,114] |
| Release of cTns as a result of apoptosis of MCs | It has been proven that apoptosis of cardiomyocytes (without signs of necrosis) is accompanied by an increase in the serum concentration of cTns. Thus, any physiological (physical activity, old age) and pathological (heart failure, arterial hypertension, chronic obstructive pulmonary disease, etc.) conditions that enhance apoptosis may be accompanied by the release of cTns from cardiomyocytes and an increase in serum levels. | [117,118,119,120,127,128,129,130,131,132,133,134,135,136] |
| Release of cTns as a result of the formation of membrane vesicles on the surface of MCs | Membrane vesicles (blebbing vesicles) formed on the surface of the plasma membrane of cardiomyocytes, hypothetically, may contain cytoplasmic proteins, including cTns. The number of membrane vesicles increases during ischemia of MCs and may be associated with the release of cTns into the bloodstream. | [137,138,139,140,141,142] |
| Intracellular proteolytic degradation of cTns molecules into small fragments and the release of the latter through the intact membrane of MCs | Molecules of cTns can be fragmented/destroyed by the action of certain proteolytic enzymes: calpain, thrombin, matrix metalloproteinases. As a result of the action of these enzymes, there can form small fragments of troponin molecules, which, due to their size, have a higher probability of release from the cell. This mechanism may have high clinical significance: for example, all those physiological and pathological conditions and/or drugs that affect the activity of these proteolytic enzymes can also affect the release of cTns and their concentration in the bloodstream. | [147,148,149,150,151,152,153,154,155] |
| Release of cTns as a result of increased membrane permeability of MCs | An increase in the release of cTns molecules into the bloodstream is observed in case of an increase in the membrane permeability of MCs, which is characteristic of myocardial ischemia, an increase in preload and stretching of the heart wall. | [159,160,161,162,163] |
| Release of cTns as a result of small-scale (subclinical) necrosis of cardiomyocytes | The death of a small number of cardiomyocytes may not manifest itself clinically and instrumentally (since these are relatively low-sensitivity methods), but highly sensitive methods of detection can register such subclinical lesions. Possible causes of subclinical necrosis of cardiomyocytes are ischemia, inflammatory-toxic processes and imbalances in the neuroendocrine system. | [164,165,166,167,168,169,170,171,172,173,174,175] |
| Release of cTns from non-cardiac cells | This is a controversial mechanism of increased levels of cTns in the bloodstream. In the literature, there are works confirming the expression of cTns in skeletal muscle tissue in patients with CRF and hereditary skeletal myopathies, as well as studies that refute this hypothesis. | [103,104,176,177,178,179,180,181,182] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaulin, A.M. Biology of Cardiac Troponins: Emphasis on Metabolism. Biology 2022, 11, 429. https://doi.org/10.3390/biology11030429
Chaulin AM. Biology of Cardiac Troponins: Emphasis on Metabolism. Biology. 2022; 11(3):429. https://doi.org/10.3390/biology11030429
Chicago/Turabian StyleChaulin, Aleksey M. 2022. "Biology of Cardiac Troponins: Emphasis on Metabolism" Biology 11, no. 3: 429. https://doi.org/10.3390/biology11030429
APA StyleChaulin, A. M. (2022). Biology of Cardiac Troponins: Emphasis on Metabolism. Biology, 11(3), 429. https://doi.org/10.3390/biology11030429