
Oncometabolites—A Link between Cancer Cells and Tumor Microenvironment

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Lactate
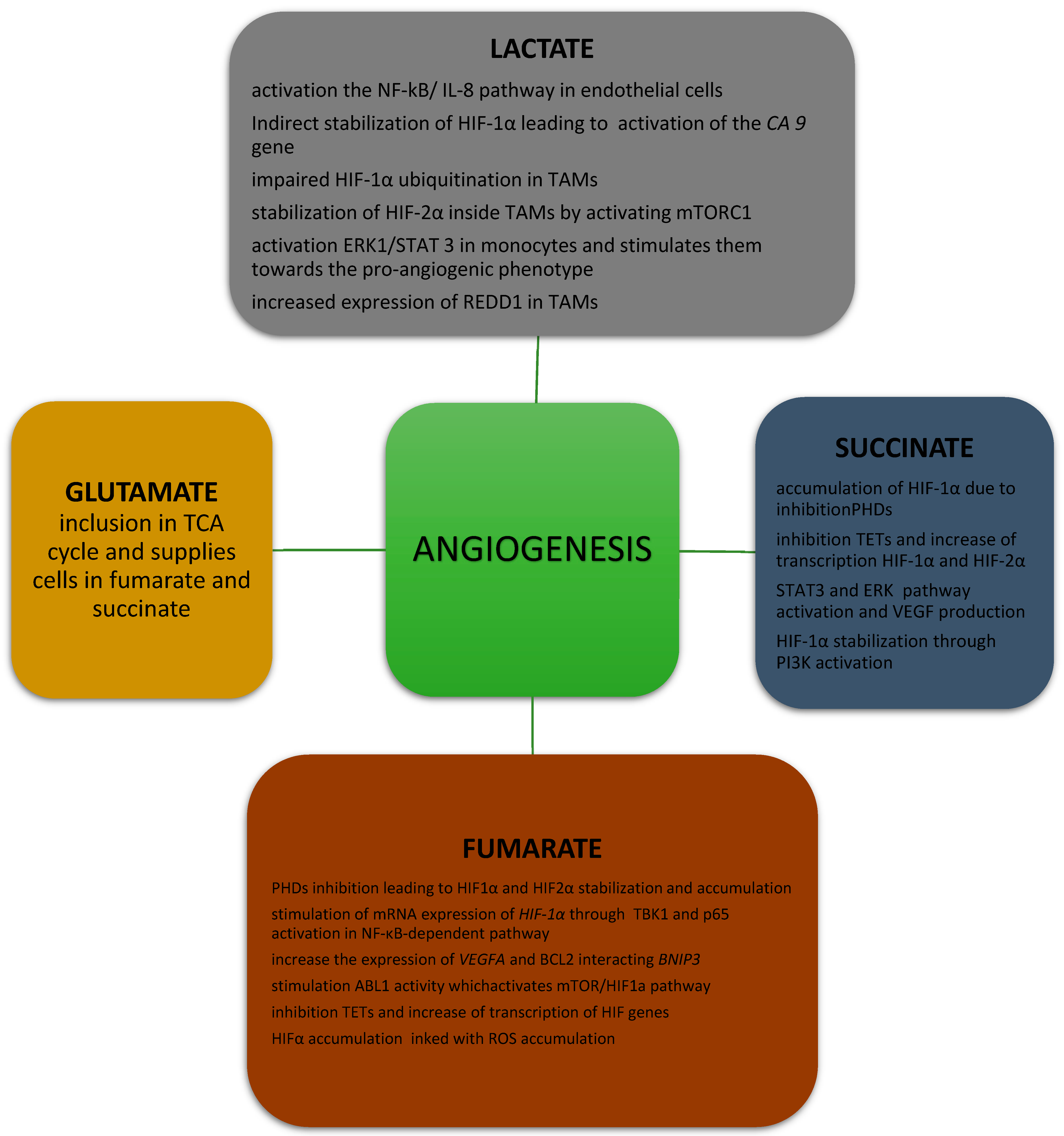
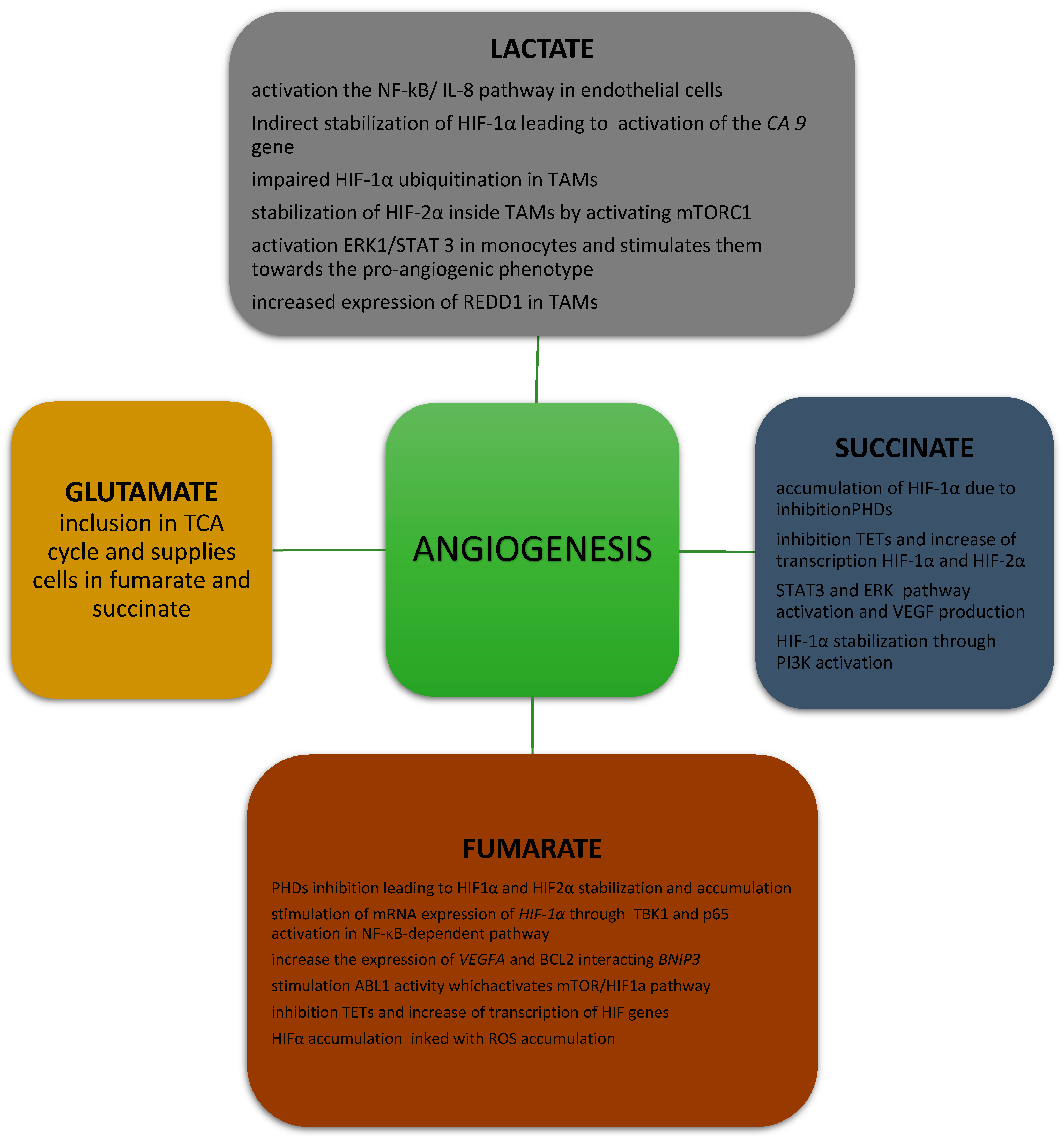
2.1. Effects of Lactate on Angiogenesis
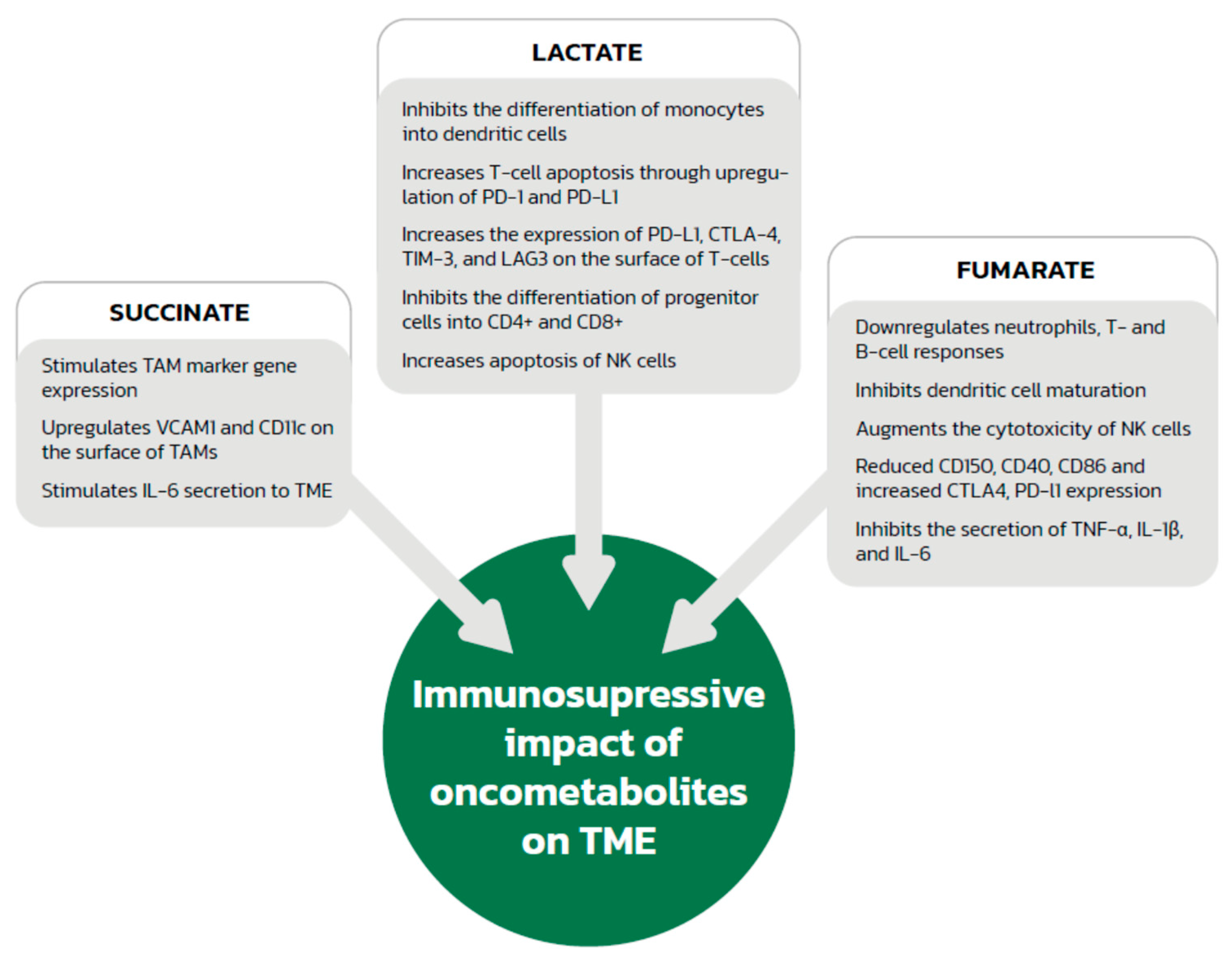
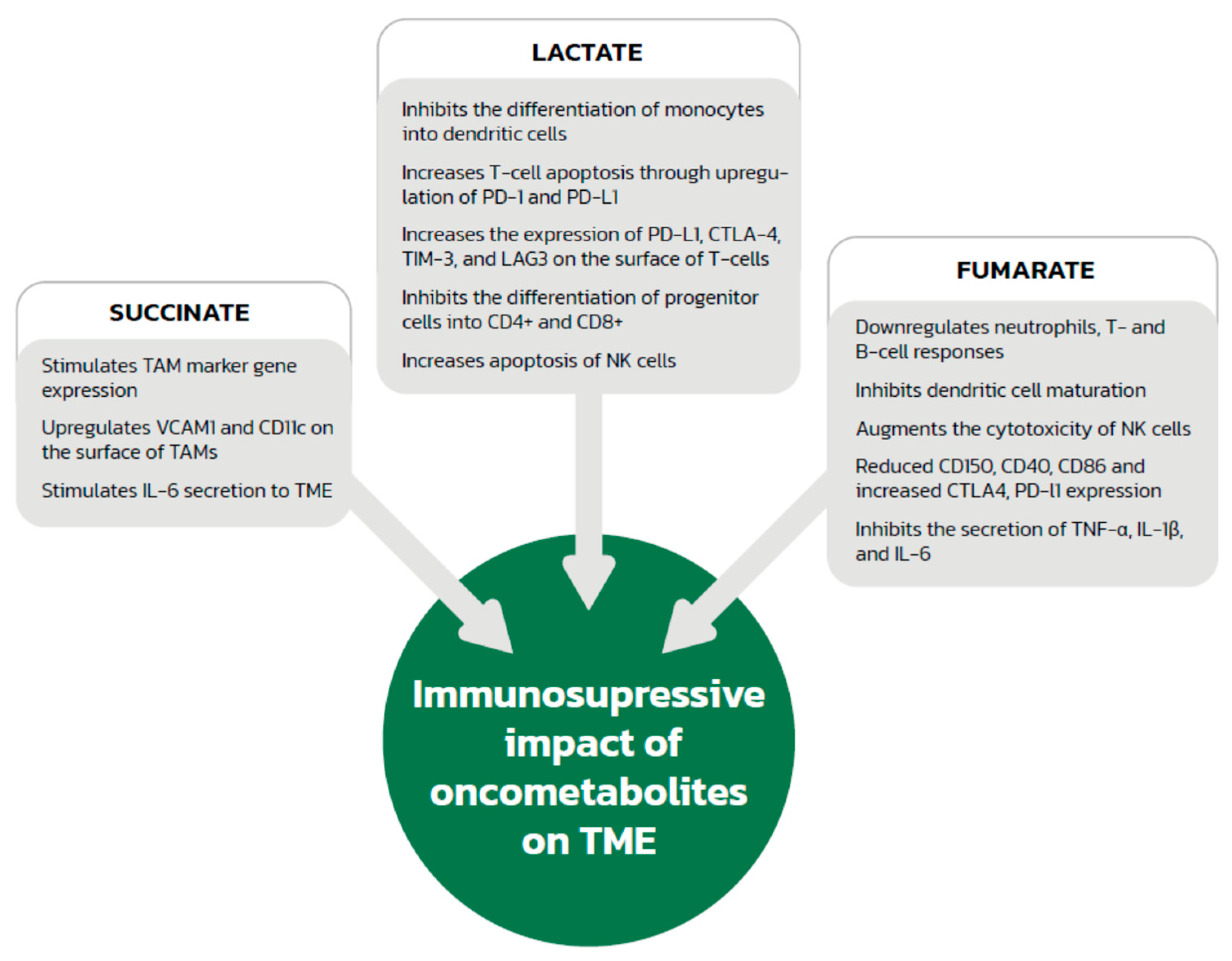
2.2. Immunosuppressive Effect of the Acidic Environment and Lactate
2.3. Other Effects of Lactate in Shaping the Microenvironment
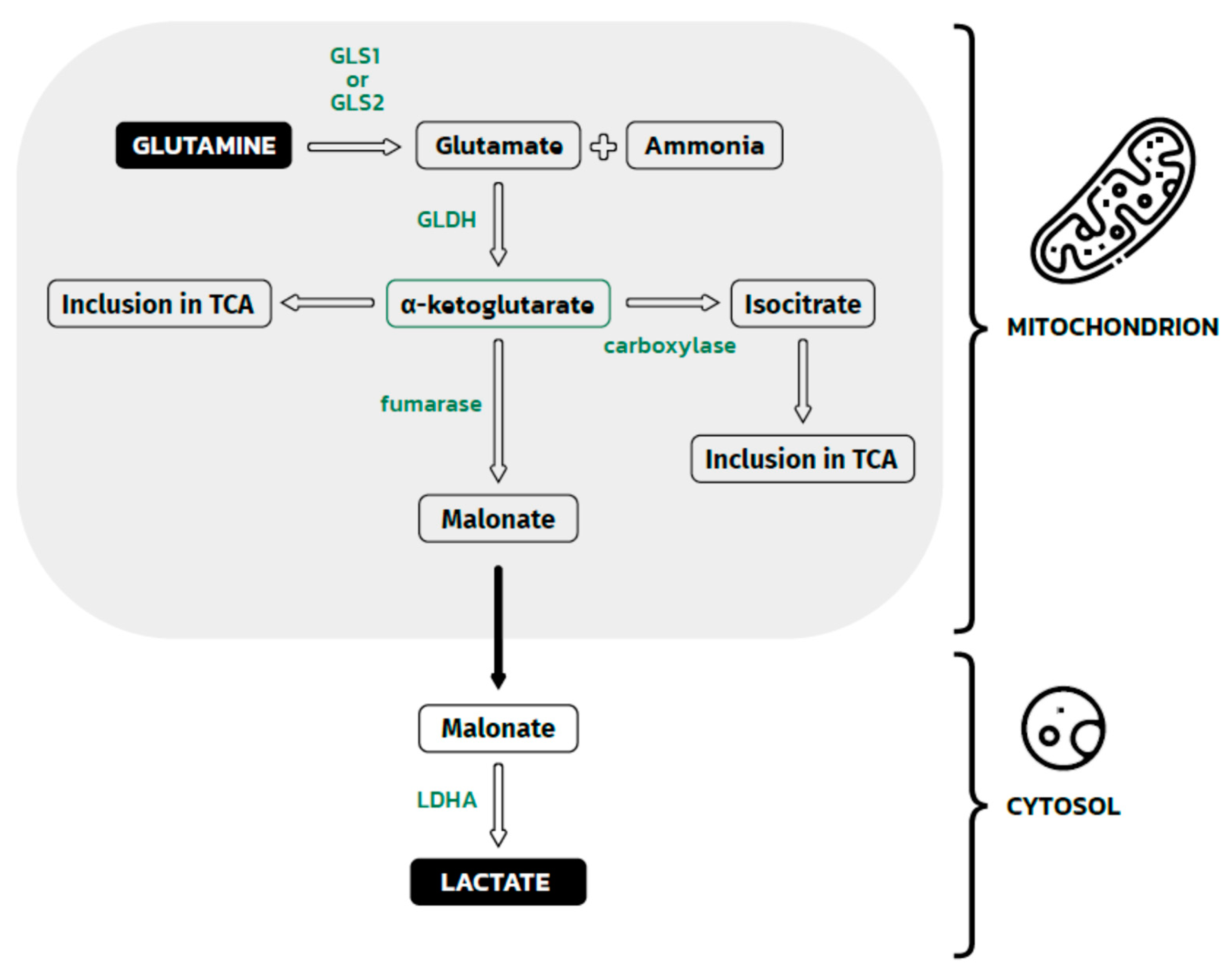
3. Glutamine
4. Succinate
4.1. Effects of Succinate on Angiogenesis
4.2. Effect of Succinate on Immune System Cells
4.3. The Role of Succinate in Neoplastic Stroma
5. Fumarate
5.1. Effect of Fumarate on Angiogenesis
5.2. Fumarate and Immune System Modification
5.3. DNA Damage Response in FH-Deficient Cells
5.4. Other Effects of Fumarate in Shaping the Microenvironment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lactate-modified pathways |
|
| Fumarate-modified pathways |
|
| Succinate-modified pathways |
|
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Egelbad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell. 2010, 18, 884–901. [Google Scholar] [CrossRef] [Green Version]
- Tameemi, W.A.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 2019, 7, 4–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldewijns, M.M.; van Vlodrop, I.J.H.; Vermeulen, P.B.; Soetekouw, P.M.; van Engeland, M.; de Bruïne, A.P. VHL and HIF signalling in renal cell carcinogenesis. J. Pathol. 2010, 221, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Xu, H.; Dai, J.; Peng, J.; Wang, W.; Xia, L.; Zhou, F. Prognostic significance of serum lactic acid, lactate dehydrogenase, and albumin levels in patients with metastatic colorectal cancer. Biomed. Res. Int. 2018, 2018, 1804086. [Google Scholar] [CrossRef] [Green Version]
- Vlachostergios, P.J.; Oikonomou, K.G.; Gibilaro, E.; Apergis, G. Elevated lactic acid is a negative prognostic factor in metastatic lung cancer. Cancer Biomark. 2015, 15, 725–734. [Google Scholar] [CrossRef]
- García-Cañaveras, J.C.; Agustín Lahoz, A. Tumor microenvironment-derived metabolites: A guide to find new metabolic therapeutic targets and biomarkers. Cancers 2021, 13, 3230. [Google Scholar] [CrossRef]
- Yong, C.; Stewart, G.D.; Frezza, C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 2020, 16, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Eskandari, R.; Ray, C.; Granlund, K.L.; Santos-Cunha, L.D.; Miloushev, V.Z.; Tee, S.S.; Jeong, S.; Aras, O.; Chen, Y.B.; et al. Hyperpolarized MRI visualizes Warburg effects and predicts treatment response to mTOR inhibitors in patient-derived ccRCC xenograft models. Cancer Res. 2019, 79, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, R.T.; McLean, M.A.; Madhu, B.; Challis, B.G.; Ten Hoopen, R.; Roberts, T.; Clark, G.R.; Pittfield, D.; Simpson, H.L.; Bulusu, V.R.; et al. Translating in vivo metabolomic analysis of succinate dehydrogenase deficient tumours into clinical utility. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolisis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [Green Version]
- De Groof, J.C.; te Lindert, M.M.; van Dommelen, M.M.T.; Wu, M.; Willemse, M.; Smift, A.L.; Winer, M.; Oerlemans, F.; Pluk, H.; Fransen, J.A.; et al. Increased OXPHOS activity precedes rise in glycolytic rate in H-RasV12/E1A transformed fibroblasts that develop a Warburg phenotype. Mol. Cancer 2009, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Beckert, S.; Farrahi, F.; Aslam, R.S.; Scheuenstuhl, H.; Königsrainer, A.; Hussain, M.Z.; Hunt, T.K. Lactate stimulates endothelial cell migration. Wound Repair Regen. 2006, 14, 321–324. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Simopoulos, C.; Polychronidis, A.; Sivridis, E. Lactate dehydrogenase 5 (LDH5) relates to up- regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis. 2005, 22, 25–30. [Google Scholar] [CrossRef] [PubMed]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not I Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef] [Green Version]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports and NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counillon, L.; Bouret, Y.; Marchiq, I.; Pouysségur, J. Na+/H+ antiporter (NHE1) and lactate/H+ symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta 2016, 1863, 2465–2480. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, D.; Xu, T.; Liu, P.; Cao, Y.; Wang, Y.; Yang, X.; Xu, X.; Wang, X.; Niu, H. Bladder cancer cells re-educate TAMs through lactate shuttling in the microfluidic cancer microenvironment. Oncotarget 2015, 6, 39196–39210. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, T.; Chen, S.; Chen, X.; Wu, T.; Yang, Y.; Li, S.; Ma, J.; Zhao, J.; Lin, W.; Li, W.; et al. M2-TAM subsets altered by lactic acid promote T-cell apoptosis through the PD-L1/PD-1 pathway. Oncol. Rep. 2020, 44, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Airley, R.E.; Loncaster, J.; Raleigh, J.A.; Harris, A.L.; Davidson, S.E.; Hunter, R.D.; West, C.M.; Stratford, I.J. GLUT-1 and CAIX as intrinsic markers of hypoxia in carcinoma of the cervix; relationship to pimonidazole binding. Int. J. Cancer 2003, 104, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Luo, J.; Kuang, D.; Xu, S.; Duan, Y.; Xia, Y.; Wei, Z.; Xie, X.; Yin, B.; Chen, F.; et al. Lactate inhibits ATP6V0D2 expression in tumor-associated macrophages to promote HIF-2α–mediated tumor progression. J. Clin. Investig. 2019, 129, 631–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Gatenbee, C.; Robertson-Tessi, M.; Bravo, R.; Dhillon, J.; Balagurunathan, Y.; Berglund, A.; Vishvakarma, N.; Ibrahim-Hashim, A.; Choi, J.; et al. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br. J. Cancer 2019, 121, 556–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Kikuchi, K.; Kusama, K.; Sano, M.; Nakanishi, Y.; Ishige, T.; Ohni, S.; Oinuma, T.; Nemoto, N. Vascular endothelial growth factor and dendritic cells in human squamous cell carcinoma of the oral cavity. Anticancer Res. 2006, 26, 1833–1848. [Google Scholar] [PubMed]
- Li, Y.L.; Zhao, H.; Ren, X.B. Relationship of VEGF/VEGFR with immune and cancer cells: Staggering or forward? Cancer Biol. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell- mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelki, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+T cell in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Wada, J.; Suzuki, H.; Fuchino, R.; Yamasaki, A.; Nagai, S.; Yanai, K.; Koga, K.; Nakamura, M.; Tanaka, M.; Morisaki, T.; et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cell in malignant effusions. Anticancer Res. 2009, 29, 881–888. [Google Scholar]
- Huang, Y.; Chen, X.; Dikov, M.M.; Novitskiy, S.V.; Mosse, C.A.; Yang, L.; Carbone, D.P. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood 2007, 110, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varney, M.L.; Johansson, S.L.; Singh, R.K. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: Role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Res. 2005, 15, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 7, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Liu, P.; Zhu, H.; Gong, H.; Yao, J.; Sun, Y.; Geng, G.; Wang, T.; Feng, S.; Han, M.; et al. Extracellular acidification acts as a key modulator of neutrophil apoptosis and functions. PLoS ONE 2015, 10, e0137221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2018, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi, T.; Mariani, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, R.; Shuster, S.; Neudecker, B.A.; Formby, B. Lactate stimulates fibroblast expression of hyaluronan and CD44: The Warburg effect revisited. Exp. Cell Res. 2002, 276, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lan, T.; Cai, H.; Lu, A.; Yu, W. Meta-analysis of serum lactate dehydrogenase and prognosis for osteosarcoma. Medicine 2018, 97, e0741. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Wang, W.; Yang, Z.; Pan, J.; Zheng, L.; Yin, L. Prognostic value of pretreatment serum lactate dehydrogenase level in pancreatic cancer patients: A meta-analysis of 18 observational studies. Medicine 2018, 97, e13151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Yan, X.; Song, Q.; Wang, G.; Hu, Y.; Jiao, S.; Wang, J. Pretreatment lactate dehydrogenase may predict outcome of advanced non small-cell lung cancer patients treated with immune checkpoint inhibitors: A meta-analysis. Cancer Med. 2019, 8, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Yuan, S.; Zhao, D.; Liu, X.J.; Wu, X.A. LDH-A promotes malignant behavior via activation of epithelial-to-mesenchymal transition in lung adenocarcinoma. Biosci. Rep. 2019, 39, BSR20181476. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perroud, B.; Ishimaru, T.; Borowsky, A.D.; Weiss, R.H. Grade-dependent proteomics characterization of kidney cancer. Mol. Cell. Proteom. 2009, 8, 971–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Heuvel, A.P.J.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Xie, G.; Liu, C.; Zhou, J.; Chen, J.; Yu, S.; Li, J.; Pang, X.; Shi, H.; Liang, H. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim. Biophys. Acta 2013, 1833, 2996–3005. [Google Scholar] [CrossRef] [Green Version]
- Cassago, A.; Ferreira, A.P.S.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.; Greene, K.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.; Ambrosio, A.L. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Pérez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; López-Larrubia, P.; et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Souba, W.W. Glutamine and cancer. Ann. Surg. 1993, 218, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.A.; Sánchez-Jiménez, F.; Márquez, J.; Rodríguez Quesada, A.; Núñez de Castro, I. Relevance of glutamine metabolism to tumor cell growth. Mol. Cell Biochem. 1992, 113, 1–15. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Neppala, P.; Banerjee, S.; Fanta, P.T.; Yerba, M.; Porras, K.A.; Burgoyne, A.M.; Sicklick, J.K. Current management of succinate dehydrogenase–deficient gastrointestinal stromal tumors. Cancer Metastasis Rev. 2019, 38, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M.; Venkatachalam, M.A.; Roeser, N.F.; Nissim, I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. USA 2000, 97, 2826–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, A.S.; Bayley, J.P. The role of complex II in disease. Biochim. Biophys. Acta 2013, 1827, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, S.; Ebara, S.; Ando, A.; Baba, Y.; Satomi, Y.; Soga, T.; Hara, T. Succinate dehydrogenase B-deficient cancer cells are highly sensitive to bromodomain and extra-terminal inhibitors. Oncotarget 2017, 8, 28922–28938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratcliffe, P.J. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell 2020, 77, 213–227.e5. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Zhao, T.; Xu, C.; Shi, W.; Geng, B.; Shen, J.; Zhang, C.; Pan, J.; Yang, J.; Hu, S.; et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 2017, 8, 13174–13185. [Google Scholar] [CrossRef] [PubMed]
- Mavria, G.; Vercoulen, Y.; Yeo, M.; Paterson, H.; Karasarides, M.; Marais, R.; Bird, D.; Marshall, C.J. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell 2006, 9, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiawen, Z.; Qinyi, Z.; Yongbin, Y.; Qingying, W. Association between succinate receptor SUCNR1 expression and immune infiltrates in ovarian cancer. Front. Mol. Biosci. 2020, 7, 150–164. [Google Scholar] [CrossRef]
- Terra, X.; Ceperuelo-Mallafré, V.; Merma, C.; Benaiges, E.; Bosch, R.; Castillo, P.; Flores, J.C.; León, X.; Valduvieco, I.; Basté, N.; et al. Succinate pathway in head and neck squamous cell carcinoma: Potential as a diagnostic and prognostic marker. Cancers 2021, 13, 1653. [Google Scholar] [CrossRef]
- Harber, K.J.; de Goede, K.E.; Verberk, S.G.S.; Meinster, E.; de Vries, H.E.; van Weeghel, M.; de Winther, M.P.J.; Van den Bossche, J. Succinate is an inflammation-induced immunoregulatory metabolite in macrophages. Metabolites 2020, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.K.; Luchtel, R.A.; Machha, V.; Tischer, A.; Zou, Y.; Pradhan, K.; Ashai, N.; Ramachandra, N.; Albanese, J.M.; Yang, J.I.; et al. Functional succinate dehydrogenase deficiency is a pathognomonic adverse feature of clear cell renal cancer. bioRxiv 2020, 118, e2106947118. [Google Scholar] [CrossRef]
- Cervera, A.M.; Bayley, J.P.; Devilee, P.; McCreath, K.J. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol. Cancer 2009, 8, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspuria, P.J.P.; Lunt, S.Y.; Väremo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 21. [Google Scholar] [CrossRef]
- Frezza, C.; Pollard, P.J.; Gottlieb, E. Inborn and acquired metabolic defects in cancer. J. Mol. Med. 2011, 89, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.; Sciacovelli, M.; Frezza, C. Fumarate hydratase in cancer: A multifaceted tumour suppressor. Semin. Cell Dev. Biol. 2019, 98, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ohad, Y.; Orli, Y.; Esti, S.; Shaulian, E.; Goldberg, M.; Fox, T.D.; Pines, O. Fumarase: A mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response. PLoS Biol. 2010, 8, e1000328. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Garcia-Martinez, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The proto-oncometabolite fumarate binds glutathione to amplify ROS dependent signaling. Mol. Cell 2013, 51, 236–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signaling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Shanmugasundaram, K.; Nayak, B.; Shim, E.H.; Livi, C.B.; Block, K.; Sudarshan, S. The oncometabolite fumarate promotes pseudohypoxia through noncanonical activation of NF-κB signaling. J. Biol. Chem. 2014, 289, 24691–24699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Furge, K.A. Fumarate hydratase inactivation in renal tumors: HIF1α, NRF2, and “cryptic targets” of transcription factors. Chin. J. Cancer 2012, 31, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; Garavelli, S.; Carbone, F.; Di Silvestre, D.; La Rocca, C.; Greco, D.; Colamatteo, A.; Lepore, M.T.; Russo, C.; De Rosa, G.; et al. Signals of pseudo-starvation unveil the amino acid transporter SLC7A11 as key determinant in the control of Treg cell proliferative potential. Immunity 2021, 54, 1543–1560.e6. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.H.; Masjedi, A.; Baradaran, B.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Anvari, E.; Jadidi-Niaragh, F. Dimethyl fumarate: Regulatory effects on the immune system in the treatment of multiple sclerosis. J. Cell. Physiol. 2019, 234, 9943–9955. [Google Scholar] [CrossRef] [PubMed]
- Afshin, D.; Asadzadeh, Z.; Safarpour, H.; Leone, P.; Shadbad, M.A.; Heydari, A.; Baradaran, B.; Racanelli, V. Regulation of CTLA-4 and PD-L1 expression in relapsing-remitting multiple sclerosis patients after treatment with fingolimod, IFNβ-1α, glatiramer acetate, and dimethyl fumarate drugs. J. Pers. Med. 2021, 11, 721. [Google Scholar] [CrossRef]
- Alaghehbandan, R.; Stehlik, J.; Trpkov, K.; Magi-Galluzzi, C.; Condom Mundo, E.; Pane Foix, M.; Berney, D.; Sibony, M.; Suster, S.; Agaimy, A.; et al. Programmed death-1 (PD-1) receptor/PD-1 ligand (PD-L1) expression in fumarate hydratase-deficient renal cell carcinoma. Ann. Diagn. Pathol. 2017, 29, 17–22. [Google Scholar] [CrossRef]
- Mitsuko, F.; Yasuhiro, I.; Yoji, N.; Kambe, N.; Ohe, C.; Kinoshita, H.; Sato, C.; Kishida, T.; Okubo, Y.; Numakura, K.; et al. Clinicopathological and molecular features of hereditary leiomyomatosis and renal cell cancer-associated renal cell carcinomas. J. Clin. Pathol. 2020, 73, 819–825. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Fay, A.P.; Gray, K.P.; Callea, M.; Ho, T.H.; Albiges, L.; Bellmunt, J.; Song, J.; Carvo, I.; Lampron, M.; et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann. Oncol. 2014, 25, 2178–2184. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, T.; Komohara, Y.; Ma, C.; Dewi, A.K.; Noguchi, H.; Yamada, S.; Nakayama, T.; Kitada, S.; Kawano, Y.; Takahashi, W.; et al. PD-L1 expression in papillary renal cell carcinoma. BMC Urol. 2017, 17, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Qian, X.; Shen, J.; Wang, Y.; Li, X.; Liu, R.; Xia, Y.; Chen, Q.; Peng, G.; Lin, S.Y.; et al. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat. Cell Biol. 2015, 17, 1158–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.I.; Costa, A.S.H.; Ferguson, A.N.; Frezza, C. Fumarate hydratase loss promotes mitotic entry in the presence of DNA damage after ionising radiation. Cell Death Dis. 2018, 9, 913. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Fumarate drives EMT in renal cancer. Cell Death Differ. 2017, 24, 1–2. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baryła, M.; Semeniuk-Wojtaś, A.; Róg, L.; Kraj, L.; Małyszko, M.; Stec, R. Oncometabolites—A Link between Cancer Cells and Tumor Microenvironment. Biology 2022, 11, 270. https://doi.org/10.3390/biology11020270
Baryła M, Semeniuk-Wojtaś A, Róg L, Kraj L, Małyszko M, Stec R. Oncometabolites—A Link between Cancer Cells and Tumor Microenvironment. Biology. 2022; 11(2):270. https://doi.org/10.3390/biology11020270
Chicago/Turabian StyleBaryła, Maksymilian, Aleksandra Semeniuk-Wojtaś, Letycja Róg, Leszek Kraj, Maciej Małyszko, and Rafał Stec. 2022. "Oncometabolites—A Link between Cancer Cells and Tumor Microenvironment" Biology 11, no. 2: 270. https://doi.org/10.3390/biology11020270
APA StyleBaryła, M., Semeniuk-Wojtaś, A., Róg, L., Kraj, L., Małyszko, M., & Stec, R. (2022). Oncometabolites—A Link between Cancer Cells and Tumor Microenvironment. Biology, 11(2), 270. https://doi.org/10.3390/biology11020270