
A New Approach: Determining cyt b G143A Allele Frequency in Zymoseptoria tritici by Digital Droplet PCR


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Isolates
2.2. DNA Extraction
2.3. Sanger Sequencing
2.4. ddPCR Reaction Setup and Equipment
2.5. Evaluation of DNA Amplification by ddPCR Assay Using Standard PCR
2.6. Preliminary Experiments of Droplet Generation
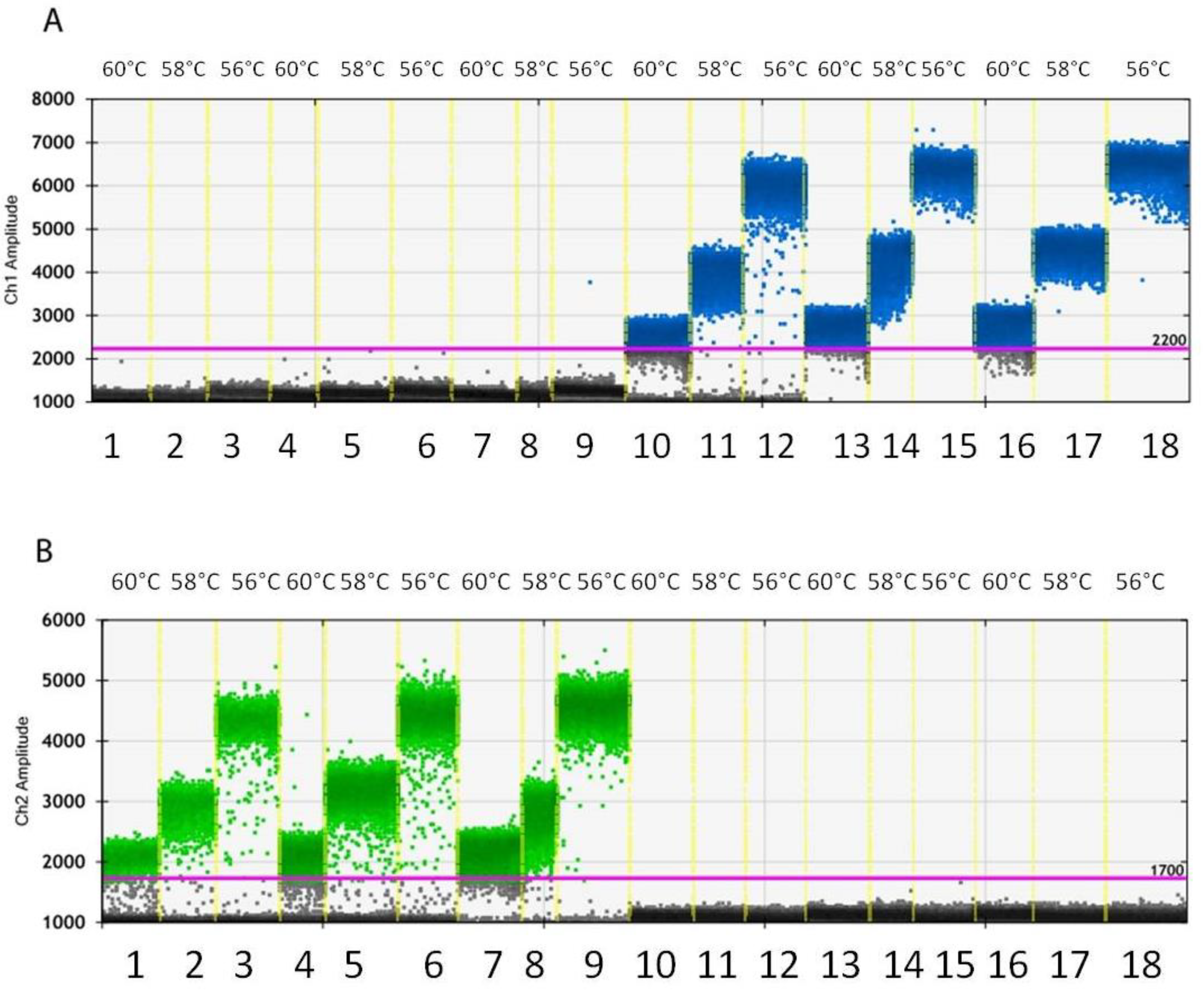
2.7. Optimization of ddPCR Conditions
2.8. Test of the ddPCR Protocol Sensitivity
2.9. Amplicon Sequencing
3. Results
3.1. Sanger Sequencing
3.2. DNA Amplification by ddPCR Assay Using Standard PCR
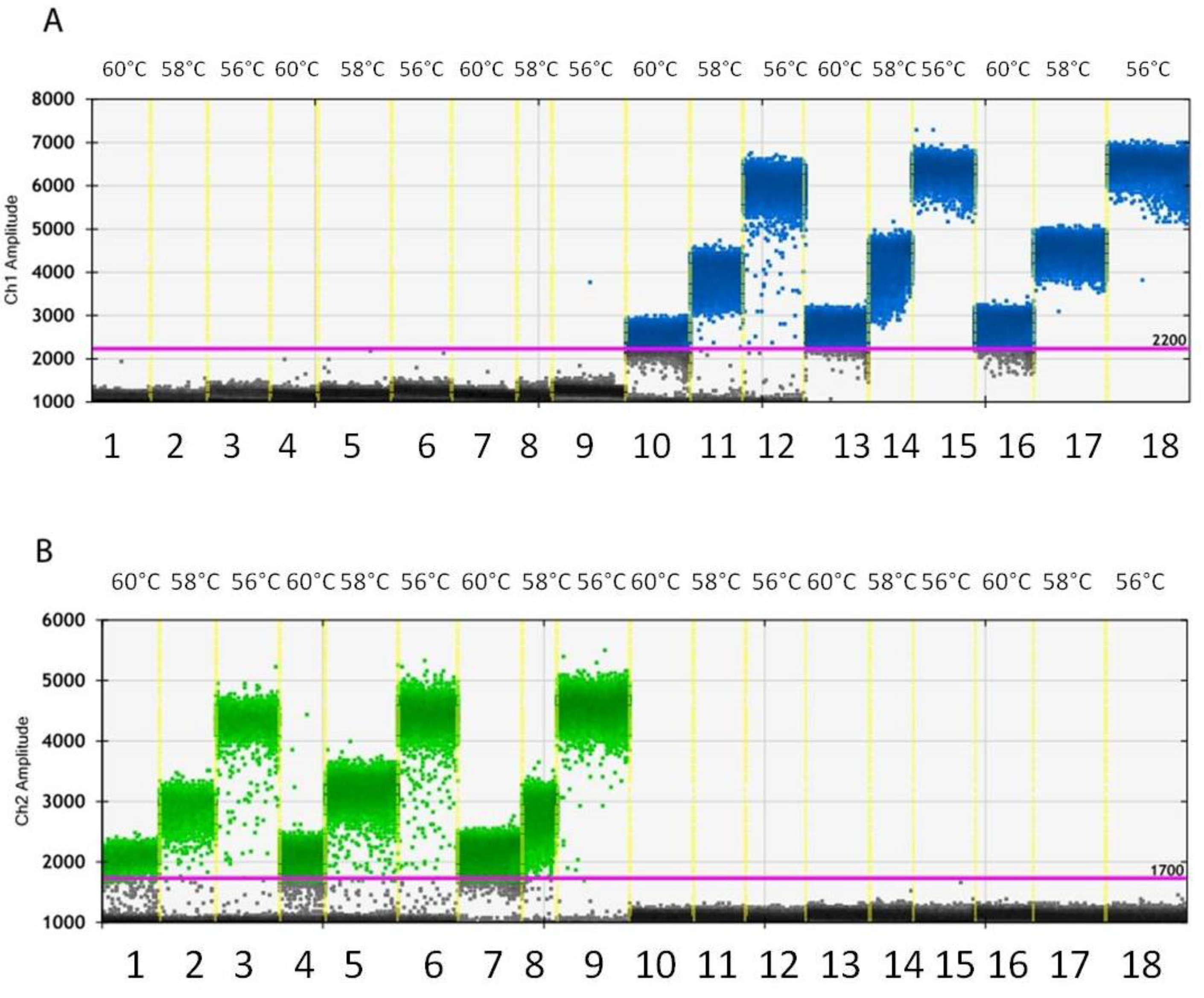
3.3. Droplet Generation and Optimization of PCR Conditions
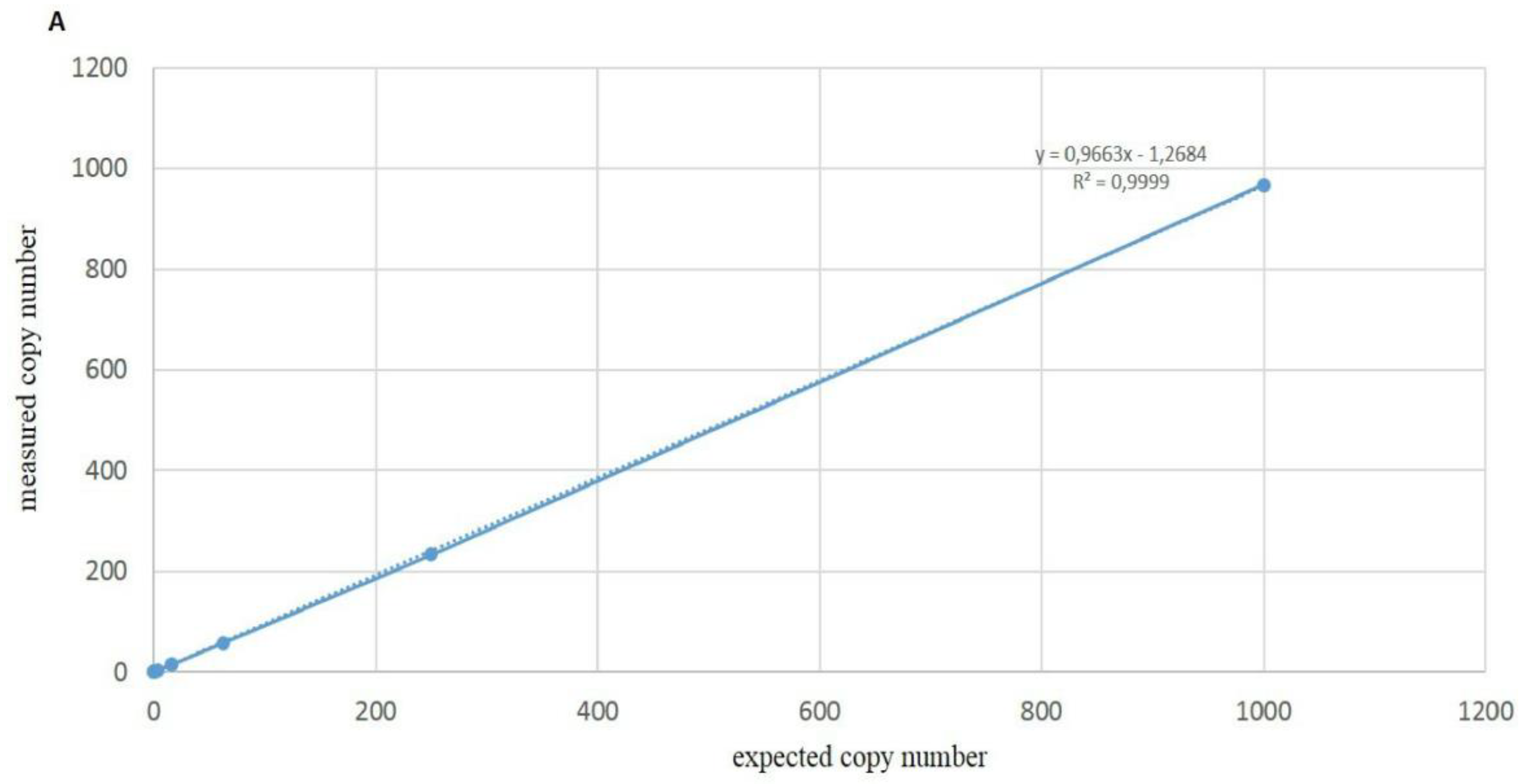
3.4. Sensitivity of ddPCR Optimized Protocol
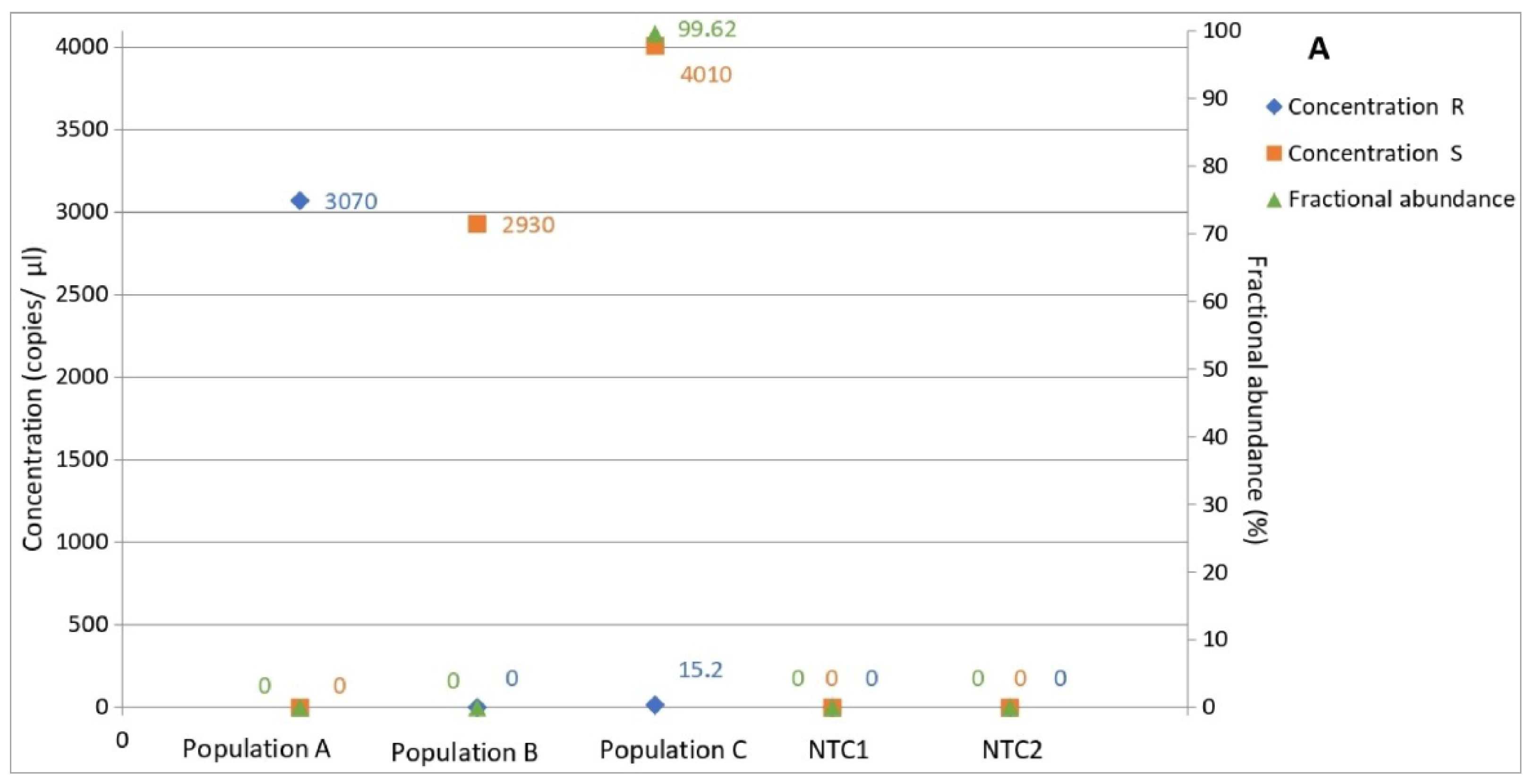
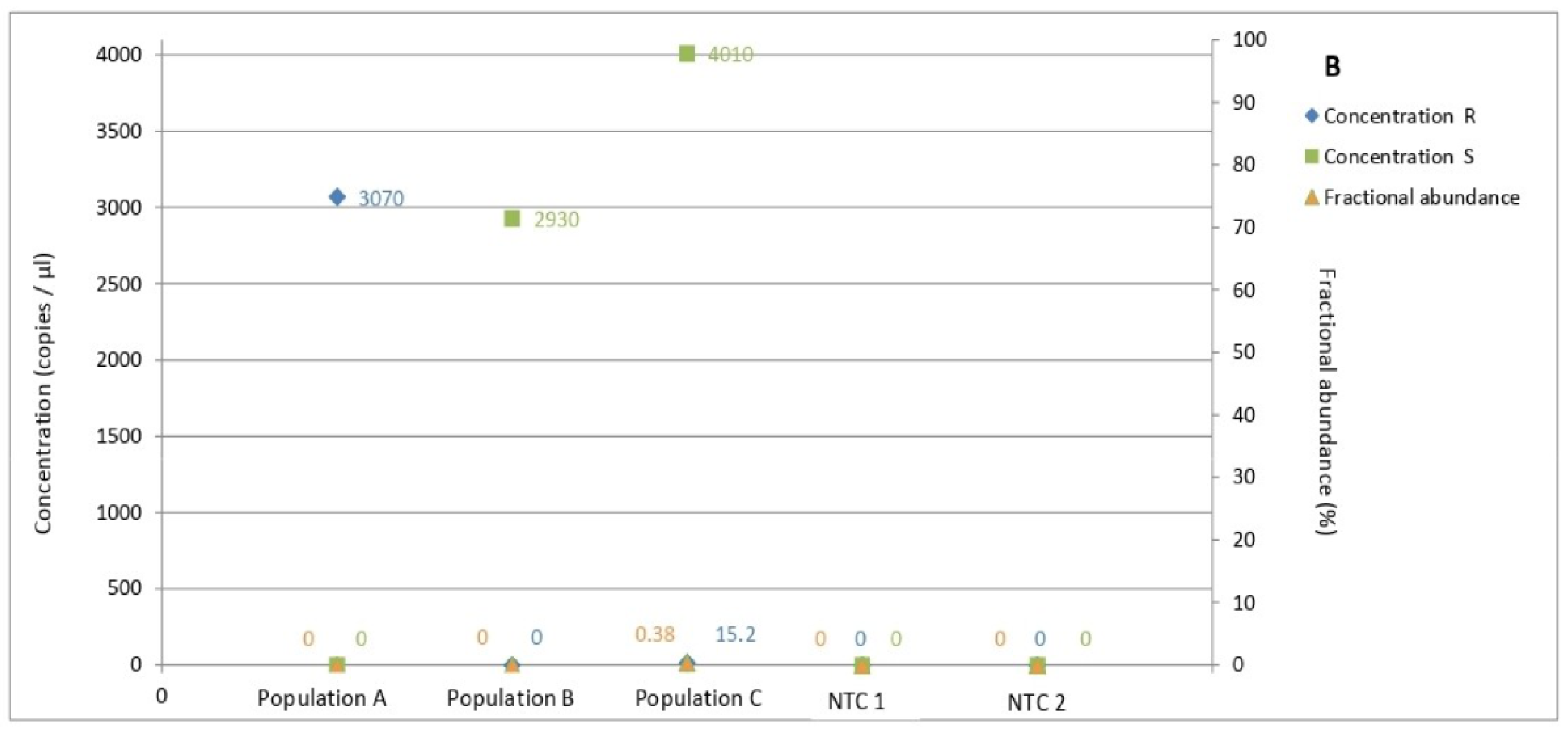
3.5. Determination of Allele Frequency in Populations
3.6. Determination of Allele Frequency in Population C by Amplicon Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pasquini, M.; Iori, A.; Nocente, F.; Sereni, L.; Cacciatori, P.; Cristofori, C.; Caprara, F.; Codianni, P.; Licciardello, S.; Martelli, M.; et al. Sensibilità dei frumenti alle principali malattie fungine. L’informatore Agrar. 2015, 39, 42–46. Available online: http://www.obiettivocereali.com/uploads/3/1/0/9/31095281/frumento_malattiefungine.pdf (accessed on 18 December 2021).
- Tonti, S.; Alvisi, G.; Pisi, A.; Nipoti, P.; Prodi, A. DNA Quantification to Assess Zymoseptoria tritici on a Susceptible Cultivar of Durum Wheat to Establish the Best Timing for Fungicide Application in an Italian Environment. Cereal Res. Commun. 2019, 47, 304–313. [Google Scholar] [CrossRef]
- Torriani, S.F.F.; Melichar, J.P.E.; Mills, C.; Paino, N.; Sierotzki, H.; Courbot, M. Zymoseptoria tritici: A major threat to wheat production, integrated approaches to control. Fungal Genet. Biol. 2015, 79, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Clough, J.M.; Godwin, J.R.; Hall, A.A.; Hamer, M.; Parr-Dobrzanski, B. The strobilurin fungicides. Pest. Manag. Sci. 2002, 58, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Fungicide Resistance Action Committee (FRAC). Protocol of the Discussions and Use Recommendations of the QoI Working Group of the Fungicide Resistance Action. Available online: https://www.frac.info/frac-teams/working-groups/qol-fungicides/recommendations-for-qoi (accessed on 18 December 2021).
- Fraaije, B.A.; Lucas, J.A.; Clark, W.S.; Burnett, F.J. QoI resistance development in populations of cereal pathogens in the UK. In Proceedings of the BCPC Inter- national Congress ed by BCPC, Alton, UK. Crop Sci. Technol. 2003, 2, 689–694. [Google Scholar]
- Gisi, U.; Pavic, L.; Stanger, C.; Hugelshofer, U.; Sierotzki, H. Dynamics of Mycosphaerella graminicola Populations in Response to Selection by different Fungicides. In Modern Fungicide and Antifungal Compounds IV; Dehne, H.W., Gisi, U., Kuck, K.H., Russell, P.E., Lyr, H., Eds.; BCPC: Alton, UK, 2005; pp. 73–80. [Google Scholar]
- Torriani, S.F.F.; Brunner, P.C.; McDonald, B.A.; Sierotzki, H. QoI resistance emerged independently at least 4 times in European populations of Mycosphaerella graminicola. Pest. Manag. Sci. 2009, 65, 155–162. [Google Scholar] [CrossRef]
- Estep, L.K.; Zala, M.; Anderson, N.P.; Sackett, K.E.; Flowers, M.D.; McDonald, B.A.; Mundt, C.C. First report of Resistance to Qoi Fungicides in North American Populations of Zymoseptoria tritici, Causal Agent of Septoria Tritici Blotch of Wheat. Plant Dis. 2013, 97, 1511. [Google Scholar] [CrossRef]
- Stewart, T.M.; Perry, A.J.; Evans, M.J. Resistance of Zymoseptoria tritici to azoxystrobin and epoxiconazole in the lower North Island of New Zeland. N. Z. Plant Protect. 2014, 67, 304–313. [Google Scholar] [CrossRef]
- Heick, T.M.; Justesen, A.F.; Jorgensen, L.N. Resistance of wheat pathogen Zymoseptoria tritici to DMI and Qoi fungicides in the Nordic-Batic region-a status. Eur. J. Plant Pathol. 2017, 149, 669–682. [Google Scholar] [CrossRef]
- Alvisi, G.; Cristiani, C.; Ponti, D.; Betti, C.; Ferrari, A. Grano duro: Lotta alla septoriosi, quale strategia di difesa usare. L’informatore Agrar. 2014, 9, 20–24. [Google Scholar]
- Ponti, D.; Alvisi, G.; Spagna, M.; Varotti, M.; Cristiani, C.; Antonacci, L.; Bugiani, R. Complesso della septoriosi su grano duro, come contenerla. L’informatore Agrar. 2018, 16, 77–80. Available online: http://www.caemilia.it/immagini/allegati/pdf_articolo_frumento_162.pdf (accessed on 18 December 2021).
- Battistini, G.; Gazzetti, K.; Menghini, M.; Frey, R.; Torriani, S.F.F.; Collina, M. Sensitivity of Italian Zymoseptoria tritici isolates to azoxystrobin and pyraclostrobin. In Modern Fungicides and Antifungal Compounds, Proceedings of the 19th International Reinhardsbrunn-Symposium, Friedrichroda, Germany, 7–11 April 2019; Deising, H.B., Fraaije, B., Mehl, A., Oerke, E.C., Sierotzki, H., Stammler, G., Eds.; Deutsche Phytomedizinische Gesellschaft: Braunschweig, Germany, 2020; Volume IX, pp. 39–40. ISBN 978-3-941261-16-7. [Google Scholar]
- Battistini, G.; Gazzetti, K.; Menghini, M.; Frey, R.; Torriani, S.F.F.; Collina, M.; Corresponding author affiliation: Department of Agricultural and Food Sciences, University of Bologna, Viale, G. Fanin 42, 40127 Bologna, Italy. 2021; (manuscript in preparation).
- Morley, A.A. Digital PCR: A brief history. Biomol. Detect. Quantif. 2014, 1, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Cui, X.; Hu, J.; Li, Z.; Choi, J.R.; Yang, Q.; Lin, M.; Hui, L.Y.; Xu, F. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosens. Bioelectron. 2014, 90, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of droplet Digital Polymerase Chain Reaction Format for DNA Copy Number Quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef]
- Chen, D.F.; Zhang, L.J.; Tan, K.; Jing, Q. Application of droplet digital PCR in quantitative detection of the cell-free circulating circRNAs. Biotechnol. Biotech. Eq. 2018, 32, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Jeschke, A.; Becks, L. Use of ddPCR in experimental evolution studies. Methods Ecol. Evol. 2016, 7, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, L.; Iwobi, A.; Busch, U.; Pecoraro, S. Optimization of digital droplet polymerase reaction for quantification of genetically modified organism. Biomol. Detect. Quantif. 2016, 7, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Paternò, A.; Verginelli, D.; Bonini, P.; Misto, M.; Quarchioni, C.; Dainese, E.; Peddis, S.; Fusco, C.; Vinciguerra, D.; Marchesi, U. In-House validation and comparison of two Wheat (Triticum aestivum) Taxon-Specific Real-time PCR Methods for GMO Quantification Supported by Droplet Digital PCR. Food Anal. Methods 2018, 11, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Demeke, T.; Dobnik, D. Critical assessment of digital PCR for the detection and quantification of genetically modified organisms. Anal. Bioanal. Chem. 2018, 410, 4039–4050. [Google Scholar] [CrossRef] [Green Version]
- Di Francesco, A.; Calassanzio, M.; Ratti, C.; Mari, M.; Folchi, A.; Baraldi, E. Molecular characterization of the two postharvest biological control agents Aureobasidium pullulans L1 and L8. Biol. Control 2018, 123, 53–59. [Google Scholar] [CrossRef]
- Mehle, N.; Dobnik, D.; Ravnikar, M.; Pompe Novak, M. Validated reverse transcription droplet digital PCR serves as higher order method for absolute quantification of Potato virus Y strains. Anal. Bioanal. Chem. 2018, 410, 3815–3825. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.D.H.; Wist, T.J.; Bekkaoui, D.R.; Hegedus, D.D.; Olivier, C.Y. Aster leafhopper survival and reproduction, and Aster yellows transmission under static and fuctuating temperatures, using ddPCR for phytoplasma quantification. Nature 2018, 8, 227. [Google Scholar] [CrossRef]
- Zulak, K.G.; Cox, B.A.; Tucker, M.A.; Oliver, R.P.; Lopez-Ruiz, F.J. Improve Detection and Monitoring of fungicide resistance in Blumeria graminis f. sp. hordei with High-Throughput Genotype Quantification by Digital PCR. Front. Microbiol. 2018, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.D.; Neill, T.M.; Colle, M.; Warneke, B.; Robinson, G.; Stergiopoulos, I.; Mahaffee, W.F. Allele-specific detection methods for QoI fungicide-resistant Erysiphe necator in vineyard. Plant Dis. 2021, 105, 175–182. [Google Scholar] [CrossRef]
- Prodi, A.; Purahong, W.; Tonti, S.; Salomoni, D.; Nipoti, P.; Covarelli, L.; Pisi, A. Difference in chemotype composition of Fusarium graminearum populations isolated from durum wheat in adjacent areas separated by the Apennines in Northern-Central Italy. J. Plant. Pathol. 2011, 27, 354–356. [Google Scholar] [CrossRef] [Green Version]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.R.; Garson, J.; Haynes, R.J.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The Digital MIQE Guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- The dMIQE Group. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clin. Chem. 2020, 66, 1012–1029. [Google Scholar] [CrossRef]
- Bio-Rad Droplet Digital Assay. Available online: https://www.bio-rad.com/digital-assays (accessed on 18 December 2021).
- Bio-Rad ddPCR Mutation Detection Assays. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/10000050422.pdf (accessed on 18 December 2021).
- Bustin, A.S.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M. Primer Sequence Disclosure: A Clarification of the MIQE Guidelines. Clin. Chem. 2011, 57, 6. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Through Put Sequencing Reads. EMB Net J. 2011. Available online: http://journal.embnet.org/index.php/embnetjournal/article/view/200/458 (accessed on 18 December 2021).
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Edgar, R.C. SEARCH_16S: A New Algorithm for Identifying 16S Ribosomal RNA Genes in Contigs and Chromosomes. Available online: https://www.biorxiv.org/content/biorxiv/early/2017/04/04/124131.full.pdf (accessed on 18 December 2021).
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, 2584. [Google Scholar] [CrossRef]
- Mc Cartney, C.; Mercer, P.C.; Cooke, L.R.; Fraaije, B.A. Effects of a strobilurin-based spray programme on disease control, green leaf area, yield and development of fungicide-resistance in Mycosphaerella graminicola in Northern Ireland. Crop. Prot. 2007, 26, 1272–1280. [Google Scholar] [CrossRef]
- Taher, K.; Graf, S.; Fakhfakh, M.M.; Salah, H.B.H.; Yahyaoul, A.; Rezgui, S.; Nazraoui, B.; Stammler, G. Sensitivity of Zymoseptoria tritici Isolates from Tunisia to Pyraclostrobin, Fluxapiroxad, Epoxiconazole, Metconazole, Prochloraz and Tebuconazole. J. Phytopathol. 2014, 162, 442–448. [Google Scholar] [CrossRef]
- Bio-Rad, Droplet Digital Applications Guide. Available online: https://www.biorad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf (accessed on 18 December 2021).
- Bio-Rad Rare Mutation Detection Best Practices Guides. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6628.pdf (accessed on 18 December 2021).
- Wang, M.; Yang, J.; Gai, Z.; Huo, S.; Zhu, J.; Li, J.; Wang, R.; Xing, S.; Shi, G.; Sci, F.; et al. Comparison between digital PCR and real- time PCR in detection of Salmonella typhimurium in milk. Int. J. Food Mycrobiol. 2018, 266, 251–256. [Google Scholar] [CrossRef]
- Fraaije, B.A.; Cools, H.J.; Fountaine, J.; Lovell, D.J.; Motteram, J.; West, J.S.; Lucas, J.A. Role of Ascospore in further Spread of Qoi resistant Cytochrome b Alleles (G143A) in field Populations of Mycosphaerella graminicola. Ecol. Epidemiol. 2005, 95, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benlloch, S.; Payà, A.; Alenda, C.; Llor, X.; Aranda, F.I.; Massutì, B. Detection of BRA V600E mutation in colorectal cancer: Comparison of automatic sequencing and real-time chemistry methodology. J. Mol. Diagn. 2006, 8, 540–543. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M. The JAK2 exon 12 mutations: A comprehensive review. Am. J. Hematol. 2011, 86, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Whitehall, V.; Tran, K.; Umapathy, A.; Grieu, F.; Hewitt, C.; Evans, T.J.; Ismail, T.; Li, W.Q.; Collins, P.; Ravetto, P.; et al. A multicenter blinded study to evaluate KRAS mutation testing methodologies in clinical setting. J. Mol. Diagn. 2009, 11, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Wet-lab Validated ddPCR Assay for Mutation Detection and Copy Number Variation. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_7144.pdf (accessed on 18 December 2021).
- Dodhia, K.N.; Cox, B.A.; Olivers, R.P.; Lopez-Ruiz, F.J. Rapid in situ quantification of the strobilurin resistance mutation G143A in the wheat pathogen Blumeria graminis f. sp. graminis. Sci. Rep. 2021, 11, 4526. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Site | Cultivar (Durum Wheat) | Fungicide Applications | |||
|---|---|---|---|---|---|---|
| Nr | Active Ingredients (Formulate) | Dose Rate/ha | Phenological Growth Stage | |||
| A | Dugliolo, Budrio (BO) (trial site) | San Carlo | from plots with 1 application | Pyraclostrobin + epoxiconazole (Opera New) | 2 L | BBCH39 |
| B | Sassoleone, Casalfiumanese (BO) | Miradoux | 0 | untreated | - | - |
| C | Medicina (BO) | San Carlo | 3 | tebuconazole + prochloraz (Orius P) | 1.7 L | BBCH32 |
| bixafen + tebuconazole (Zantara) | 1.5 L | BBCH39 | ||||
| prochloraz + propiconazole (Novel Duo) | 1.1 L | BBCH61 | ||||
| Groups | Copy Number of Alleles for Each Sample (S/R) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 | Sample 6 | Sample 7 | Sample 8 | Sample 9 | |
| Group 1 | 1000/1000 | 1000/250 | 1000/62.5 | 1000/15.6 | 1000/3.9 | 1000/0.9 | 1000/0.22 | 1000/0.056 | 1000/0 |
| Group 2 | 1000/1000 | 250/1000 | 62.5/1000 | 15.6/1000 | 3.9/1000 | 0.9/1000 | 0.22/1000 | 0.056/1000 | 0/1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battistini, G.; Gazzetti, K.; Collina, M. A New Approach: Determining cyt b G143A Allele Frequency in Zymoseptoria tritici by Digital Droplet PCR. Biology 2022, 11, 240. https://doi.org/10.3390/biology11020240
Battistini G, Gazzetti K, Collina M. A New Approach: Determining cyt b G143A Allele Frequency in Zymoseptoria tritici by Digital Droplet PCR. Biology. 2022; 11(2):240. https://doi.org/10.3390/biology11020240
Chicago/Turabian StyleBattistini, Greta, Katia Gazzetti, and Marina Collina. 2022. "A New Approach: Determining cyt b G143A Allele Frequency in Zymoseptoria tritici by Digital Droplet PCR" Biology 11, no. 2: 240. https://doi.org/10.3390/biology11020240
APA StyleBattistini, G., Gazzetti, K., & Collina, M. (2022). A New Approach: Determining cyt b G143A Allele Frequency in Zymoseptoria tritici by Digital Droplet PCR. Biology, 11(2), 240. https://doi.org/10.3390/biology11020240