
Evolutionary History of RNA Modifications at N6-Adenosine Originating from the R-M System in Eukaryotes and Prokaryotes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Similarity Searches
2.2. Alignment Generation and Phylogenetic Analyzes
2.3. Structural Inferion and Similarity Comparison
2.4. Structure-Based Alignments
3. Results
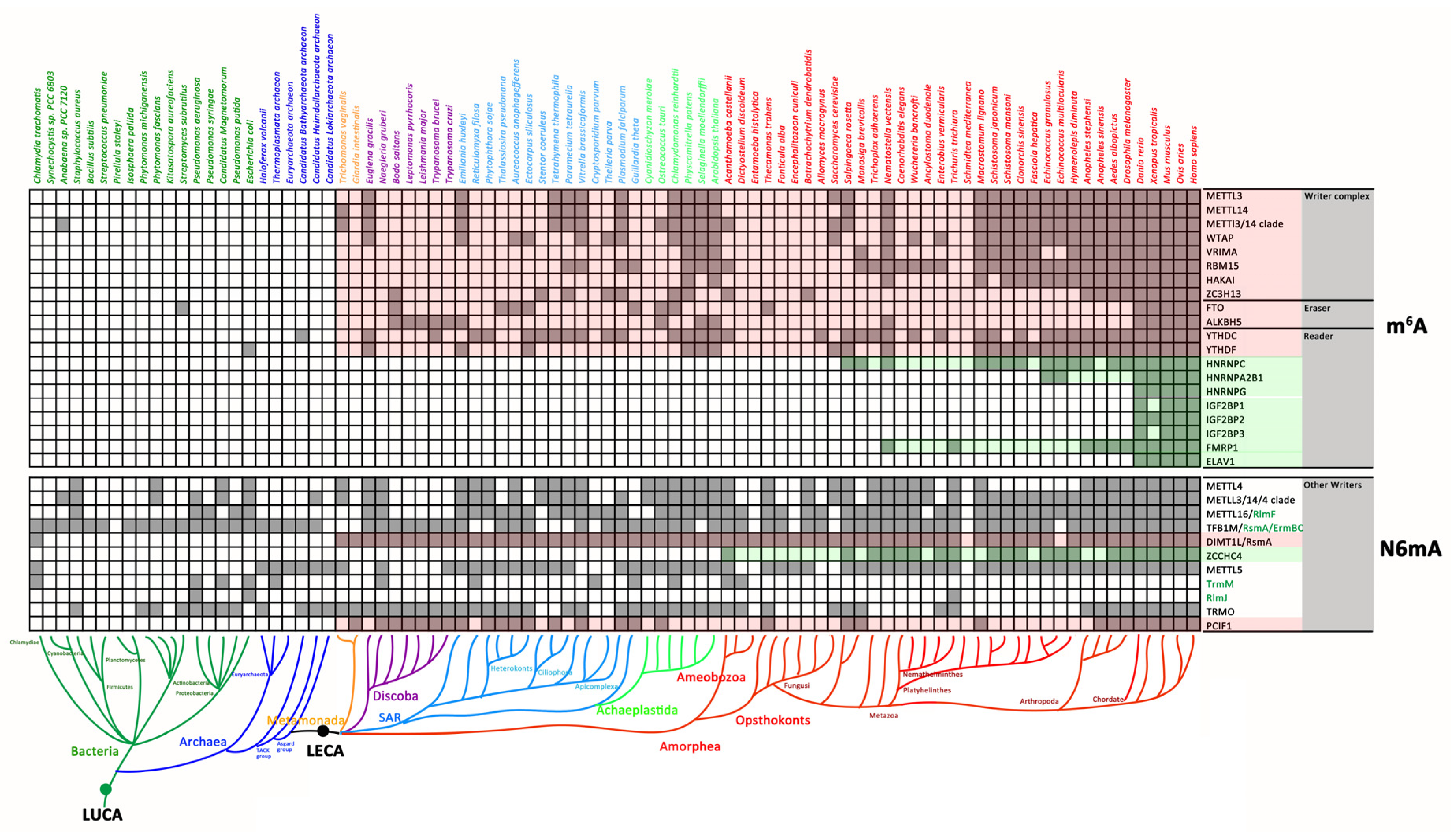
3.1. The Distribution of N6mA Modification Components in Prokaryotes and Eukaryotes
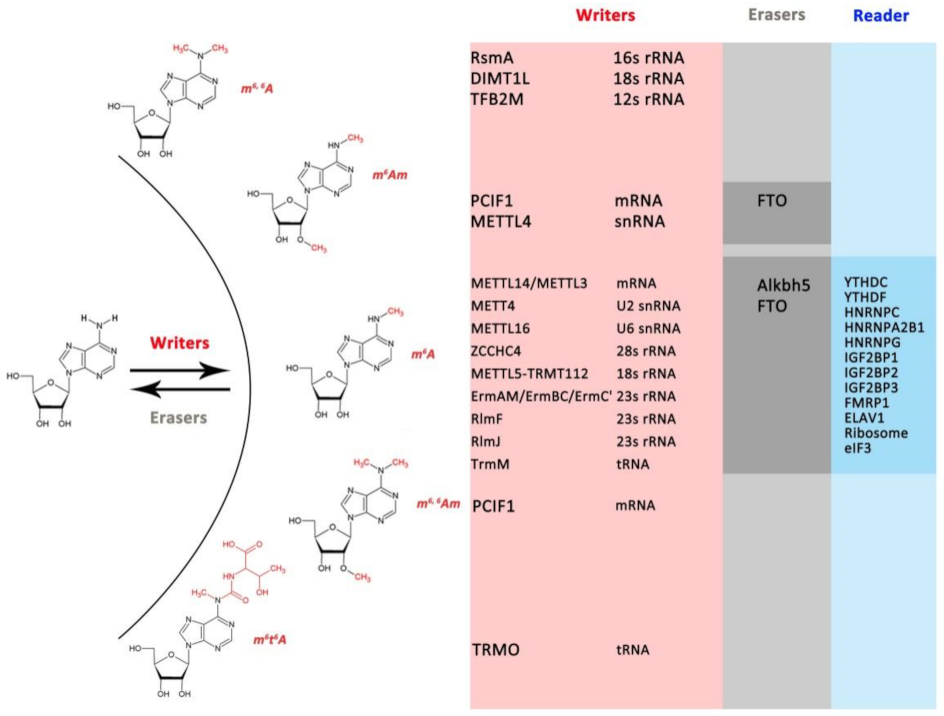
3.1.1. mRNA m6A Modification Components in Prokaryotes and Eukaryotes
3.1.2. The Distribution of Other N6mA-MTases in Prokaryotes and Eukaryotes
3.2. The Evolutionary History of the RNA N6mA Modification Machinery
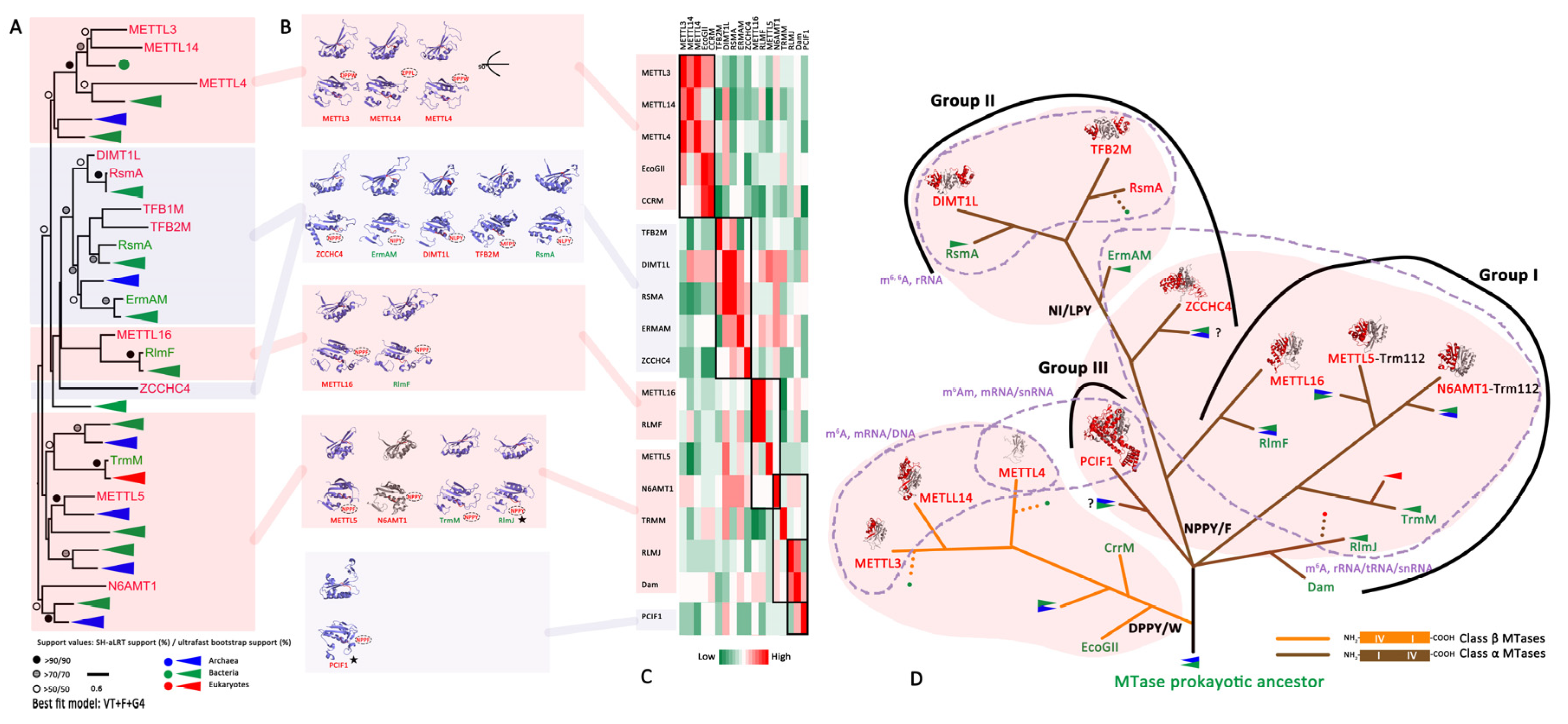
3.2.1. The Writer: RNA N6mA-MTases
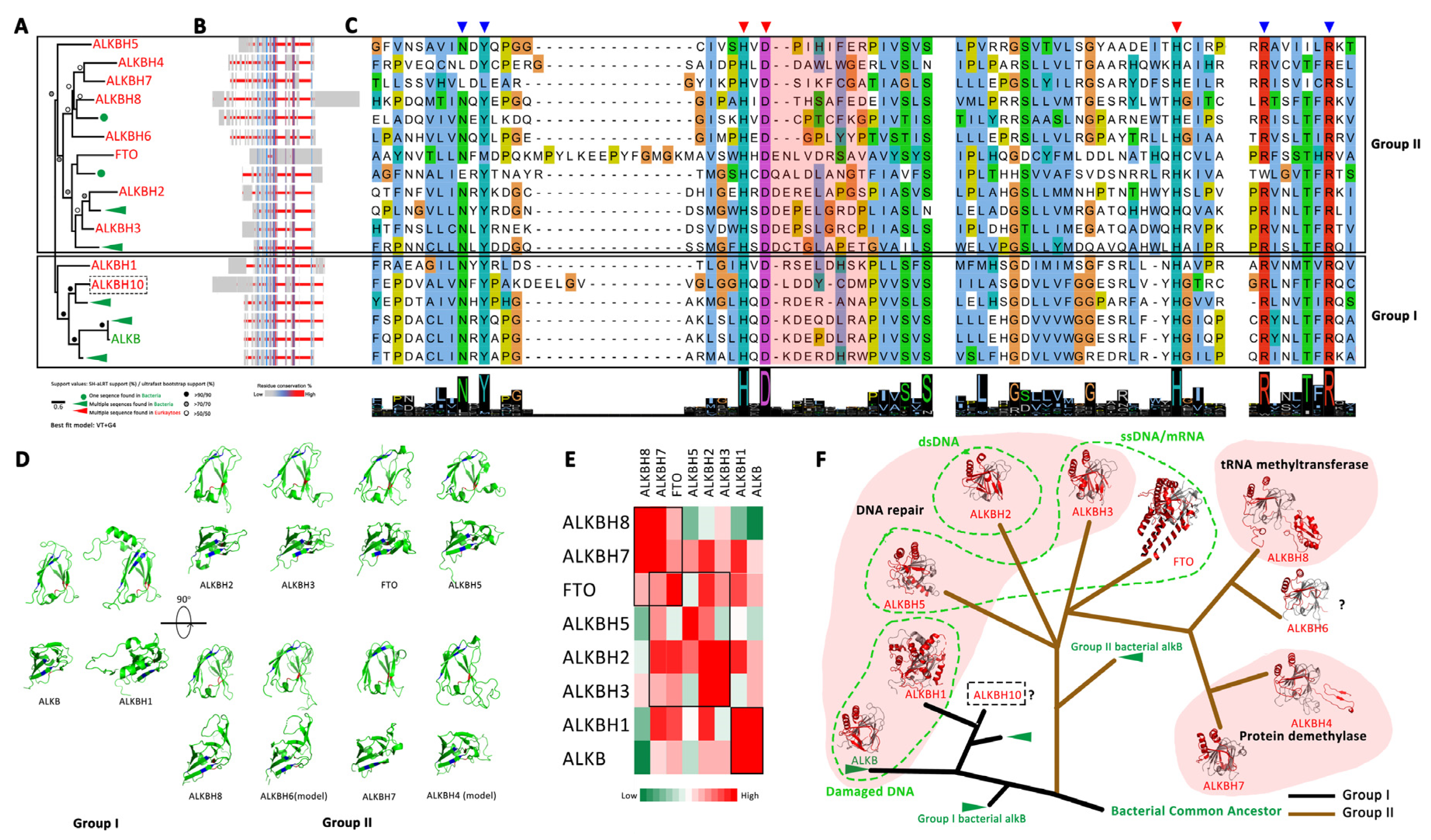
3.2.2. The Eraser: ALKBH Family Demethylases
3.2.3. The Reader: YTH Family
4. Discussion
4.1. Eukaryotic mRNA m6A Modification Components Originating from LECA
4.2. The Concerto of Gene Losses and Gains Involved in the mRNA m6A Modification Machinery
4.3. The Evolutionary History of the RNA N6mA Modification Machinery
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Balacco, D.L.; Soller, M. The m(6)A Writer: Rise of a Machine for Growing Tasks. Biochemistry 2019, 58, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Doudna, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F. RNA evolution and the origins of life. Nature 1989, 338, 217–224. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Song, J.; Liu, Y.; Song, C.X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [Green Version]
- Nachtergaele, S.; He, C. The emerging biology of RNA post-transcriptional modifications. RNA Biol. 2017, 14, 156–163. [Google Scholar] [CrossRef]
- Dubin, D.T.; Taylor, R.H. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. 1975, 2, 1653–1668. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Li, H.B.; Yin, Z.; Flavell, R.A. Recent advances in dynamic m6A RNA modification. Open Biol. 2016, 6, 160003. [Google Scholar] [CrossRef] [Green Version]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [Green Version]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Goh, Y.T.; Koh, C.W.Q.; Sim, D.Y.; Roca, X.; Goh, W.S.S. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 2020, 48, 9250–9261. [Google Scholar] [CrossRef]
- Sendinc, E.; Valle-Garcia, D.; Dhall, A.; Chen, H.; Henriques, T.; Navarrete-Perea, J.; Sheng, W.; Gygi, S.P.; Adelman, K.; Shi, Y. PCIF1 Catalyzes m6Am mRNA Methylation to Regulate Gene Expression. Mol. Cell 2019, 75, 620–630.e9. [Google Scholar] [CrossRef] [PubMed]
- Garcias Morales, D.; Reyes, J.L. A birds’-eye view of the activity and specificity of the mRNA m(6) A methyltransferase complex. Wiley Interdiscip. Rev. RNA 2021, 12, e1618. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Chen, J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Pandey, R.R.; Delfino, E.; Homolka, D.; Roithova, A.; Chen, K.M.; Li, L.; Franco, G.; Vagbo, C.B.; Taillebourg, E.; Fauvarque, M.O.; et al. The Mammalian Cap-Specific m(6)Am RNA Methyltransferase PCIF1 Regulates Transcript Levels in Mouse Tissues. Cell Rep. 2020, 32, 108038. [Google Scholar] [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m(6)A, m(6)Am, and m(1)A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol. Cell 2018, 71, 973–985.e5. [Google Scholar] [CrossRef] [Green Version]
- Pinto, R.; Vagbo, C.B.; Jakobsson, M.E.; Kim, Y.; Baltissen, M.P.; O’Donohue, M.F.; Guzman, U.H.; Malecki, J.M.; Wu, J.; Kirpekar, F.; et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 2020, 48, 830–846. [Google Scholar] [CrossRef]
- Rong, B.; Zhang, Q.; Wan, J.; Xing, S.; Dai, R.; Li, Y.; Cai, J.; Xie, J.; Song, Y.; Chen, J.; et al. Ribosome 18S m(6)A Methyltransferase METTL5 Promotes Translation Initiation and Breast Cancer Cell Growth. Cell Rep. 2020, 33, 108544. [Google Scholar] [CrossRef]
- Bangs, J.D.; Crain, P.F.; Hashizume, T.; McCloskey, J.A.; Boothroyd, J.C. Mass spectrometry of mRNA cap 4 from trypanosomatids reveals two novel nucleosides. J. Biol. Chem. 1992, 267, 9805–9815. [Google Scholar] [CrossRef]
- Seo, K.W.; Kleiner, R.E. Mechanisms of epitranscriptomic gene regulation. Biopolymers 2021, 112, e23403. [Google Scholar] [CrossRef]
- Woodcock, C.B.; Horton, J.R.; Zhang, X.; Blumenthal, R.M.; Cheng, X. Beta class amino methyltransferases from bacteria to humans: Evolution and structural consequences. Nucleic Acids Res. 2020, 48, 10034–10044. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, V.; Koonin, E.V.; Aravind, L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002, 30, 1427–1464. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Rendon, J.; Cardenas-Brito, S.; Pineda, J.D.; Corredor, M.; Benitez-Paez, A. Evolutionary and sequence-based relationships in bacterial AdoMet-dependent non-coding RNA methyltransferases. BMC Res. Notes 2014, 7, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujnicki, J.M.; Feder, M.; Radlinska, M.; Blumenthal, R.M. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J. Mol. Evol. 2002, 55, 431–444. [Google Scholar] [CrossRef]
- Iyer, L.M.; Zhang, D.; Aravind, L. Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. BioEssays 2016, 38, 27–40. [Google Scholar] [CrossRef]
- Xu, B.; Liu, D.; Wang, Z.; Tian, R.; Zuo, Y. Multi-substrate selectivity based on key loops and non-homologous domains: New insight into ALKBH family. Cell. Mol. Life Sci. CMLS 2021, 78, 129–141. [Google Scholar] [CrossRef]
- Degner, E.C.; Ahmed-Braimah, Y.H.; Borziak, K.; Wolfner, M.F.; Harrington, L.C.; Dorus, S. Proteins, Transcripts, and Genetic Architecture of Seminal Fluid and Sperm in the Mosquito Aedes aegypti. Mol. Cell. Proteom. MCP 2019, 18 (Suppl. S1), S6–S22. [Google Scholar] [CrossRef]
- Leismann, J.; Spagnuolo, M.; Pradhan, M.; Wacheul, L.; Vu, M.A.; Musheev, M.; Mier, P.; Andrade-Navarro, M.A.; Graille, M.; Niehrs, C.; et al. The 18S ribosomal RNA m(6) A methyltransferase Mettl5 is required for normal walking behavior in Drosophila. EMBO Rep. 2020, 21, e49443. [Google Scholar] [CrossRef]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Stover, B.C.; Muller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version Open-Source. Available online: https://github.com/schrodinger/pymol-open-source (accessed on 13 March 2021).
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Miyauchi, K.; Ikeuchi, Y.; Thiaville, P.C.; Crecy-Lagard, V.; Suzuki, T. Discovery of the beta-barrel-type RNA methyltransferase responsible for N6-methylation of N6-threonylcarbamoyladenosine in tRNAs. Nucleic Acids Res. 2014, 42, 9350–9365. [Google Scholar] [CrossRef] [Green Version]
- Bheemanaik, S.; Reddy, Y.V.; Rao, D.N. Structure, function and mechanism of exocyclic DNA methyltransferases. Biochem. J. 2006, 399, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Goedecke, K.; Pignot, M.; Goody, R.S.; Scheidig, A.J.; Weinhold, E. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat. Struct. Biol. 2001, 8, 121–125. [Google Scholar] [CrossRef]
- Casadesus, J. Bacterial DNA Methylation and Methylomes. Adv. Exp. Med. Biol. 2016, 945, 35–61. [Google Scholar] [CrossRef]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. MMBR 2013, 77, 53–72. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. Regulation of genetic flux between bacteria by restriction-modification systems. Proc. Natl. Acad. Sci. USA 2016, 113, 5658–5663. [Google Scholar] [CrossRef] [Green Version]
- Murray, I.A.; Morgan, R.D.; Luyten, Y.; Fomenkov, A.; Correa, I.R., Jr.; Dai, N.; Allaw, M.B.; Zhang, X.; Cheng, X.; Roberts, R.J. The non-specific adenine DNA methyltransferase M.EcoGII. Nucleic Acids Res. 2018, 46, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar] [PubMed]
- Woodcock, C.B.; Yu, D.; Hajian, T.; Li, J.; Huang, Y.; Dai, N.; Correa, I.R., Jr.; Wu, T.; Vedadi, M.; Zhang, X.; et al. Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 2019, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizabal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA Methylation on N6-Adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, Z.; Zhang, Q.; Li, B.; Lu, C.; Li, W.; Cheng, T.; Xia, Q.; Zhao, P. DNA methylation on N6-adenine in lepidopteran Bombyx mori. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 815–825. [Google Scholar] [CrossRef]
- Malone, T.; Blumenthal, R.M.; Cheng, X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995, 253, 618–632. [Google Scholar] [CrossRef]
- van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Shi, Y.; Zhang, T.; Ye, J.; Ding, J. Structural insight into human N6amt1-Trm112 complex functioning as a protein methyltransferase. Cell Discov. 2019, 5, 51. [Google Scholar] [CrossRef]
- Woodcock, C.B.; Yu, D.; Zhang, X.; Cheng, X. Human HemK2/KMT9/N6AMT1 is an active protein methyltransferase, but does not act on DNA in vitro, in the presence of Trm112. Cell Discov. 2019, 5, 50. [Google Scholar] [CrossRef]
- Akichika, S.; Hirano, S.; Shichino, Y.; Suzuki, T.; Nishimasu, H.; Ishitani, R.; Sugita, A.; Hirose, Y.; Iwasaki, S.; Nureki, O.; et al. Cap-specific terminal N (6)-methylation of RNA by an RNA polymerase II-associated methyltransferase. Science 2019, 363, 6423. [Google Scholar] [CrossRef]
- Van Deuren, V.; Plessers, S.; Robben, J. Structural determinants of nucleobase modification recognition in the AlkB family of dioxygenases. DNA Repair 2020, 96, 102995. [Google Scholar] [CrossRef] [PubMed]
- Alemu, A.E.; He, C.; Klungland, A. ALKBHs-facilitated RNA modifications and de-modifications. DNA Repair 2016, 44, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB Family of Fe(II)/alpha-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J. Biol. Chem. 2015, 290, 20734–20742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.; Meng, B. Prediction of the molecular boundary and functionality of novel viral AlkB domains using homology modelling and principal component analysis. J. Gen. Virol. 2019, 100, 691–703. [Google Scholar] [CrossRef]
- Li, M.M.; Nilsen, A.; Shi, Y.; Fusser, M.; Ding, Y.H.; Fu, Y.; Liu, B.; Niu, Y.; Wu, Y.S.; Huang, C.M.; et al. ALKBH4-dependent demethylation of actin regulates actomyosin dynamics. Nat. Commun. 2013, 4, 1832. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; He, Q.; Feng, C.; Liu, Y.; Deng, Z.; Qi, X.; Wu, W.; Mei, P.; Chen, Z. The atomic resolution structure of human AlkB homolog 7 (ALKBH7), a key protein for programmed necrosis and fat metabolism. J. Biol. Chem. 2014, 289, 27924–27936. [Google Scholar] [CrossRef] [Green Version]
- van den Born, E.; Vagbo, C.B.; Songe-Moller, L.; Leihne, V.; Lien, G.F.; Leszczynska, G.; Malkiewicz, A.; Krokan, H.E.; Kirpekar, F.; Klungland, A.; et al. ALKBH8-mediated formation of a novel diastereomeric pair of wobble nucleosides in mammalian tRNA. Nat. Commun. 2011, 2, 172. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Liao, S.; Sun, H.; Xu, C. YTH Domain: A Family of N(6)-methyladenosine (m(6)A) Readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef]
- Zhang, Z.; Theler, D.; Kaminska, K.H.; Hiller, M.; de la Grange, P.; Pudimat, R.; Rafalska, I.; Heinrich, B.; Bujnicki, J.M.; Allain, F.H.; et al. The YTH domain is a novel RNA binding domain. J. Biol. Chem. 2010, 285, 14701–14710. [Google Scholar] [CrossRef] [Green Version]
- Stoilov, P.; Rafalska, I.; Stamm, S. YTH: A new domain in nuclear proteins. Trends Biochem. Sci. 2002, 27, 495–497. [Google Scholar] [CrossRef]
- Hosford, C.J.; Bui, A.Q.; Chappie, J.S. The structure of the Thermococcus gammatolerans McrB N-terminal domain reveals a new mode of substrate recognition and specificity among McrB homologs. J. Biol. Chem. 2020, 295, 743–756. [Google Scholar] [CrossRef]
- Woodcock, C.B.; Horton, J.R.; Zhou, J.; Bedford, M.T.; Blumenthal, R.M.; Zhang, X.; Cheng, X. Biochemical and structural basis for YTH domain of human YTHDC1 binding to methylated adenine in DNA. Nucleic Acids Res. 2020, 48, 10329–10341. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, K.; Ahmed, H.; Loppnau, P.; Schapira, M.; Min, J. Structural Basis for the Discriminative Recognition of N6-Methyladenosine RNA by the Human YT521-B Homology Domain Family of Proteins. J. Biol. Chem. 2015, 290, 24902–24913. [Google Scholar] [CrossRef] [Green Version]
- Daubin, V.; Szollosi, G.J. Horizontal Gene Transfer and the History of Life. Cold Spring Harb. Perspect. Biol. 2016, 8, a018036. [Google Scholar] [CrossRef] [Green Version]
- Husnik, F.; McCutcheon, J.P. Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol. 2018, 16, 67–79. [Google Scholar] [CrossRef]
- Lallemand, T.; Leduc, M.; Landes, C.; Rizzon, C.; Lerat, E. An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice. Genes 2020, 11, 1046. [Google Scholar] [CrossRef]
- A von der Dunk, S.H.; Snel, B. Recurrent sequence evolution after independent gene duplication. BMC Evol. Biol. 2020, 20, 98. [Google Scholar] [CrossRef]
- De Grassi, A.; Lanave, C.; Saccone, C. Genome duplication and gene-family evolution: The case of three OXPHOS gene families. Gene 2008, 421, 1–6. [Google Scholar] [CrossRef]
- Sheridan, P.O.; Raguideau, S.; Quince, C.; Holden, J.; Zhang, L.; Thames, C.; Williams, T.A.; Gubry-Rangin, C. Gene duplication drives genome expansion in a major lineage of Thaumarchaeota. Nat. Commun. 2020, 11, 5494. [Google Scholar] [CrossRef]
- Albalat, R.; Canestro, C. Evolution by gene loss. Nat. Rev. Genet. 2016, 17, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gu, L.; Orellana, E.A.; Wang, Y.; Guo, J.; Liu, Q.; Wang, L.; Shen, Z.; Wu, H.; Gregory, R.I.; et al. METTL4 is an snRNA m(6)Am methyltransferase that regulates RNA splicing. Cell Res. 2020, 30, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Wang, L.; Chen, H.; Hong, J.; Shen, Z.; Dhall, A.; Lao, T.; Liu, C.; Wang, Z.; Xu, Y.; et al. CG14906 (mettl4) mediates m(6)A methylation of U2 snRNA in Drosophila. Cell Discov. 2020, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, S.; Jiang, H.; Zhang, Y.; Guo, X.; Wang, Y. Differential m6A methylomes between two major life stages allows potential regulations in Trypanosoma brucei. Biochem. Biophys. Res. Commun. 2019, 508, 1286–1290. [Google Scholar] [CrossRef]
- Lence, T.; Soller, M.; Roignant, J.Y. A fly view on the roles and mechanisms of the m(6)A mRNA modification and its players. RNA Biol. 2017, 14, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Iranzo, J.; Wolf, Y.I.; Koonin, E.V.; Sela, I. Gene gain and loss push prokaryotes beyond the homologous recombination barrier and accelerate genome sequence divergence. Nat. Commun. 2019, 10, 5376. [Google Scholar] [CrossRef] [Green Version]
- Chauve, C.; Doyon, J.P.; El-Mabrouk, N. Gene family evolution by duplication, speciation, and loss. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2008, 15, 1043–1062. [Google Scholar] [CrossRef] [Green Version]
- Huelsmann, M.; Hecker, N.; Springer, M.S.; Gatesy, J.; Sharma, V.; Hiller, M. Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Sci. Adv. 2019, 5, eaaw6671. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Chen, K.; Luo, G.Z.; Weng, X.; Ji, Q.; Zhou, T.; He, C. Widespread occurrence of N6-methyladenosine in bacterial mRNA. Nucleic Acids Acids Res. 2015, 43, 6557–6567. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Cao, J.; Zhang, H.; Yin, J. Evolutionary History of RNA Modifications at N6-Adenosine Originating from the R-M System in Eukaryotes and Prokaryotes. Biology 2022, 11, 214. https://doi.org/10.3390/biology11020214
Liu C, Cao J, Zhang H, Yin J. Evolutionary History of RNA Modifications at N6-Adenosine Originating from the R-M System in Eukaryotes and Prokaryotes. Biology. 2022; 11(2):214. https://doi.org/10.3390/biology11020214
Chicago/Turabian StyleLiu, Congshan, Jianping Cao, Haobing Zhang, and Jianhai Yin. 2022. "Evolutionary History of RNA Modifications at N6-Adenosine Originating from the R-M System in Eukaryotes and Prokaryotes" Biology 11, no. 2: 214. https://doi.org/10.3390/biology11020214
APA StyleLiu, C., Cao, J., Zhang, H., & Yin, J. (2022). Evolutionary History of RNA Modifications at N6-Adenosine Originating from the R-M System in Eukaryotes and Prokaryotes. Biology, 11(2), 214. https://doi.org/10.3390/biology11020214