
Combined Treatment with KV Channel Inhibitor 4-Aminopyridine and either γ-Cystathionine Lyase Inhibitor β-Cyanoalanine or Epinephrine Restores Blood Pressure, and Improves Survival in the Wistar Rat Model of Anaphylactic Shock

 ,
,  , , , ,
, , , ,  ,
,  ,
, 
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
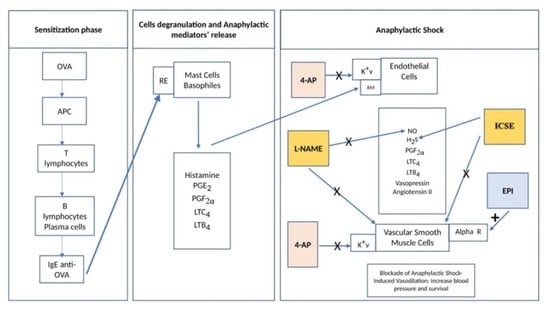
1. Introduction
2. Materials and Methods
2.1. Anaphylactic Shock Model
2.2. Treatment Groups
2.3. Plasma Mediator Concentrations
2.4. Statistical Analysis
3. Results
3.1. Inhibitors of NO Biosynthesis, H2S Biosynthesis, and Kv Channels Attenuated Systemic Hypotension and Increased Survival in AS
3.2. NO and H2S Inhibitors Enhance Early Protective Effects of 4-AP in Anaphylactic Shock
3.3. Combined 4-AP Treatments Increase Time-Integrated MAP (AUC-MAP)
3.4. Metabolic Acidosis Was Not Rescued by Treatment
3.5. NO, H2S and Kv Inhibitors Differentially Alter Oxidative Stress
3.6. Inhibitors of NO, H2S Biosynthesis, or Kv Channels Differentially Alter Plasma Inflammatory Mediators
3.7. AP Increases Angiotensin II and Vasopressin Plasma Levels in the Rat AS Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Warren, C.M.; Smith, B.M.; Jiang, J.; Blumenstock, J.A.; Davis, M.M.; Schleimer, R.P.; Nadeau, K.C. Prevalence and severity of food allergies among US adults. JAMA Netw. Open 2019, 2, e185630. [Google Scholar] [CrossRef] [PubMed]
- Takazawa, T.; Mitsuhata, H.; Mertes, P.M. Sugammadex and rocuronium-induced anaphylaxis. J. Anesth. 2016, 30, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Galletly, D.; Treuren, B. Anaphylactoid reactions during anaesthesia: Seven years’ experience of intradermal testing. Anaesthesia 1985, 40, 329–333. [Google Scholar] [CrossRef]
- Savic, L.C.; Kaura, V.; Yusaf, M.; Hammond-Jones, A.-M.; Jackson, R.; Howell, S.; Savic, S.; Hopkins, P.M. Incidence of suspected perioperative anaphylaxis: A multicenter snapshot study. J. Allergy Clin. Immunol. Pract. 2015, 3, 454–455.e1. [Google Scholar] [CrossRef]
- Gibbs, N.; Sadleir, P.; Clarke, R.; Platt, P. Survival from perioperative anaphylaxis in Western Australia 2000–2009. Br. J. Anaesth. 2013, 111, 589–593. [Google Scholar] [CrossRef]
- Charuluxananan, S.; Punjasawadwong, Y.; Suraseranivongse, S.; Srisawasdi, S.; Kyokong, O.; Chinachoti, T.; Chanchayanon, T.; Rungreungvanich, M.; Thienthong, S.; Sirinan, C. The Thai Anesthesia Incidents Study (THAI Study) of anesthetic outcomes: II. Anesthetic profiles and adverse events. J. Med. Assoc. Thail. Chotmaihet Thangphaet 2005, 88, S14–S29. [Google Scholar]
- Escolano, F.; Valero, A.; Huguet, J.; Baxarias, P.; De Molina, M.; Castro, A.; Granel, C.; Sanosa, J.; Bartolomé, B. Prospective epidemiologic study of perioperative anaphylactoid reactions occurring in Catalonia (1996-7). Rev. Esp. Anestesiol. Reanim. 2002, 49, 286–293. [Google Scholar]
- Mitsuhata, H.; Matsumoto, S.; Hasegawa, J. The epidemiology and clinical features of anaphylactic and anaphylactoid reactions in the perioperative period in Japan. Masui. Jpn. J. Anesthesiol. 1992, 41, 1664–1669. [Google Scholar]
- Laxenaire, M.C.; Moneret-Vautrin, D.A.; Boileau, S.; Moeller, R. Adverse reactions to intravenous agents in anaesthesia in France. Klin. Wochenschr. 1982, 60, 1006–1009. [Google Scholar] [CrossRef]
- Watkins, J. Adverse anaesthetic reactions: An update from a proposed national reporting and advisory service. Anaesthesia 1985, 40, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Tacquard, C.; Collange, O.; Gomis, P.; Malinovsky, J.M.; Petitpain, N.; Demoly, P.; Nicoll, S.; Mertes, P. Anaesthetic hypersensitivity reactions in France between 2011 and 2012: The 10th GERAP epidemiologic survey. Acta Anaesthesiol. Scand. 2017, 61, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Mertes, P.; Petitpain, N.; Hasdenteufel, F.; Malinovsky, J. Hypersensitivity reactions during anesthesia. Results from the ninth French survey (2005–2007). Minerva Anestesiol. 2012, 78, 868. [Google Scholar] [PubMed]
- Volcheck, G.W.; Mertes, P.M. Local and general anesthetics immediate hypersensitivity reactions. Immunol. Allergy Clin. 2014, 34, 525–546. [Google Scholar] [CrossRef] [PubMed]
- Mertes, P.M.; Volcheck, G.W.; Garvey, L.H.; Takazawa, T.; Platt, P.R.; Guttormsen, A.B.; Tacquard, C. Epidemiology of perioperative anaphylaxis. La Presse Médicale 2016, 45, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.M.; More, D. The epidemiology and clinical features of anaphylactic reactions in anaesthesia. Anaesth. Intensive Care 1981, 9, 226–234. [Google Scholar] [CrossRef]
- Harper, N.; Cook, T.; Garcez, T.; Farmer, L.; Floss, K.; Marinho, S.; Torevell, H.; Warner, A.; Ferguson, K.; Hitchman, J. Anaesthesia, surgery, and life-threatening allergic reactions: Epidemiology and clinical features of perioperative anaphylaxis in the 6th National Audit Project (NAP6). Br. J. Anaesth. 2018, 121, 159–171. [Google Scholar] [CrossRef]
- Reitter, M.; Petitpain, N.; Latarche, C.; Cottin, J.; Massy, N.; Demoly, P.; Gillet, P.; Mertes, P.; French Network of Regional Pharmacovigilance Centres. Fatal anaphylaxis with neuromuscular blocking agents: A risk factor and management analysis. Allergy 2014, 69, 954–959. [Google Scholar] [CrossRef]
- Hepner, D.L.; Castells, M.C. Anaphylaxis during the perioperative period. Anesth. Analg. 2003, 97, 1381–1395. [Google Scholar] [CrossRef]
- Garvey, L.H.; Belhage, B.; Krøigaard, M.; Husum, B.; Malling, H.-J.; Mosbech, H. Treatment with epinephrine (adrenaline) in suspected anaphylaxis during anesthesia in Denmark. J. Am. Soc. Anesthesiol. 2011, 115, 111–116. [Google Scholar] [CrossRef]
- Garvey, L.H.; Dewachter, P.; Hepner, D.L.; Mertes, P.M.; Voltolini, S.; Clarke, R.; Cooke, P.; Garcez, T.; Guttormsen, A.B.; Ebo, D.G. Management of suspected immediate perioperative allergic reactions: An international overview and consensus recommendations. Br. J. Anaesth. 2019, 123, e50–e64. [Google Scholar] [CrossRef] [PubMed]
- Triggiani, M.; Patella, V.; Staiano, R.; Granata, F.; Marone, G. Allergy and the cardiovascular system. Clin. Exp. Immunol. 2008, 153 (Suppl. S1), 7–11. [Google Scholar] [CrossRef] [PubMed]
- Golden, D.B. What is anaphylaxis? Curr. Opin. Allergy Clin. Immunol. 2007, 7, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Low, I.; Stables, S. Anaphylactic deaths in Auckland, New Zealand: A review of coronial autopsies from 1985 to 2005. Pathology 2006, 38, 328–332. [Google Scholar] [CrossRef] [PubMed]
- De Bisschop, M.B.; Bellou, A. Anaphylaxis. Curr. Opin. Crit. Care 2012, 18, 308–317. [Google Scholar] [CrossRef]
- Bellou, A.; Manel, J.; Samman-Kaakaji, H.; De Korwin, J.D.; Moneret-Vautrin, D.A.; Bollaert, P.E.; Lambert, H. Spectrum of acute allergic diseases in an emergency department: An evaluation of one years’ experience. Emerg. Med. 2003, 15, 341–347. [Google Scholar] [CrossRef]
- Brown, S.G.; Stone, S.F.; Fatovich, D.M.; Burrows, S.A.; Holdgate, A.; Celenza, A.; Coulson, A.; Hartnett, L.; Nagree, Y.; Cotterell, C. Anaphylaxis: Clinical patterns, mediator release, and severity. J. Allergy Clin. Immunol. 2013, 132, 1141–1149.e5. [Google Scholar] [CrossRef]
- Worm, M.; Moneret-Vautrin, A.; Scherer, K.; Lang, R.; Fernandez-Rivas, M.; Cardona, V.; Kowalski, M.; Jutel, M.; Poziomkowska-Gesicka, I.; Papadopoulos, N. First European data from the network of severe allergic reactions (NORA). Allergy 2014, 69, 1397–1404. [Google Scholar] [CrossRef]
- Muñoz-Cano, R.; Pascal, M.; Araujo, G.; Goikoetxea, M.; Valero, A.L.; Picado, C.; Bartra, J. Mechanisms, cofactors, and augmenting factors involved in anaphylaxis. Front. Immunol. 2017, 8, 1193. [Google Scholar] [CrossRef]
- Daff, S. NO synthase: Structures and mechanisms. Nitric Oxide 2010, 23, 1–11. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.; DeSantiago, B.; Lee, D.Y.; Yang, G.; Kim, J.Y.; Foster, D.B.; Chan-Li, Y.; Horton, M.R.; Panettieri, R.A.; Wang, R. H2S relaxes isolated human airway smooth muscle cells via the sarcolemmal KATP channel. Biochem. Biophys. Res. Commun. 2014, 446, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Calvert, J.W.; Butler, J.; Lefer, D.J. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica 2014, 2014, 768607. [Google Scholar] [CrossRef]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L.; Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, S.; Nemmar, A.; Aburawi, E.H.; Kazzam, E.E.; Abdulle, A.; Bellou, M.; Bellou, A. Glyburide, a K+ ATP channel blocker, improves hypotension and survival in anaphylactic shock induced in Wistar rats sensitized to ovalbumin. Eur. J. Pharmacol. 2013, 720, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Bellou, A.; Al-Hammadi, S.; Aburawi, E.H.; Dhanasekaran, S.; Nemmar, A.; Oulhaj, A.; Shafiuallah, M.; Zerrouki, M.; Yasin, J.; Bellou, L. 4-Aminopyridine, a blocker of Voltage-dependent K+ channels, restores blood pressure and improves survival in the Wistar rat model of anaphylactic shock. Crit. Care Med. 2016, 44, e1082–e1089. [Google Scholar] [CrossRef]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol.-Cell Physiol. 1995, 268, C799–C822. [Google Scholar] [CrossRef] [PubMed]
- Köhler, R.; Ruth, P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflügers Arch.-Eur. J. Physiol. 2010, 459, 969–976. [Google Scholar] [CrossRef]
- Beech, D.; Bolton, T. A voltage-dependent outward current with fast kinetics in single smooth muscle cells isolated from rabbit portal vein. J. Physiol. 1989, 412, 397–414. [Google Scholar] [CrossRef]
- Dongo, E.; Beliczai-Marosi, G.; Dybvig, A.S.; Kiss, L. The mechanism of action and role of hydrogen sulfide in the control of vascular tone. Nitric Oxide 2018, 81, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Roviezzo, F.; Bertolino, A.; Sorrentino, R.; Terlizzi, M.; Matteis, M.; Calderone, V.; Mattera, V.; Martelli, A.; Spaziano, G.; Pinto, A. Hydrogen sulfide inhalation ameliorates allergen induced airway hypereactivity by modulating mast cell activation. Pharmacol. Res. 2015, 100, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lambden, S.; Creagh-Brown, B.C.; Hunt, J.; Summers, C.; Forni, L.G. Definitions and pathophysiology of vasoplegic shock. Crit. Care 2018, 22, 174. [Google Scholar] [CrossRef] [PubMed]
- Afroze, T.; Yang, G.; Khoshbin, A.; Tanwir, M.; Tabish, T.; Momen, A.; Husain, M. Calcium efflux activity of plasma membrane Ca2+ ATPase-4 (PMCA4) mediates cell cycle progression in vascular smooth muscle cells. J. Biol. Chem. 2014, 289, 7221–7231. [Google Scholar] [CrossRef]
- Shaikh, S.; Sarkar, J.; Pramanik, A.; Karmakar, K.; Chakraborti, S. Effect of m-calpain in PKCα-mediated proliferation of pulmonary artery smooth muscle cells by low dose of ouabain. Indian J. Biochem. Biophys. 2013, 50, 419–427. [Google Scholar]
- Fan, G.; Cui, Y.; Gollasch, M.; Kassmann, M. Elementary calcium signaling in arterial smooth muscle. Channels 2019, 13, 505–519. [Google Scholar] [CrossRef]
- Dinenno, F.A.; Joyner, M.J. α-Adrenergic control of skeletal muscle circulation at rest and during exercise in aging humans. Microcirculation 2006, 13, 329–341. [Google Scholar] [CrossRef]
- Petrof, B.J. Molecular pathophysiology of myofiber injury in deficiencies of the dystrophin-glycoprotein complex. Am. J. Phys. Med. Rehabil. 2002, 81, S162–S174. [Google Scholar] [CrossRef]
- Raven, P. Recent advances in baroreflex control of blood pressure during exercise in humans: An overview. Med. Sci. Sport. Exerc. 2008, 40, 2033. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, D.R.; March, K.L.; Lash, J.A.; Adam, L.P.; Wilensky, R.L. Vascular smooth muscle. A review of the molecular basis of contractility. Circulation 1991, 83, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Busse, R. Signal transduction of eNOS activation. Cardiovasc. Res. 1999, 43, 532–541. [Google Scholar] [CrossRef]
- Vallance, P.; Collier, J.; Moncada, S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 1989, 334, 997–1000. [Google Scholar] [CrossRef]
- Moncada, S. Nitric oxide: Physiology, pathophysiology and pharmacology. Pharm. Rev. 1991, 43, 109–142. [Google Scholar]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the⋅ NO/cGMP signaling pathway. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1411, 334–350. [Google Scholar] [CrossRef]
- Kimmoun, A.; Ducrocq, N.; Levy, B. Mechanisms of vascular hyporesponsiveness in septic shock. Curr. Vasc. Pharmacol. 2013, 11, 139–149. [Google Scholar]
- Bae, S.W.; Kim, H.S.; Cha, Y.N.; Park, Y.S.; Jo, S.A.; Jo, I. Rapid increase in endothelial nitric oxide production by bradykinin is mediated by protein kinase A signaling pathway. Biochem. Biophys. Res. Commun. 2003, 306, 981–987. [Google Scholar] [CrossRef]
- Butt, E.; Bernhardt, M.; Smolenski, A.; Kotsonis, P.; Fröhlich, L.G.; Sickmann, A.; Meyer, H.E.; Lohmann, S.M.; Schmidt, H.H. Endothelial nitric-oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J. Biol. Chem. 2000, 275, 5179–5187. [Google Scholar] [CrossRef]
- Perinotto, P.; Biggi, A.; Carra, N.; Orrico, A.; Valmadre, G.; Dall’aglio, P.; Novarini, A.; Montanari, A. Angiotensin II and prostaglandin interactions on systemic and renal effects of L-NAME in humans. J. Am. Soc. Nephrol. 2001, 12, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Lamas, S.; Marsden, P.A.; Li, G.K.; Tempst, P.; Michel, T. Endothelial nitric oxide synthase: Molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc. Natl. Acad. Sci. USA 1992, 89, 6348–6352. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhata, H.; Saitoh, J.; Hasome, N.; Takeuchi, H.; Horiguchi, Y.; Shimizu, R. Nitric oxide synthase inhibition is detrimental to cardiac function and promotes bronchospasm in anaphylaxis in rabbits. Shock 1995, 4, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Cauwels, A.; Janssen, B.; Buys, E.; Sips, P.; Brouckaert, P. Anaphylactic shock depends on PI3K and eNOS-derived NO. J. Clin. Investig. 2006, 116, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Okamoto, Y.; Yoshioka, K.; Du, W.; Takuwa, N.; Zhang, W.; Asano, M.; Shibamoto, T.; Takuwa, Y. Sphingosine-1-phosphate receptor 2 protects against anaphylactic shock through suppression of endothelial nitric oxide synthase in mice. J. Allergy Clin. Immunol. 2013, 132, 1205–1214.e9. [Google Scholar] [CrossRef] [PubMed]
- Bellou, A.; Lambert, H.; Gillois, P.; Montémont, C.; Gerard, P.; Vauthier, E.; Sainte-Laudy, J.; Longrois, D.; Gueant, J.L.; Mallié, J.P. Constitutive nitric oxide synthase inhibition combined with histamine and serotonin receptor blockade improves the initial ovalbumin-induced arterial hypotension but decreases the survival time in brown norway rats anaphylactic shock. Shock 2003, 19, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Vrankova, S.; Parohova, J.; Barta, A.; Janega, P.; Simko, F.; Pechanova, O. Effect of nuclear factor kappa B inhibition on L-NAME-induced hypertension and cardiovascular remodelling. J. Hypertens. 2010, 28, S45–S49. [Google Scholar] [CrossRef]
- Laukevičienė, A.; Uginčius, P.; Korotkich, I.; Lažauskas, R.; Kėvelaitis, E. Anaphylaxis of small arteries: Putative role of nitric oxide and prostanoids. Medicina 2010, 46, 38. [Google Scholar] [CrossRef]
- Al-Salam, S.; Aburawi, E.H.; Al-Hammadi, S.; Dhanasekaran, S.; Shafiuallah, M.; Yasin, J.; Sudhadevi, M.; Awwad, A.; Alper, S.L.; Kazzam, E.E. Cellular and Immunohistochemical Changes in Anaphylactic Shock Induced in the Ovalbumin-Sensitized Wistar Rat Model. Biomolecules 2019, 9, 101. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef]
- Brozovich, F.; Nicholson, C.; Degen, C.; Gao, Y.Z.; Aggarwal, M.; Morgan, K. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Miyahara, N.; Gelfand, E.W. The role of leukotriene B4 in allergic diseases. Allergol. Int. 2008, 57, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Hydrogen sulfide: A new EDRF. Kidney Int. 2009, 76, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 2000, 267, 129–133. [Google Scholar] [CrossRef]
- San Cheang, W.; Wong, W.T.; Shen, B.; Lau, C.W.; Tian, X.Y.; Tsang, S.Y.; Yao, X.; Chen, Z.Y.; Huang, Y. 4-aminopyridine-sensitive K+ channels contributes to NaHS-induced membrane hyperpolarization and relaxation in the rat coronary artery. Vasc. Pharmacol. 2010, 53, 94–98. [Google Scholar] [CrossRef]
- Hedegaard, E.; Nielsen, B.; Kun, A.; Hughes, A.; Krøigaard, C.; Mogensen, S.; Matchkov, V.; Fröbert, O.; Simonsen, U. K V7 channels are involved in hypoxia-induced vasodilatation of porcine coronary arteries. Br. J. Pharmacol. 2014, 171, 69–82. [Google Scholar] [CrossRef]
- Mok, Y.-Y.P.; Atan, M.S.B.M.; Ping, C.Y.; Jing, W.Z.; Bhatia, M.; Moochhala, S.; Moore, P.K. Role of hydrogen sulphide in haemorrhagic shock in the rat: Protective effect of inhibitors of hydrogen sulphide biosynthesis. Br. J. Pharmacol. 2004, 143, 881. [Google Scholar] [CrossRef]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen sulfide and cellular redox homeostasis. Oxidative Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef]
- Searcy, D.G.; Whitehead, J.P.; Maroney, M.J. Interaction of Cu, Zn superoxide dismutase with hydrogen sulfide. Arch. Biochem. Biophys. 1995, 318, 251–263. [Google Scholar] [CrossRef]
- Jeney, V.; Komódi, E.; Nagy, E.; Zarjou, A.; Vercellotti, G.M.; Eaton, J.W.; Balla, G.; Balla, J. Supression of hemin-mediated oxidation of low-density lipoprotein and subsequent endothelial reactions by hydrogen sulfide (H2S). Free. Radic. Biol. Med. 2009, 46, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Kan, J.; Liu, X.; Ma, F.; Tran, B.H.; Zou, Y.; Wang, S.; Zhu, Y.Z. Cardioprotective effects of a novel hydrogen sulfide agent–controlled release formulation of s-propargyl-cysteine on heart failure rats and molecular mechanisms. PLoS ONE 2013, 8, e69205. [Google Scholar] [CrossRef] [PubMed]
- Laggner, H.; Muellner, M.K.; Schreier, S.; Sturm, B.; Hermann, M.; Exner, M.; Laggner, H.; Muellner, M.K.; Schreier, S.; Sturm, B. Hydrogen sulphide: A novel physiological inhibitor of LDL atherogenic modification by HOCl. Free. Radic. Res. 2007, 41, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Shibamoto, T.; Kuda, Y.; Sun, L.; Tanida, M.; Kurata, Y. Angiotensin II and vasopressin are involved in the defense system against anaphylactic hypotension in anesthetized rats. Eur. J. Pharmacol. 2014, 731, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Umino, T.; Kusano, E.; Muto, S.; Akimoto, T.; Yanagiba, S.; Ono, S.; Amemiya, M.; Ando, Y.; Homma, S.; Ikeda, U. AVP inhibits LPS-and IL-1β-stimulated NO and cGMP via V1 receptor in cultured rat mesangial cells. Am. J. Physiol.-Ren. Physiol. 1999, 276, F433–F441. [Google Scholar] [CrossRef]
- Noguera, I.; Medina, P.; Segarra, G.; Martinez, M.; Aldasoro, M.; Vila, J.; Lluch, S. Potentiation by vasopressin of adrenergic vasoconstriction in the rat isolated mesenteric artery. Br. J. Pharmacol. 1997, 122, 431–438. [Google Scholar] [CrossRef]
- Iversen, B.M.; Arendshorst, W.J. ANG II and vasopressin stimulate calcium entry in dispersed smooth muscle cells of preglomerular arterioles. Am. J. Physiol.-Ren. Physiol. 1998, 274, F498–F508. [Google Scholar] [CrossRef]
- Wakatsuki, T.; Nakaya, Y.; Inoue, I. Vasopressin modulates K+-channel activities of cultured smooth muscle cells from porcine coronary artery. Am. J. Physiol.-Heart Circ. Physiol. 1992, 263, H491–H496. [Google Scholar] [CrossRef]
- Li, X.; Kribben, A.; Wieder, E.D.; Tsai, P.; Nemenoff, R.A.; Schrier, R.W. Inhibition of vasopressin action in vascular smooth muscle by the V1 antagonist OPC-21268. Hypertension 1994, 23, 217–222. [Google Scholar] [CrossRef]
- Hayabuchi, Y.; Standen, N.; Davies, N. Angiotensin II inhibits and alters kinetics of voltage-gated K+ channels of rat arterial smooth muscle. Am. J. Physiol.-Heart Circ. Physiol. 2001, 281, H2480–H2489. [Google Scholar] [CrossRef]
- Gronich, J.; Bonventre, J.; Nemenoff, R.A. Identification and characterization of a hormonally regulated form of phospholipase A2 in rat renal mesangial cells. J. Biol. Chem. 1988, 263, 16645–16651. [Google Scholar] [CrossRef]
- Welsh, C.J.; Schmeichel, K.; Cao, H.-T.; Chabbott, H. Vasopressin stimulates phospholipase D activity against phosphatidylcholine in vascular smooth muscle cells. Lipids 1990, 25, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Billah, M.; Eckel, S.; Mullmann, T.; Egan, R.; Siegel, M. Phosphatidylcholine hydrolysis by phospholipase D determines phosphatidate and diglyceride levels in chemotactic peptide-stimulated human neutrophils: Involvement of phosphatidate phosphohydrolase in signal transduction. J. Biol. Chem. 1989, 264, 17069–17077. [Google Scholar] [CrossRef]
- Khanna, A.; English, S.W.; Wang, X.S.; Ham, K.; Tumlin, J.; Szerlip, H.; Busse, L.W.; Altaweel, L.; Albertson, T.E.; Mackey, C. Angiotensin II for the treatment of vasodilatory shock. N. Engl. J. Med. 2017, 377, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Golub, M.; Nozawa, K.; Berger, M.; Knoll, E.; Yanagawa, N.; Natarajan, R.; Nadler, J.; Tuck, M. Selective inhibition of angiotensin II-mediated vasoconstriction by lipoxygenase blockade. Am. J. Physiol.-Heart Circ. Physiol. 1989, 257, H434–H443. [Google Scholar] [CrossRef] [PubMed]
- Stanke-Labesque, F.; Devillier, P.; Bedouch, P.; Cracowski, J.L.; Chavanon, O.; Bessard, G. Angiotensin II-induced contractions in human internal mammary artery: Effects of cyclooxygenase and lipoxygenase inhibition. Cardiovasc. Res. 2000, 47, 376–383. [Google Scholar] [CrossRef][Green Version]
- Stanke-Labesque, F.; Devillier, P.; Veitl, S.; Caron, F.; Cracowski, J.-L.; Bessard, G. Cysteinyl leukotrienes are involved in angiotensin II-induced contraction of aorta from spontaneously hypertensive rats. Cardiovasc. Res. 2001, 49, 152–160. [Google Scholar] [CrossRef]
- Shastri, S.; McNeill, J.R.; Wilson, T.W.; Poduri, R.; Kaul, C.; Gopalakrishnan, V. Cysteinyl leukotrienes mediate enhanced vasoconstriction to angiotensin II but not endothelin-1 in SHR. Am. J. Physiol.-Heart Circ. Physiol. 2001, 281, H342–H349. [Google Scholar] [CrossRef]
- Sade, K.; Schwartz, I.; Etkin, S.; Schwartzenberg, S.; Levo, Y.; Kivity, S. Expression of inducible nitric oxide synthase in a mouse model of anaphylaxis. J. Investig. Allergol. Clin. Immunol. 2007, 17, 379. [Google Scholar]
 ), L-NG-Nitroarginine methyl ester (L-NAME, filled triangles,
), L-NG-Nitroarginine methyl ester (L-NAME, filled triangles,  ), with 4-aminopyridine (4-AP, ★), epinephrine (EPI,
), with 4-aminopyridine (4-AP, ★), epinephrine (EPI,  ) or combination drug treatment BCA+4AP (
) or combination drug treatment BCA+4AP ( ), DPG+4AP (
), DPG+4AP ( ), L-NAME+4AP (Δ), EPI+4-AP (
), L-NAME+4AP (Δ), EPI+4-AP ( ) administered 1 min after induction of AS in sensitized rats by IV ovalbumin (OVA). MAP and HR were recorded every minute for 60 min post-OVA injection. Values are Mean ± S.E.M. The difference between NA and AS rats was significant (p < 0.0001, two-way analysis of variance, ANOVA) (A,B). The difference observed between AS and all treated groups except AS+ LNAME was significant (p < 0.0001) (A). The differences between AS and single drug treatments (4-AP, BCA, DPG, EPI) and two-drug treatments (BCA+4-AP, DPG+4-AP, EPI+4-AP) were significant (p < 0.0001) with L-NAME (p < 0.01) (B,C) Survival for each group, NA
) administered 1 min after induction of AS in sensitized rats by IV ovalbumin (OVA). MAP and HR were recorded every minute for 60 min post-OVA injection. Values are Mean ± S.E.M. The difference between NA and AS rats was significant (p < 0.0001, two-way analysis of variance, ANOVA) (A,B). The difference observed between AS and all treated groups except AS+ LNAME was significant (p < 0.0001) (A). The differences between AS and single drug treatments (4-AP, BCA, DPG, EPI) and two-drug treatments (BCA+4-AP, DPG+4-AP, EPI+4-AP) were significant (p < 0.0001) with L-NAME (p < 0.01) (B,C) Survival for each group, NA  , AS
, AS  , AS+EPI
, AS+EPI  , AS+LNAME
, AS+LNAME  , AS+BCA
, AS+BCA  , AS+DPG
, AS+DPG  , AS+4AP
, AS+4AP  , AS+ EPI+4AP
, AS+ EPI+4AP  , AS+DPG+4AP
, AS+DPG+4AP  , AS+LNAME+4AP
, AS+LNAME+4AP  , AS+BCA+4AP
, AS+BCA+4AP  . NA, non-allergic rats; AS, allergic rats; BCA, β-cyanoalanine; DPG, dl-propargylglycine; L-NAME, L-NG-Nitroarginine methyl ester; 4-AP, 4-aminopyridine; EPI, epinephrine. BCA+4-AP, β-cyanoalanine+4-aminopyridine; DPG+4-AP, dl-propargylglycine+4-aminopyridine; L-NAME+4-AP, L-NG-Nitroarginine methyl ester+4-aminopyridine; EPI+4-AP, epinephrine+4-aminopyridine.
), L-NG-Nitroarginine methyl ester (L-NAME, filled triangles, ), with 4-aminopyridine (4-AP, ★), epinephrine (EPI, ) or combination drug treatment BCA+4AP (), DPG+4AP (), L-NAME+4AP (Δ), EPI+4-AP () administered 1 min after induction of AS in sensitized rats by IV ovalbumin (OVA). MAP and HR were recorded every minute for 60 min post-OVA injection. Values are Mean ± S.E.M. The difference between NA and AS rats was significant (p < 0.0001, two-way analysis of variance, ANOVA) (A,B). The difference observed between AS and all treated groups except AS+ LNAME was significant (p < 0.0001) (A). The differences between AS and single drug treatments (4-AP, BCA, DPG, EPI) and two-drug treatments (BCA+4-AP, DPG+4-AP, EPI+4-AP) were significant (p < 0.0001) with L-NAME (p < 0.01) (B,C) Survival for each group, NA , AS , AS+EPI , AS+LNAME , AS+BCA , AS+DPG , AS+4AP , AS+ EPI+4AP , AS+DPG+4AP , AS+LNAME+4AP , AS+BCA+4AP . NA, non-allergic rats; AS, allergic rats; BCA, β-cyanoalanine; DPG, dl-propargylglycine; L-NAME, L-NG-Nitroarginine methyl ester; 4-AP, 4-aminopyridine; EPI, epinephrine. BCA+4-AP, β-cyanoalanine+4-aminopyridine; DPG+4-AP, dl-propargylglycine+4-aminopyridine; L-NAME+4-AP, L-NG-Nitroarginine methyl ester+4-aminopyridine; EPI+4-AP, epinephrine+4-aminopyridine.
. NA, non-allergic rats; AS, allergic rats; BCA, β-cyanoalanine; DPG, dl-propargylglycine; L-NAME, L-NG-Nitroarginine methyl ester; 4-AP, 4-aminopyridine; EPI, epinephrine. BCA+4-AP, β-cyanoalanine+4-aminopyridine; DPG+4-AP, dl-propargylglycine+4-aminopyridine; L-NAME+4-AP, L-NG-Nitroarginine methyl ester+4-aminopyridine; EPI+4-AP, epinephrine+4-aminopyridine.
), L-NG-Nitroarginine methyl ester (L-NAME, filled triangles, ), with 4-aminopyridine (4-AP, ★), epinephrine (EPI, ) or combination drug treatment BCA+4AP (), DPG+4AP (), L-NAME+4AP (Δ), EPI+4-AP () administered 1 min after induction of AS in sensitized rats by IV ovalbumin (OVA). MAP and HR were recorded every minute for 60 min post-OVA injection. Values are Mean ± S.E.M. The difference between NA and AS rats was significant (p < 0.0001, two-way analysis of variance, ANOVA) (A,B). The difference observed between AS and all treated groups except AS+ LNAME was significant (p < 0.0001) (A). The differences between AS and single drug treatments (4-AP, BCA, DPG, EPI) and two-drug treatments (BCA+4-AP, DPG+4-AP, EPI+4-AP) were significant (p < 0.0001) with L-NAME (p < 0.01) (B,C) Survival for each group, NA , AS , AS+EPI , AS+LNAME , AS+BCA , AS+DPG , AS+4AP , AS+ EPI+4AP , AS+DPG+4AP , AS+LNAME+4AP , AS+BCA+4AP . NA, non-allergic rats; AS, allergic rats; BCA, β-cyanoalanine; DPG, dl-propargylglycine; L-NAME, L-NG-Nitroarginine methyl ester; 4-AP, 4-aminopyridine; EPI, epinephrine. BCA+4-AP, β-cyanoalanine+4-aminopyridine; DPG+4-AP, dl-propargylglycine+4-aminopyridine; L-NAME+4-AP, L-NG-Nitroarginine methyl ester+4-aminopyridine; EPI+4-AP, epinephrine+4-aminopyridine.






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellou, A.; Sennoun, N.; Aburawi, E.H.; Jayaraj, R.L.; Alper, S.L.; Alfaki, I.A.; Yasin, J.; Sekar, S.; Shafiuallah, M.; Al-Salam, S.; et al. Combined Treatment with KV Channel Inhibitor 4-Aminopyridine and either γ-Cystathionine Lyase Inhibitor β-Cyanoalanine or Epinephrine Restores Blood Pressure, and Improves Survival in the Wistar Rat Model of Anaphylactic Shock. Biology 2022, 11, 1455. https://doi.org/10.3390/biology11101455
Bellou A, Sennoun N, Aburawi EH, Jayaraj RL, Alper SL, Alfaki IA, Yasin J, Sekar S, Shafiuallah M, Al-Salam S, et al. Combined Treatment with KV Channel Inhibitor 4-Aminopyridine and either γ-Cystathionine Lyase Inhibitor β-Cyanoalanine or Epinephrine Restores Blood Pressure, and Improves Survival in the Wistar Rat Model of Anaphylactic Shock. Biology. 2022; 11(10):1455. https://doi.org/10.3390/biology11101455
Chicago/Turabian StyleBellou, Abdelouahab, Nacira Sennoun, Elhadi H. Aburawi, Richard L. Jayaraj, Seth L. Alper, Ibrahim Abdallah Alfaki, Javed Yasin, Subramanian Sekar, Mohamed Shafiuallah, Suhail Al-Salam, and et al. 2022. "Combined Treatment with KV Channel Inhibitor 4-Aminopyridine and either γ-Cystathionine Lyase Inhibitor β-Cyanoalanine or Epinephrine Restores Blood Pressure, and Improves Survival in the Wistar Rat Model of Anaphylactic Shock" Biology 11, no. 10: 1455. https://doi.org/10.3390/biology11101455
APA StyleBellou, A., Sennoun, N., Aburawi, E. H., Jayaraj, R. L., Alper, S. L., Alfaki, I. A., Yasin, J., Sekar, S., Shafiuallah, M., Al-Salam, S., Nemmar, A., Kazzam, E., Mertes, P. M., & Al-Hammadi, S. (2022). Combined Treatment with KV Channel Inhibitor 4-Aminopyridine and either γ-Cystathionine Lyase Inhibitor β-Cyanoalanine or Epinephrine Restores Blood Pressure, and Improves Survival in the Wistar Rat Model of Anaphylactic Shock. Biology, 11(10), 1455. https://doi.org/10.3390/biology11101455