
Evaluation of the Effectiveness of Derived Features of AlphaFold2 on Single-Sequence Protein Binding Site Prediction

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
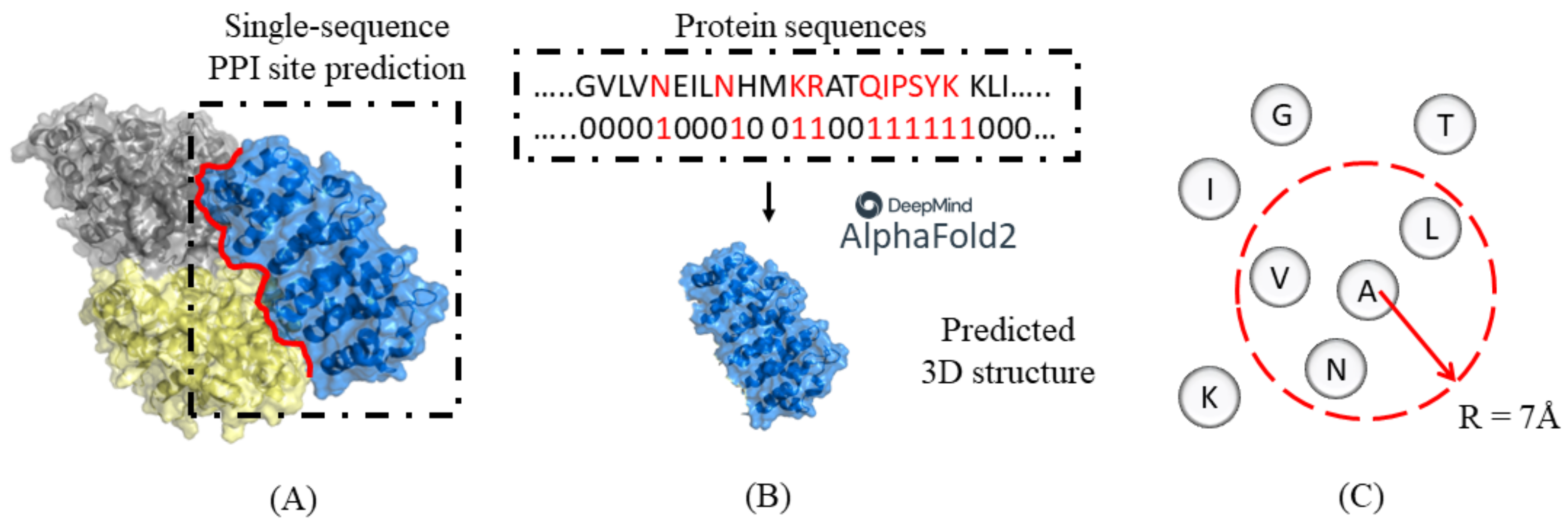
2. Materials and Methods
2.1. Datasets
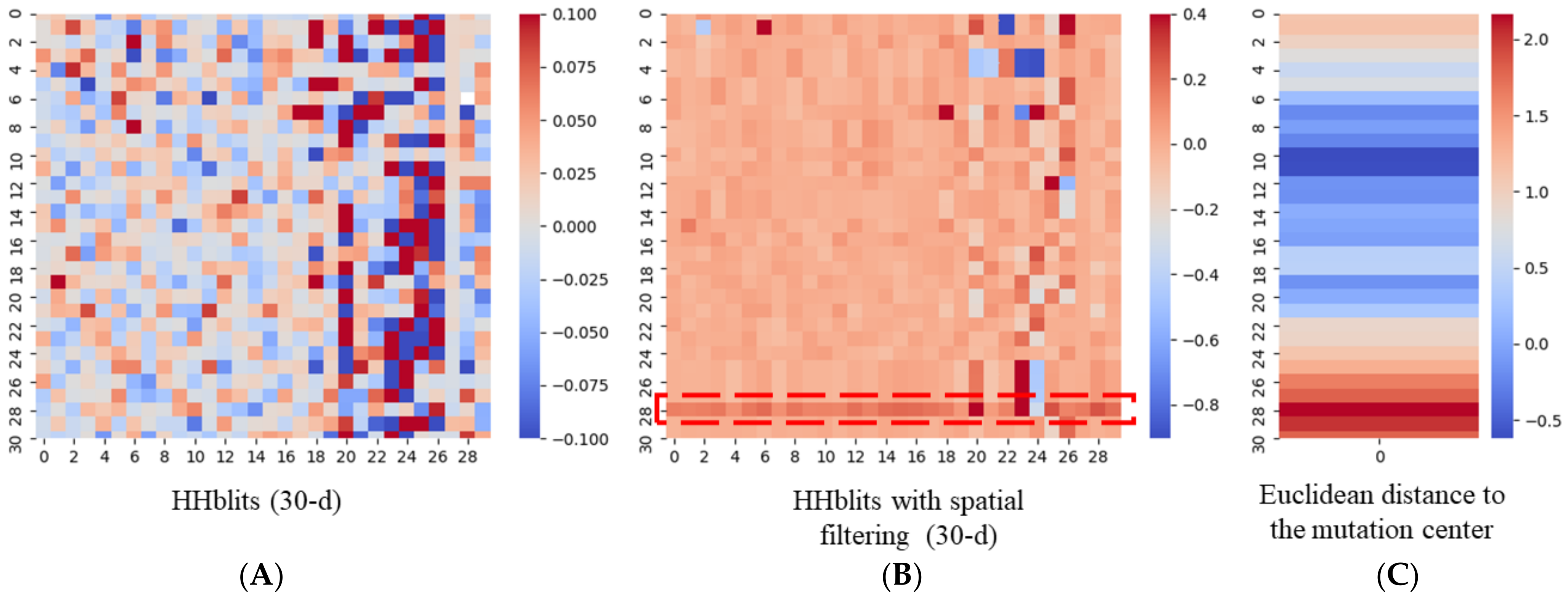
2.2. Spatial Filtering with HHBlits
2.3. Feature Engineering
2.3.1. OneHot and HHblits Profiles
2.3.2. SVD of the Distance Map
2.3.3. The Secondary Structure
2.3.4. The Relative Accessible Surface Area
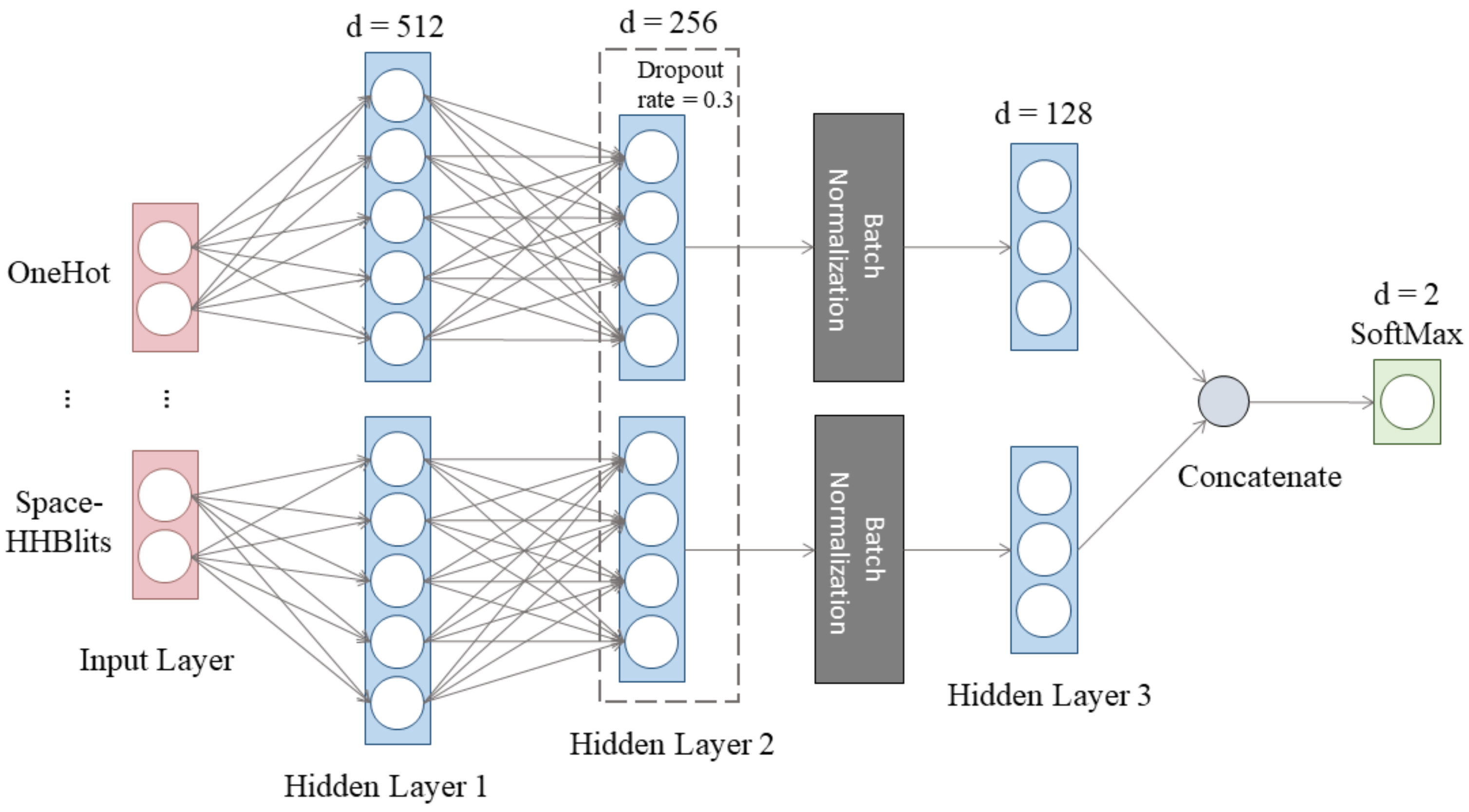
2.4. Deep Learning Network and Training Strategy
2.4.1. The Architecture of the Deep Artificial Neural Network
2.4.2. Performance Evaluation
3. Results
3.1. Comparison between Various Spatial Filter Radiuses with HHblits
3.2. Ablation Study of AlphaFold2-Derived Features on Single-Sequence PPI Site Prediction
3.3. Evaluation of AlphaFold2-Derived Features on Membrane Protein-Metal Binding Site Prediction
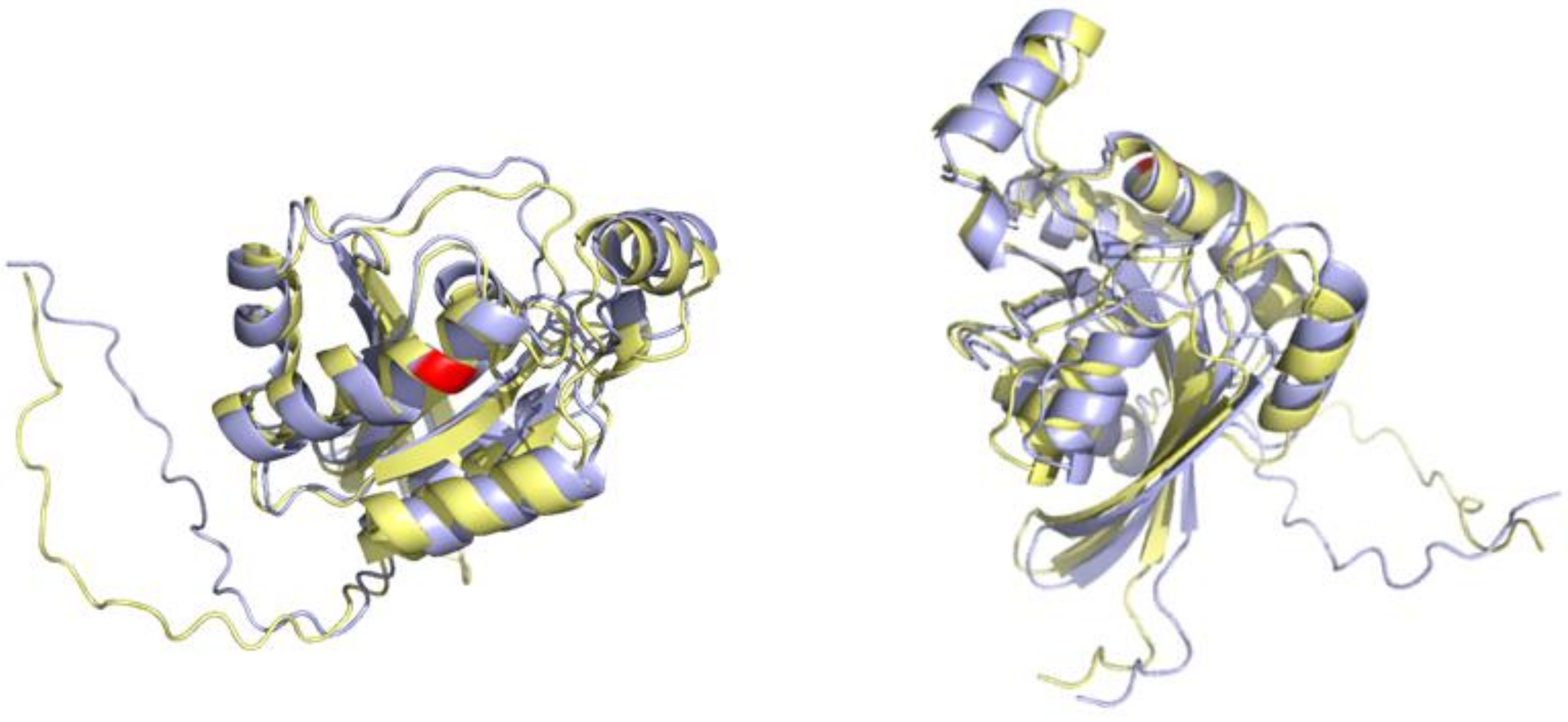
3.4. An Example of a Protein Carrying Variant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tubiana, J.; Schneidman-Duhovny, D.; Wolfson, H.J. ScanNet: An interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods 2022, 19, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhang, C.; Liu, S.; Zhou, Y. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006, 34, 3698–3707. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Zhang, F.; Wu, F.-X.; Li, Y.; Wang, J.; Li, M. Protein-protein interaction site prediction through combining local and global features with deep neural networks. Bioinformatics 2020, 36, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fang, Y.; Li, X.; Yang, Y.; Shen, H.-B. RBPsuite: RNA-protein binding sites prediction suite based on deep learning. BMC Genom. 2020, 21, 884. [Google Scholar] [CrossRef]
- Amos-Binks, A.; Patulea, C.; Pitre, S.; Schoenrock, A.; Gui, Y.; Green, J.R.; Golshani, A.; Dehne, F. Binding Site Prediction for Protein-Protein Interactions and Novel Motif Discovery using Re-occurring Polypeptide Sequences. BMC Bioinform. 2011, 12, 225. [Google Scholar] [CrossRef]
- Shoemaker, B.A.; Panchenko, A.R. Deciphering protein–protein interactions. Part I. Experimental techniques and databases. PLoS Comput. Biol. 2007, 3, e42. [Google Scholar] [CrossRef] [PubMed]
- Terentiev, A.A.; Moldogazieva, N.T.; Shaitan, K.V. Dynamic proteomics in modeling of the living cell. Protein-protein interactions. Biochemistry 2009, 74, 1586–1607. [Google Scholar] [CrossRef]
- Wodak, S.J.; Vlasblom, J.; Turinsky, A.L.; Pu, S. Protein–protein interaction networks: The puzzling riches. Curr. Opin. Struct. Biol. 2013, 23, 941–953. [Google Scholar] [CrossRef]
- Li, Y.; Golding, G.B.; Ilie, L. DELPHI: Accurate deep ensemble model for protein interaction sites prediction. Bioinformatics 2021, 37, 896–904. [Google Scholar] [CrossRef]
- Zhang, B.; Li, J.; Quan, L.; Chen, Y.; Lü, Q. Sequence-based prediction of protein-protein interaction sites by simplified long short-term memory network. Neurocomputing 2019, 357, 86–100. [Google Scholar] [CrossRef]
- Wei, Z.-S.; Han, K.; Yang, J.-Y.; Shen, H.-B.; Yu, D.-J. Protein–protein interaction sites prediction by ensembling SVM and sample-weighted random forests. Neurocomputing 2016, 193, 201–212. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; et al. MGnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 2020, 48, D570–D578. [Google Scholar] [CrossRef]
- Steinegger, M.; Mirdita, M.; Söding, J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat. Methods 2019, 16, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.W.; Green, T.; Hassabis, D. Protein complex prediction with AlphaFold-Multimer. BioRxiv 2022. [CrossRef]
- Zheng, S.; Li, Y.; Chen, S.; Xu, J.; Yang, Y. Predicting drug–protein interaction using quasi-visual question answering system. Nat. Mach. Intell. 2020, 2, 134–140. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Liu, Y.; Gong, W.; Yang, Z.; Li, C. SNB-PSSM: A spatial neighbor-based PSSM used for protein–RNA binding site prediction. J. Mol. Recognit. 2021, 34, e2887. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Z.; Zhang, E.; He, F.; Ma, Z.; Wang, H. MPLs-Pred: Predicting Membrane Protein-Ligand Binding Sites Using Hybrid Sequence-Based Features and Ligand-Specific Models. Int. J. Mol. Sci. 2019, 20, 3120. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, É.; Elbattah, M.; Gignon, M.; Dequen, G. NLP-Based Prediction of Medical Specialties at Hospital Admission Using Triage Notes. In Proceedings of the 2021 IEEE 9th International Conference on Healthcare Informatics (ICHI), Victoria, BC, Canada, 9–12 August 2021. [Google Scholar]
- Tanwar, S.; Ramani, T.; Tyagi, S. Dimensionality reduction using PCA and SVD in big data: A comparative case study. In International Conference on Future Internet Technologies and Trends; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Miller, S.M.; Lesk, A.; Jo, E. The accessible surface area and stability of oligomeric proteins. Nature 1987, 328, 834–836. [Google Scholar] [CrossRef]
- Liu, Z.; Gong, Y.; Bao, Y.; Guo, Y.; Wang, H.; Lin, G.N. TMPSS: A Deep Learning-Based Predictor for Secondary Structure and Topology Structure Prediction of Alpha-Helical Transmembrane Proteins. Front. Bioeng. Biotechnol. 2021, 8, 1514. [Google Scholar] [CrossRef]
- Fang, C.; Shang, Y.; Xu, N. Improving Protein Gamma-Turn Prediction Using Inception Capsule Networks. Sci. Rep. 2018, 8, 15741. [Google Scholar] [CrossRef]
- Singh, J.; Litfin, T.; Singh, J.; Paliwal, K.; Zhou, Y. SPOT-Contact-LM: Improving single-sequence-based prediction of protein contact map using a transformer language model. Bioinformatics 2022, 38, 1888–1894. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. DSSP: Definition of secondary structure of proteins given a set of 3D coordinates. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Tien, M.Z.; Meyer, A.G.; Sydykova, D.K.; Spielman, S.J.; Wilke, C.O. Maximum Allowed Solvent Accessibilites of Residues in Proteins. PLoS ONE 2013, 8, e80635. [Google Scholar] [CrossRef]
- Ketkar, N. Introduction to keras. In Deep Learning with Python; Springer: Berlin/Heidelberg, Germany, 2017; pp. 97–111. [Google Scholar]
- Abadi, M. TensorFlow: Learning functions at scale. In Proceedings of the 21st ACM SIGPLAN International Conference on Functional Programming, Nara, Japan, 18–24 September 2016. [Google Scholar]
- Fang, C. Applications of Deep Neural Networks to Protein Structure Prediction. University of Missouri-Columbia, Columbia, MO, USA. 2018. [Google Scholar]
- Lu, C.; Gong, Y.; Liu, Z.; Guo, Y.; Ma, Z.; Wang, H. TM-ZC: A Deep Learning-Based Predictor for the Z-Coordinate of Residues in α-Helical Transmembrane Proteins. IEEE Access 2020, 8, 40129–40137. [Google Scholar] [CrossRef]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins Struct. Funct. Bioinform. 2007, 66, 630–645. [Google Scholar] [CrossRef]
- Grinsztajn, L.; Oyallon, E.; Varoquaux, G. Why do tree-based models still outperform deep learning on tabular data? arXiv 2022, arXiv:2207.08815. [Google Scholar]
- Almén, M.S.; Nordström, K.J.; Fredriksson, R.; Schiöth, H.B. Mapping the human membrane proteome: A majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef]
- Suresh, M.X.; Gromiha, M.M.; Suwa, M. Development of a Machine Learning Method to Predict Membrane Protein-Ligand Binding Residues Using Basic Sequence Information. Adv. Bioinform. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Kerrien, S.; Abbani, S.; Aranda, B.; Bhate, J.; Bidwell, S.; Bridge, A.; Briganti, L.; Brinkman, F.S.L.; Cesareni, G.; et al. Protein interaction data curation: The International Molecular Exchange (IMEx) consortium. Nat. Methods 2012, 9, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.; Myung, Y.; Pires, D.E.; Ascher, D.B. mCSM-PPI2: Predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res. 2019, 47, W338–W344. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Chatzou, M.; Magis, C.; Chang, J.-M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple sequence alignment modeling: Methods and applications. Brief. Bioinform. 2015, 17, 1009–1023. [Google Scholar] [CrossRef]
- Buel, G.R.; Walters, K.J. Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 2022, 29, 1–2. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Proteins | Residues | % Binding out of Total | ||
|---|---|---|---|---|---|
| Total | Binding | Non-Binding | |||
| Training | 1682 | 970,997 | 66,143 | 904,854 | 6.8119 |
| Validation | 30 | 16,893 | 1607 | 15,286 | 9.5128 |
| Test | 30 | 20,051 | 1460 | 18,591 | 7.2814 |
| Total | 1741 | 1,007,941 | 69,210 | 938,731 | 6.8665 |
| E1 | E2 | E3 | E4 | E5 | Average | STD | |
|---|---|---|---|---|---|---|---|
| 0 Å (no filtering) | 0.344 | 0.264 | 0.250 | 0.246 | 0.276 | 0.276 | 0.035 |
| 3 Å | 0.244 | 0.245 | 0.447 | 0.314 | 0.234 | 0.297 | 0.080 |
| 5 Å | 0.282 | 0.275 | 0.231 | 0.196 | 0.427 | 0.282 | 0.079 |
| 6 Å | 0.299 | 0.503 | 0.399 | 0.515 | 0.406 | 0.424 | 0.088 |
| 7 Å | 0.623 | 0.743 | 0.508 | 0.656 | 0.618 | 0.630 | 0.075 |
| 8 Å | 0.545 | 0.472 | 0.646 | 0.603 | 0.544 | 0.562 | 0.066 |
| 9 Å | 0.575 | 0.482 | 0.369 | 0.450 | 0.378 | 0.451 | 0.075 |
| 11 Å | 0.368 | 0.445 | 0.421 | 0.616 | 0.451 | 0.460 | 0.083 |
| E1 | E2 | E3 | E4 | E5 | Average | STD | |
|---|---|---|---|---|---|---|---|
| Onehot + HHblits (Baseline) | 0.344 | 0.264 | 0.250 | 0.246 | 0.276 | 0.276 | 0.035 |
| Onehot + HHblits + SVD8 | 0.540 | 0.359 | 0.453 | 0.587 | 0.551 | 0.498 | 0.092 |
| Onehot + HHblits + SVD16 | 0.459 | 0.505 | 0.460 | 0.371 | 0.396 | 0.438 | 0.054 |
| Onehot + HHblits + SS | 0.510 | 0.348 | 0.471 | 0.594 | 0.585 | 0.502 | 0.100 |
| Onehot + HHblits + rASA | 0.479 | 0.603 | 0.673 | 0.536 | 0.681 | 0.594 | 0.078 |
| E1 | E2 | E3 | E4 | E5 | Average | STD | |
|---|---|---|---|---|---|---|---|
| Onehot + HHblits (Baseline) | 0.528 | 0.428 | 0.505 | 0.442 | 0.446 | 0.470 | 0.039 |
| Onehot + Space-HHblits (7 Å) | 0.645 | 0.601 | 0.602 | 0.620 | 0.588 | 0.611 | 0.020 |
| Onehot + HHblits + SVD8 | 0.601 | 0.630 | 0.640 | 0.653 | 0.610 | 0.627 | 0.019 |
| Onehot + HHblits + SVD16 | 0.600 | 0.629 | 0.639 | 0.606 | 0.574 | 0.610 | 0.023 |
| Onehot + HHblits + SS | 0.643 | 0.620 | 0.612 | 0.597 | 0.635 | 0.621 | 0.016 |
| Onehot + HHblits + rASA | 0.556 | 0.602 | 0.615 | 0.598 | 0.586 | 0.591 | 0.020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Pan, W.; Li, W.; Zhen, X.; Liang, J.; Cai, W.; Xu, F.; Yuan, K.; Lin, G.N. Evaluation of the Effectiveness of Derived Features of AlphaFold2 on Single-Sequence Protein Binding Site Prediction. Biology 2022, 11, 1454. https://doi.org/10.3390/biology11101454
Liu Z, Pan W, Li W, Zhen X, Liang J, Cai W, Xu F, Yuan K, Lin GN. Evaluation of the Effectiveness of Derived Features of AlphaFold2 on Single-Sequence Protein Binding Site Prediction. Biology. 2022; 11(10):1454. https://doi.org/10.3390/biology11101454
Chicago/Turabian StyleLiu, Zhe, Weihao Pan, Weihao Li, Xuyang Zhen, Jisheng Liang, Wenxiang Cai, Fei Xu, Kai Yuan, and Guan Ning Lin. 2022. "Evaluation of the Effectiveness of Derived Features of AlphaFold2 on Single-Sequence Protein Binding Site Prediction" Biology 11, no. 10: 1454. https://doi.org/10.3390/biology11101454
APA StyleLiu, Z., Pan, W., Li, W., Zhen, X., Liang, J., Cai, W., Xu, F., Yuan, K., & Lin, G. N. (2022). Evaluation of the Effectiveness of Derived Features of AlphaFold2 on Single-Sequence Protein Binding Site Prediction. Biology, 11(10), 1454. https://doi.org/10.3390/biology11101454