
ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules

 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Macroscopic Level: Clinical Features of IAHSP and Other ALS2-Related Pathologies
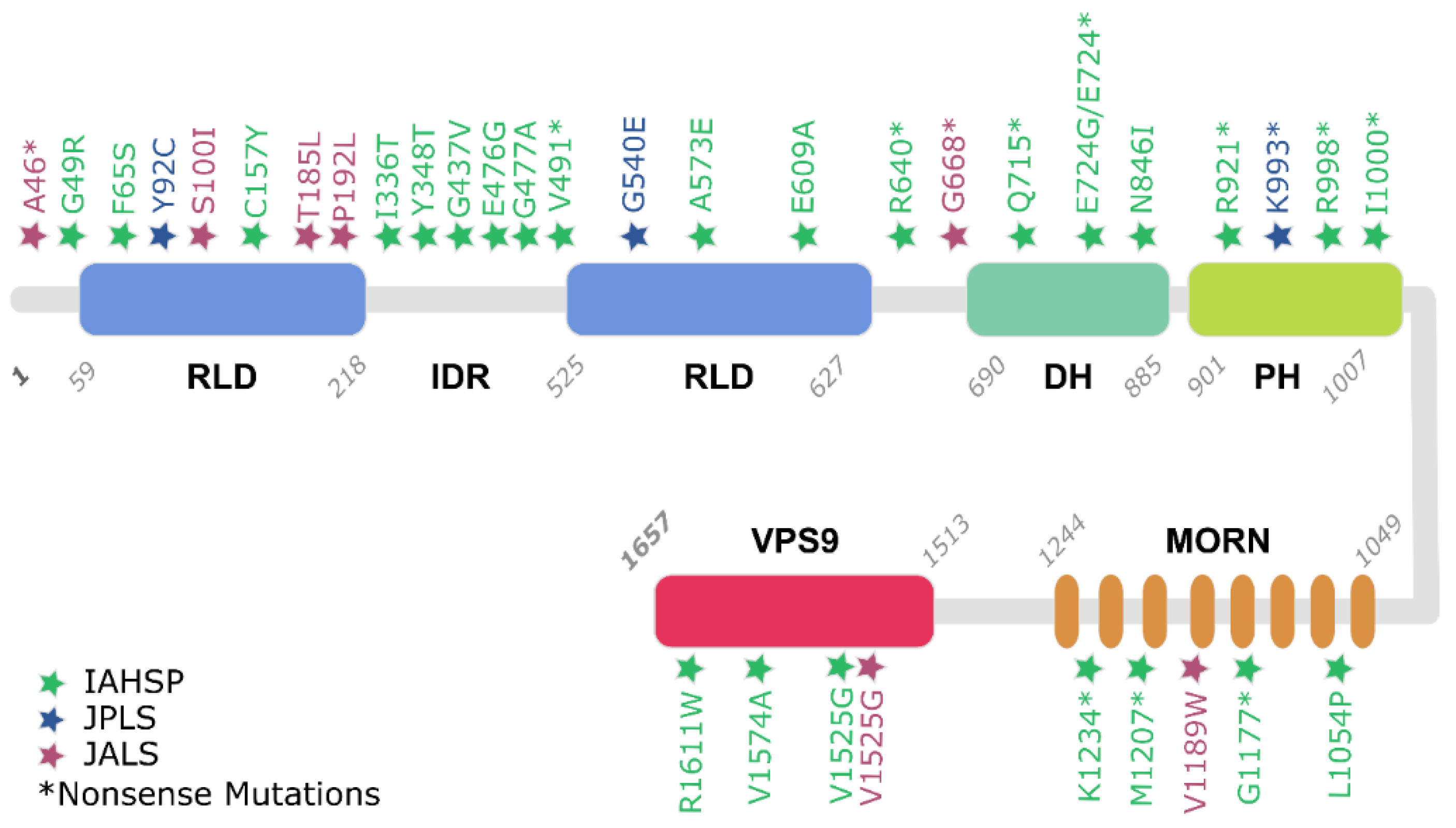
3. Microscopic Level: Molecular Features of IAHSP and Other Related ALS2 Pathologies
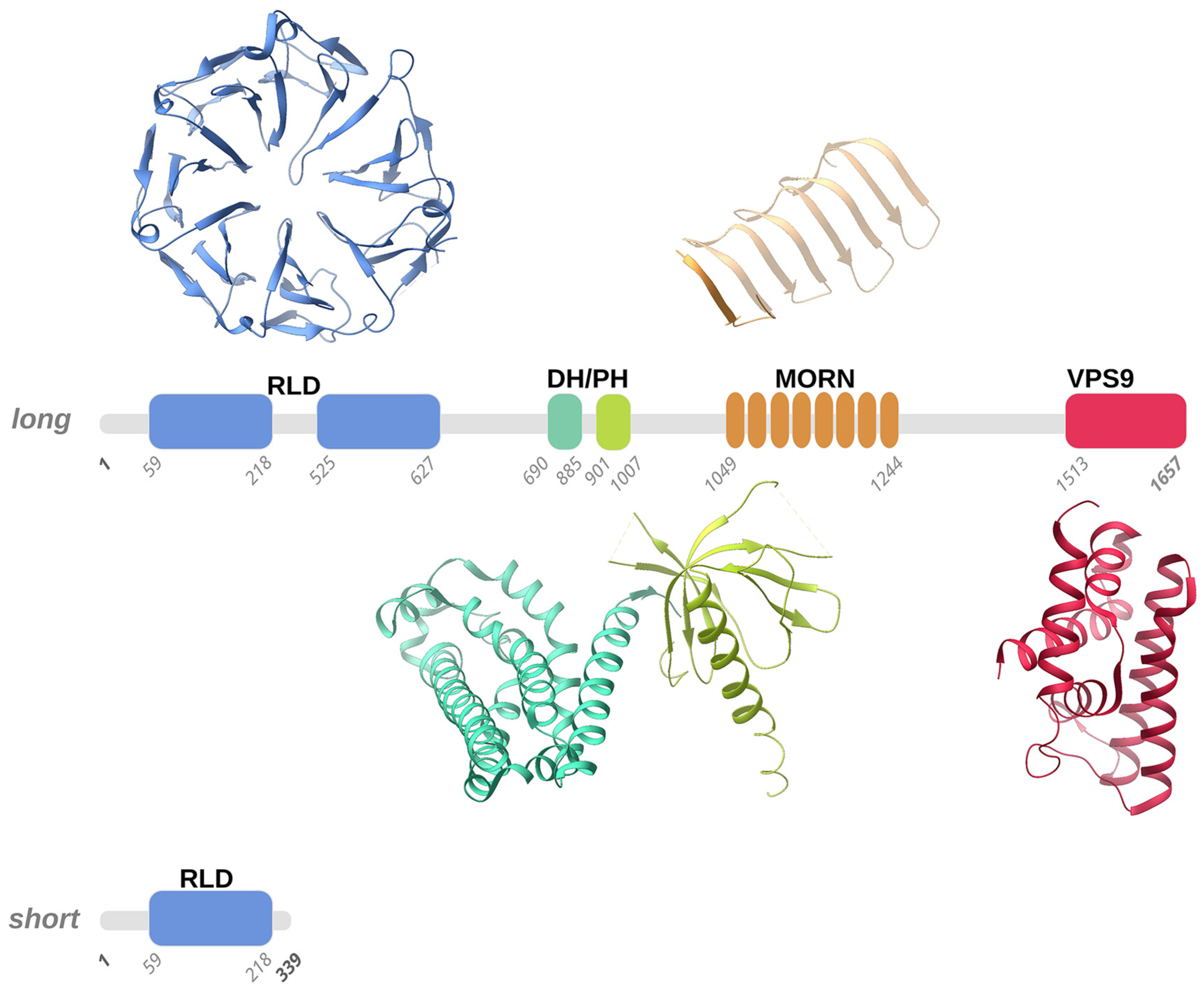
3.1. Alsin Molecular Structure
3.1.1. RLD Domain and the Intrinsically Disordered Domain
3.1.2. DH and PH Domains
3.1.3. MORN and VPS9 Domain
3.2. Alsin Mutations Lead to Signalling Pathways Alteration
3.2.1. Alsin’s RLD Domain and Altered Trafficking of AMPA Receptors
3.2.2. Alsin Neuroprotective Role against SOD1 Mutations
3.2.3. Alsin DH/PH Stimulates Rac1-PAK Binding and Neuron Growth
3.2.4. Alsin VPS9: Endosomal Trafficking and Rab5-Mediated Mechanisms
4. Population View on ALS2-Related Pathologies
5. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MND | motor neuron disease |
| HSP | Hereditary Spastic Paraplegia |
| IAHSP | Infantile-onset ascending hereditary spastic paralysis |
| JPLS | Juvenil Primary Lateral Sclerosis |
| ALS | Amyotrophic Lateral Sclerosis |
| JALS | Juvenil Amyotrophic Lateral Sclerosis |
| ALS2 | Amyotrophic Lateral Sclerosis type 2 |
| GEF | guanine exchange factor |
| GTPases | guanosine triphosphatases |
| RCC1 | Regulator of Chromosome Condensation 1 |
| RLD | RCC1-like domain |
| DH | central B cell lymphoma homology |
| PH | pleckstrin homology |
| VPS9 | vacuolar protein-sorting 9 |
| MORN | membrane occupation and recognition nexus |
| IDR | intrinsically disordered region |
| Ran | Ras-related nuclear |
| LMN | lower motor neurons |
| GRIP1 | glutamate receptor-interacting protein 1 |
| AMPA | a-amino-3-hydroxy-5-methylisoxazole-4-propio-nate |
| ALS2(−;−): | ALS2 knockout homozygous |
| SOD1 | Cu/Zn-superoxide dismutase |
| IGF1 | insulin-like growth factor |
| EEA1 | early endosome-associated protein |
| PAK1 | p21-activated kinase |
References
- Daud, S.; Kakar, N.; Goebel, I.; Hashmi, A.S.; Yaqub, T.; Nürnberg, G.; Nürnberg, P.; Morris-Rosendahl, D.J.; Wasim, M.; Volk, A.E.; et al. Identification of two novel ALS2 mutations in infantile-onset ascending hereditary spastic paraplegia. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 260–265. [Google Scholar] [CrossRef]
- Verschuuren-Bemelmans, C.C.; Winter, P.; Sival, D.A.; Elting, J.-W.; Brouwer, O.F.; Müller, U. Novel homozygous ALS2 nonsense mutation (p.Gln715X) in sibs with infantile-onset ascending spastic paralysis: The first cases from northwestern Europe. Eur. J. Hum. Genet. 2008, 16, 1407–1411. [Google Scholar] [CrossRef]
- Orrell, R.W. ALS2-Related Disorder. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20301421 (accessed on 1 November 2020).
- Helal, M.; Mazaheri, N.; Shalbafan, B.; Malamiri, R.A.; Dilaver, N.; Buchert, R.; Mohammadiasl, J.; Golchin, N.; Sedaghat, A.; Mehrjardi, M.Y.V.; et al. Clinical presentation and natural history of infantile-onset ascending spastic paralysis from three families with an ALS2 founder variant. Neurol. Sci. 2018, 39, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Löscher, W.; Quasthoff, S.; Wanschitz, J.; Auer-Grumbach, M.; Stevanin, G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012, 318, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lo Giudice, T.; Lombardi, F.; Santorelli, F.M.; Kawarai, T.; Orlacchio, A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol. 2014, 261, 518–539. [Google Scholar] [CrossRef] [PubMed]
- Salinas, S.; Proukakis, C.; Crosby, A.; Warner, T.T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol. 2008, 7, 1127–1138. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef]
- Reid, E. Pure hereditary spastic paraplegia. J. Med. Genet. 1997, 34, 499–503. [Google Scholar] [CrossRef][Green Version]
- de Souza, P.V.S.; de Rezende Pinto, W.B.V.; de Rezende Batistella, G.N.; Bortholin, T.; Oliveira, A.S.B. Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum 2017, 16, 525–551. [Google Scholar] [CrossRef]
- Hadano, S.; Benn, S.C.; Kakuta, S.; Otomo, A.; Sudo, K.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Shefner, J.M.; Cox, G.A.; et al. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 2006, 15, 233–250. [Google Scholar] [CrossRef]
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-Onset Ascending Hereditary Spastic Paralysis Is Associated with Mutations in the Alsin Gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef]
- Sato, K.; Otomo, A.; Ueda, M.T.; Hiratsuka, Y.; Suzuki-Utsunomiya, K.; Sugiyama, J.; Murakoshi, S.; Mitsui, S.; Ono, S.; Nakagawa, S.; et al. Altered oligomeric states in pathogenic ALS2 variants associated with juvenile motor neuron diseases cause loss of ALS2-mediated endosomal function. J. Biol. Chem. 2018, 293, 17135–17153. [Google Scholar] [CrossRef]
- Travaglini, L.; Aiello, C.; Stregapede, F.; D’Amico, A.; Alesi, V.; Ciolfi, A.; Bruselles, A.; Catteruccia, M.; Pizzi, S.; Zanni, G.; et al. The impact of next-generation sequencing on the diagnosis of pediatric-onset hereditary spastic paraplegias: New genotype-phenotype correlations for rare HSP-related genes. Neurogenetics 2018, 19, 111–121. [Google Scholar] [CrossRef]
- Pringle, C.E.; Hudson, A.J.; Munoz, D.G.; Kiernan, J.A.; Brown, W.F.; Ebers, G.C. Primary lateral sclerosis: Clinical features, neuropathology and diagnostic criteria. Brain 1992, 115, 495–520. [Google Scholar] [CrossRef]
- Brugman, F.; Eymard-Pierre, E.; van den Berg, L.H.; Wokke, J.H.J.; Gauthier-Barichard, F.; Boespflug-Tanguy, O. Adult-onset primary lateral sclerosis is not associated with mutations in the ALS2 gene. Neurology 2007, 69, 702–704. [Google Scholar] [CrossRef]
- Mintchev, N.; Zamba-Papanicolaou, E.; Kleopa, K.A.; Christodoulou, K. A novel ALS2 splice-site mutation in a Cypriot juvenile-onset primary lateral sclerosis family. Neurology 2009, 72, 28–32. [Google Scholar] [CrossRef]
- Dupre, N.; Valdmanis, P.N.; Bouchard, J.-P.; Rouleau, G.A. Autosomal dominant primary lateral sclerosis. Neurology 2007, 68, 1156–1157. [Google Scholar] [CrossRef] [PubMed]
- Gascon, G.; Chavis, P.; Yaghmour, A.; Stigsby, B.; Ozand, P.; Siddique, T. Familial Childhood Primary Lateral Sclerosis with Associated Gaze Paresis. Neuropediatrics 1995, 26, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hentati, A.; Deng, H.-X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.-Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Kress, J.A.; Kühnlein, P.; Winter, P.; Ludolph, A.C.; Kassubek, J.; Müller, U.; Sperfeld, A.-D. Novel mutation in theALS2 gene in juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2005, 58, 800–803. [Google Scholar] [CrossRef]
- Shaw, P.J. Genetic inroads in familial ALS. Nat. Genet. 2001, 29, 103–104. [Google Scholar] [CrossRef]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef]
- Wakil, S.M.; Ramzan, K.; Abuthuraya, R.; Hagos, S.; Al-Dossari, H.; Al-Omar, R.; Murad, H.; Chedrawi, A.; Al-Hassnan, Z.N.; Finsterer, J.; et al. Infantile-onset ascending hereditary spastic paraplegia with bulbar involvement due to the novel ALS2 mutation c.2761C > T. Gene 2014, 536, 217–220. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Lin, H.-X.; Liu, G.-L.; Tao, Q.-Q.; Ni, W.; Xiao, B.-G.; Wu, Z.-Y. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Cui, L.-Y.; Sun, Q.; Li, X.-G.; Liu, M.-S.; Xu, Y.; Zhou, Y.; Yang, X.-Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Chartier, L.; Amador, M.-D.-M.; Lam, R.; Lautrette, G.; Nicol, M.; Machat, S.; Da Barroca, S.; Moigneu, C.; Mairey, M.; et al. Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol. Aging 2017, 58, 239.e11–239.e20. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. FUS mutation is probably the most common pathogenic gene for JALS, especially sporadic JALS. Rev. Neurol. 2020. [Google Scholar] [CrossRef]
- Siddiqi, S.; Foo, J.N.; Vu, A.; Azim, S.; Silver, D.L.; Mansoor, A.; Tay, S.K.H.; Abbasi, S.; Hashmi, A.H.; Janjua, J.; et al. A Novel Splice-Site Mutation in ALS2 Establishes the Diagnosis of Juvenile Amyotrophic Lateral Sclerosis in a Family with Early Onset Anarthria and Generalized Dystonias. PLoS ONE 2014, 9, e113258. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, Z.; Shen, H.; Bing, Q.; Li, N.; Hu, J. Clinical and Genetic Features of Patients with Juvenile Amyotrophic Lateral Sclerosis with Fused in Sarcoma (FUS) Mutation. Med. Sci. Monit. 2018, 24, 8750–8757. [Google Scholar] [CrossRef]
- Camu, W.; Khoris, J.; Moulard, B.; Salachas, F.; Briolotti, V.; Rouleau, G.; Meininger, V. Genetics of familial ALS and consequences for diagnosis. J. Neurol. Sci. 1999, 165, S21–S26. [Google Scholar] [CrossRef]
- Leblond, C.S.; Webber, A.; Gan-Or, Z.; Moore, F.; Dagher, A.; Dion, P.A.; Rouleau, G.A. De novo FUS P525L mutation in Juvenile amyotrophic lateral sclerosis with dysphonia and diplopia. Neurol. Genet. 2016, 2, e63. [Google Scholar] [CrossRef]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis. Orphanet J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Cragnaz, L.; Klima, R.; De Conti, L.; Romano, G.; Feiguin, F.; Buratti, E.; Baralle, M.; Baralle, F.E. An age-related reduction of brain TBPH/TDP-43 levels precedes the onset of locomotion defects in a Drosophila ALS model. Neuroscience 2015, 311, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals Consortium. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef]
- Luigetti, M.; Lattante, S.; Conte, A.; Romano, A.; Zollino, M.; Marangi, G.; Sabatelli, M. A novel compound heterozygous ALS2 mutation in two Italian siblings with juvenile amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 470–472. [Google Scholar] [CrossRef] [PubMed]
- Orban, P.; Devon, R.S.; Hayden, M.R.; Leavitt, B.R. Chapter 15. Juvenile amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2007; Volume 82, pp. 301–312. [Google Scholar]
- Sztriha, L.; Panzeri, C.; Kálmánchey, R.; Szabó, N.; Endreffy, E.; Túri, S.; Baschirotto, C.; Bresolin, N.; Vekerdy, Z.; Bassi, M. First case of compound heterozygosity in ALS2 gene in infantile-onset ascending spastic paralysis with bulbar involvement. Clin. Genet. 2008, 73, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Ikeda, J.-E.; Hadano, S. Defective relocalization of ALS2/alsin missense mutants to Rac1-induced macropinosomes accounts for loss of their cellular function and leads to disturbed amphisome formation. FEBS Lett. 2011, 585, 730–736. [Google Scholar] [CrossRef]
- Hadano, S.; Kunita, R.; Otomo, A.; Suzuki-Utsunomiya, K.; Ikeda, J.-E. Molecular and cellular function of ALS2/alsin: Implication of membrane dynamics in neuronal development and degeneration. Neurochem. Int. 2007, 51, 74–84. [Google Scholar] [CrossRef]
- Otomo, A. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687. [Google Scholar] [CrossRef]
- Yamanaka, K.; Vande Velde, C.; Eymard-Pierre, E.; Bertini, E.; Boespflug-Tanguy, O.; Cleveland, D.W. Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor neuron disease. Proc. Natl. Acad. Sci. USA 2003, 100, 16041–16046. [Google Scholar] [CrossRef]
- Millecamps, S.; Gentil, B.J.; Gros-Louis, F.; Rouleau, G.; Julien, J.P. Alsin is partially associated with centrosome in human cells. Biochim. Biophys. Acta Mol. Cell Res. 2005, 1745, 84–100. [Google Scholar] [CrossRef][Green Version]
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki, K.; Showguchi-Miyata, J.; Yanagisawa, Y.; Hadano, S.; Ikeda, J.-E. Homo-oligomerization of ALS2 through Its Unique Carboxyl-terminal Regions Is Essential for the ALS2-associated Rab5 Guanine Nucleotide Exchange Activity and Its Regulatory Function on Endosome Trafficking. J. Biol. Chem. 2004, 279, 38626–38635. [Google Scholar] [CrossRef]
- Dasso, M. RCC1 in the cell cycle: The regulator of chromosome condensation takes on new roles. Trends Biochem. Sci. 1993, 18, 96–101. [Google Scholar] [CrossRef]
- Soares, D.C.; Barlow, P.N.; Porteous, D.J.; Devon, R.S. An interrupted beta-propeller and protein disorder: Structural bioinformatics insights into the N-terminus of alsin. J. Mol. Model. 2009, 15, 113–122. [Google Scholar] [CrossRef]
- Shimakura, K.; Sato, K.; Mitsui, S.; Ono, S.; Otomo, A.; Hadano, S. The N-terminal intrinsically disordered region mediates intracellular localization and self-oligomerization of ALS2. Biochem. Biophys. Res. Commun. 2021, 569, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Dasso, M. Running on Ran: Nuclear transport and the mitotic spindle. Cell 2001, 104, 321–324. [Google Scholar] [CrossRef]
- Topp, J.D.; Gray, N.W.; Gerard, R.D.; Horazdovsky, B.F. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 2004, 279, 24612–24623. [Google Scholar] [CrossRef]
- Hadjebi, O.; Casas-Terradellas, E.; Garcia-Gonzalo, F.R.; Rosa, J.L. The RCC1 superfamily: From genes, to function, to disease. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Shim, H.; Lai, C.; Xie, C.; Lin, X.; Yang, W.J.; Chandran, J. ALS2/Alsin Knockout Mice and Motor Neuron Diseases. Neurodegener. Dis. 2008, 5, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Worthylake, D.K.; Rossman, K.L.; Sondek, J. Crystal Structure of the DH/PH Fragment of Dbs without Bound GTPase. Structure 2004, 12, 1079–1086. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, D.S.; Shaw, R.; Winkelmann, J.C.; Shaw, G. Binding of PH domains of β-adrenergic receptor kinase and β-spectrin to WD40/β-transducin repeat containing regions of the β-subunit of trimeric G-proteins. Biochem. Biophys. Res. Commun. 1994, 203, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Kawakami, Y.; Kawakami, T. The pleckstrin homology domain of Bruton tyrosine kinase interacts with protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 9175–9179. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, B.; Zhu, K.; Kubiseski, T.J.; Liu, G.A.; Pawson, T.; Zheng, Y.; Rosen, M.K. Structure and mutagenesis of the Dbl homology domain. Nat. Struct. Biol. 1998, 5, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Soisson, S.M.; Nimnual, A.S.; Uy, M.; Bar-Sagi, D.; Kuriyan, J. Crystal structure of the Dbl and pleckstrin homology domains from the human Son of sevenless protein. Cell 1998, 95, 259–268. [Google Scholar] [CrossRef][Green Version]
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki-Utsunomiya, K.; Hadano, S.; Ikeda, J.-E. The Rab5 Activator ALS2/alsin Acts as a Novel Rac1 Effector through Rac1-activated Endocytosis. J. Biol. Chem. 2007, 282, 16599–16611. [Google Scholar] [CrossRef]
- Carney, D.S.; Davies, B.A.; Horazdovsky, B.F. Vps9 domain-containing proteins: Activators of Rab5 GTPases from yeast to neurons. Trends Cell Biol. 2006, 16, 27–35. [Google Scholar] [CrossRef]
- Delprato, A.; Merithew, E.; Lambright, D.G. Structure, exchange determinants, and family-wide Rab specificity of the tandem helical bundle and Vps9 domains of Rabex-5. Cell 2004, 118, 607–617. [Google Scholar] [CrossRef]
- Kwak, S.; Weiss, J.H. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 2006, 16, 281–287. [Google Scholar] [CrossRef]
- Lai, C.; Xie, C.; McCormack, S.G.; Chiang, H.-C.; Michalak, M.K.; Lin, X.; Chandran, J.; Shim, H.; Shimoji, M.; Cookson, M.R.; et al. Amyotrophic Lateral Sclerosis 2-Deficiency Leads to Neuronal Degeneration in Amyotrophic Lateral Sclerosis through Altered AMPA Receptor Trafficking. J. Neurosci. 2006, 26, 11798–11806. [Google Scholar] [CrossRef]
- Lai, C.; Xie, C.; Shim, H.; Chandran, J.; Howell, B.W.; Cai, H. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol. Brain 2009, 2, 23. [Google Scholar] [CrossRef]
- Lin, X.; Shim, H.; Cai, H. Deficiency in the ALS2 gene does not affect the motor neuron degeneration in SOD1G93A transgenic mice. Neurobiol. Aging 2007, 28, 1628–1630. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shaw, P.J. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Hashimoto, Y.; Niikura, T.; Aiso, S.; Matsuoka, M.; Nishimoto, I. Alsin, the Product of ALS2 Gene, Suppresses SOD1 Mutant Neurotoxicity through RhoGEF Domain by Interacting with SOD1 Mutants. J. Biol. Chem. 2004, 279, 19247–19256. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Chakrabartty, A. Structure, folding, and misfolding of Cu, Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1025–1037. [Google Scholar] [CrossRef]
- Chandran, J.; Ding, J.; Cai, H. Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol. Neurobiol. 2007, 36, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. Loss of ALS2 Function Is Insufficient to Trigger Motor Neuron Degeneration in Knock-Out Mice but Predisposes Neurons to Oxidative Stress. J. Neurosci. 2005, 25, 7567–7574. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Hashimoto, Y.; Kita, Y.; Sasabe, J.; Aiso, S.; Nishimoto, I.; Matsuoka, M. A Rac1/Phosphatidylinositol 3-Kinase/Akt3 Anti-apoptotic Pathway, Triggered by AlsinLF, the Product of the ALS2 Gene, Antagonizes Cu/Zn-superoxide Dismutase (SOD1) Mutant-induced Motoneuronal Cell Death. J. Biol. Chem. 2005, 280, 4532–4543. [Google Scholar] [CrossRef]
- Tudor, E.L.; Perkinton, M.S.; Schmidt, A.; Ackerley, S.; Brownlees, J.; Jacobsen, N.J.O.; Byers, H.L.; Ward, M.; Hall, A.; Leigh, P.N.; et al. ALS2/Alsin Regulates Rac-PAK Signaling and Neurite Outgrowth. J. Biol. Chem. 2005, 280, 34735–34740. [Google Scholar] [CrossRef]
- Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Onoe, K.; Osuga, H.; Hadano, S.; Ikeda, J.-E. ALS2/alsin deficiency in neurons leads to mild defects in macropinocytosis and axonal growth. Biochem. Biophys. Res. Commun. 2008, 370, 87–92. [Google Scholar] [CrossRef]
- Devon, R.S.; Orban, P.C.; Gerrow, K.; Barbieri, M.A.; Schwab, C.; Cao, L.P.; Helm, J.R.; Bissada, M.; Cruz-Aguado, R.; Davidson, T.L.; et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 9595–9600. [Google Scholar] [CrossRef]
- Hadano, S.; Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Akatsuka, A.; Koike, M.; Aoki, M.; Uchiyama, Y.; Itoyama, Y.; Ikeda, J.-E. Loss of ALS2/Alsin Exacerbates Motor Dysfunction in a SOD1H46R-Expressing Mouse ALS Model by Disturbing Endolysosomal Trafficking. PLoS ONE 2010, 5, e9805. [Google Scholar] [CrossRef] [PubMed]
- Eymard-Pierre, E.; Yamanaka, K.; Haeussler, M.; Kress, W.; Gauthier-Barichard, F.; Combes, P.; Cleveland, D.W.; Boespflug-Tanguy, O. Novel missense mutation in ALS2 gene results in infantile ascending hereditary spastic paralysis. Ann. Neurol. 2006, 59, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Sprute, R.; Jergas, H.; Ölmez, A.; Alawbathani, S.; Karasoy, H.; Salimi Dafsari, H.; Becker, K.; Daimagüeler, H.; Nürnberg, P.; Muntoni, F.; et al. Genotype–phenotype correlation in seven motor neuron disease families with novel ALS2 mutations. Am. J. Med. Genet. A 2020, 185, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Panzeri, C. The first ALS2 missense mutation associated with JPLS reveals new aspects of alsin biological function. Brain 2006, 129, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Devon, R.S.; Schwab, C.; Topp, J.D.; Orban, P.C.; Yang, Y.; Pape, T.D.; Helm, J.R.; Davidson, T.-L.; Rogers, D.A.; Gros-Louis, F.; et al. Cross-species characterization of the ALS2 gene and analysis of its pattern of expression in development and adulthood. Neurobiol. Dis. 2005, 18, 243–257. [Google Scholar] [CrossRef]
- Sheerin, U.-M.; Schneider, S.A.; Carr, L.; Deuschl, G.; Hopfner, F.; Stamelou, M.; Wood, N.W.; Bhatia, K.P. ALS2 mutations: Juvenile amyotrophic lateral sclerosis and generalized dystonia. Neurology 2014, 82, 1065–1067. [Google Scholar] [CrossRef]
- Gal, J.; Zhang, J.; Kwinter, D.M.; Zhai, J.; Jia, H.; Jia, J.; Zhu, H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging 2011, 32, 2323.e27–2323.e40. [Google Scholar] [CrossRef] [PubMed]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in Sarcoma (FUS) Protein Lacking Nuclear Localization Signal (NLS) and Major RNA Binding Motifs Triggers Proteinopathy and Severe Motor Phenotype in Transgenic Mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobágyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef]
- Cannariato, M.; Miceli, M.; Cavaglià, M.; Deriu, M.A. Prediction of protein-protein interactions between Alsin DH/PH and Rac1 and resulting protein dynamics. Front. Mol. Neurosci. 2021; in press. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| IAHSP | JPLS | JALS | |
|---|---|---|---|
| Inheritance | Autosomal recessive | Autosomal recessive | Autosomal recessive |
| Age of onset | 1–3 years old | 1–3 years old | 4–8 years old |
| Life expectancy | Adulthood | Adulthood | 7 months to 17 years old |
| Genetic causes | ALS2 mutation | ALS2 mutation | ALS2 mutation (Other forms are caused by SETX, FUS, UBQLN2, SPG11, SIGMAR1) |
| Neuron alterations | Degeneration of both upper and lower motor neurons | Progressive degeneration, upper motor neurons | Degeneration of both upper and lower motor neurons |
| Symptoms | Lower limb weakness and spasticity progressing towards quadriplegia, wheelchair dependence by the age of 10, followed by tetraparesis, feeding dependence on gastrostomy | Lower limb weakness and spasticity, wheelchair dependence by adolescence, motor speech impairment, saccadic eye movements | Lower limb weakness and spasticity, face muscle spasticity, bladder dysfunction, dysarthria, sensory disturbances, and sometimes mental retardation and sclerosis |
| Disease | Frameshift | Missense | Nonsense |
|---|---|---|---|
| IAHSP | 12. | 6 | 8 |
| JPLS | 2 | 1 | 0 |
| JALS | 4 | 2 | 1 |
| %TOT | 50% | 25% | 25% |
| Disease | RLD | DH/PH | MORN | VPS9 |
|---|---|---|---|---|
| IAHSP | 12 | 7 | 4 | 3 |
| JPLS | 2 | 1 | 0 | 0 |
| JALS | 4 | 1 | 1 | 1 |
| %TOT | 50% | 25% | 14% | 11% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miceli, M.; Exertier, C.; Cavaglià, M.; Gugole, E.; Boccardo, M.; Casaluci, R.R.; Ceccarelli, N.; De Maio, A.; Vallone, B.; Deriu, M.A. ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules. Biology 2022, 11, 77. https://doi.org/10.3390/biology11010077
Miceli M, Exertier C, Cavaglià M, Gugole E, Boccardo M, Casaluci RR, Ceccarelli N, De Maio A, Vallone B, Deriu MA. ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules. Biology. 2022; 11(1):77. https://doi.org/10.3390/biology11010077
Chicago/Turabian StyleMiceli, Marcello, Cécile Exertier, Marco Cavaglià, Elena Gugole, Marta Boccardo, Rossana Rita Casaluci, Noemi Ceccarelli, Alessandra De Maio, Beatrice Vallone, and Marco A. Deriu. 2022. "ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules" Biology 11, no. 1: 77. https://doi.org/10.3390/biology11010077
APA StyleMiceli, M., Exertier, C., Cavaglià, M., Gugole, E., Boccardo, M., Casaluci, R. R., Ceccarelli, N., De Maio, A., Vallone, B., & Deriu, M. A. (2022). ALS2-Related Motor Neuron Diseases: From Symptoms to Molecules. Biology, 11(1), 77. https://doi.org/10.3390/biology11010077