
Breeding for Economically and Environmentally Sustainable Wheat Varieties: An Integrated Approach from Genomics to Selection

 , , ,
, , ,  ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction: Wheat in a Fluctuating World
2. The BREEDWHEAT Project Addressed Several Issues
3. A Genomics Toolbox for Wheat Research and Breeding
3.1. Polymorphism Detection and High Throughput Genotyping
3.2. Genetic Mapping and Recombination Pattern Analyses
3.3. Sequencing the Bread Wheat Genome
4. Characterization and Exploitation of the Wheat Genetic Diversity
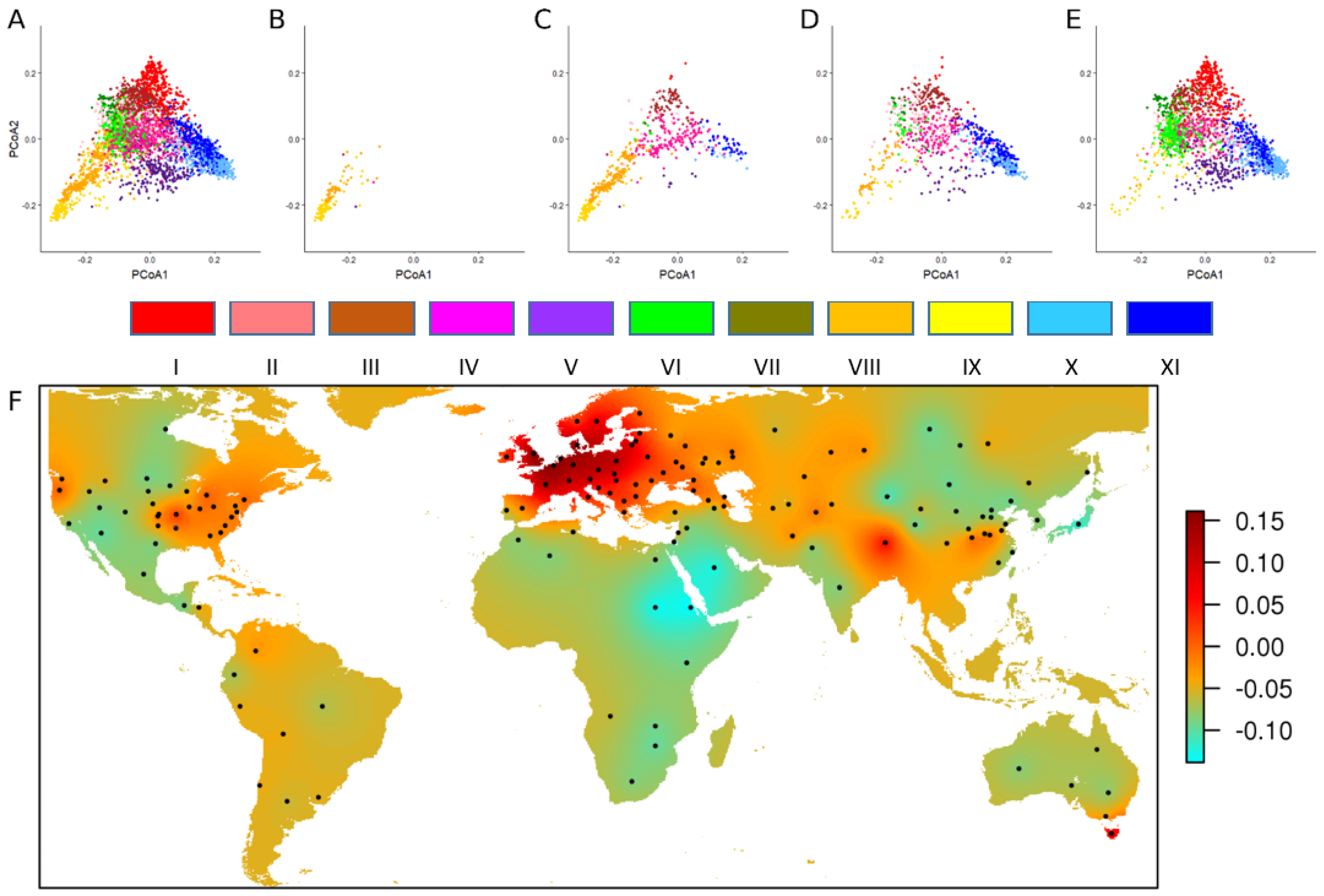
4.1. Characterizing the Worldwide Genetic Diversity
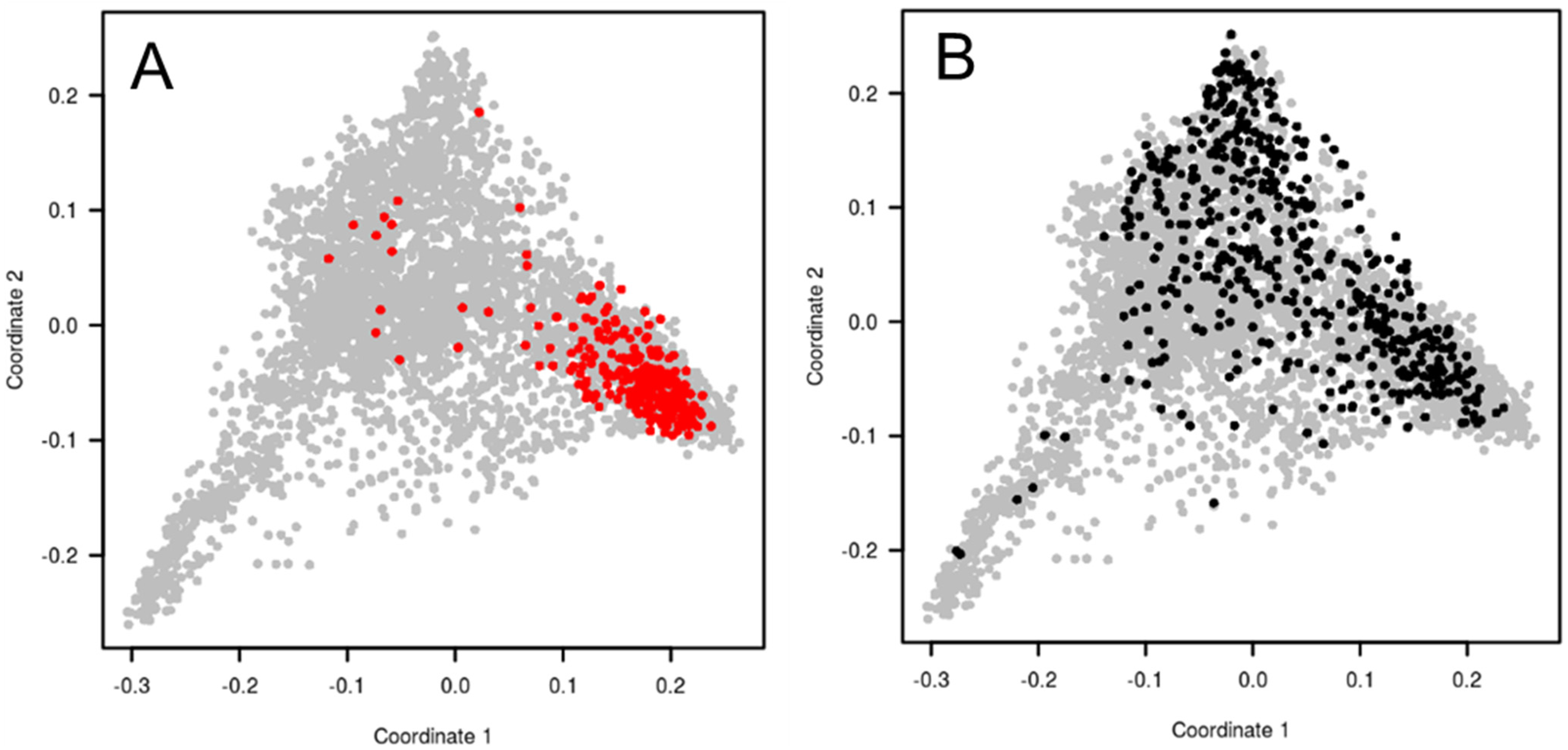
4.2. Assembling a New Pre-Breeding Panel for the European Breeding Programs
5. Genetics and Ecophysiology Studies of Wheat Adaptation to Biotic and Abiotic Stress in the Framework of Sustainable Agricultural Systems and Climate Change
5.1. Grain Composition
5.2. Adaptation to Abiotic Stress
5.2.1. Heat Stress
5.2.2. Drought Stress
5.2.3. Nitrogen Stress
5.3. Crop Modeling
5.4. High Throughput Field Phenotyping
6. Development of Innovative Methods and Cost-Efficient Breeding Platforms
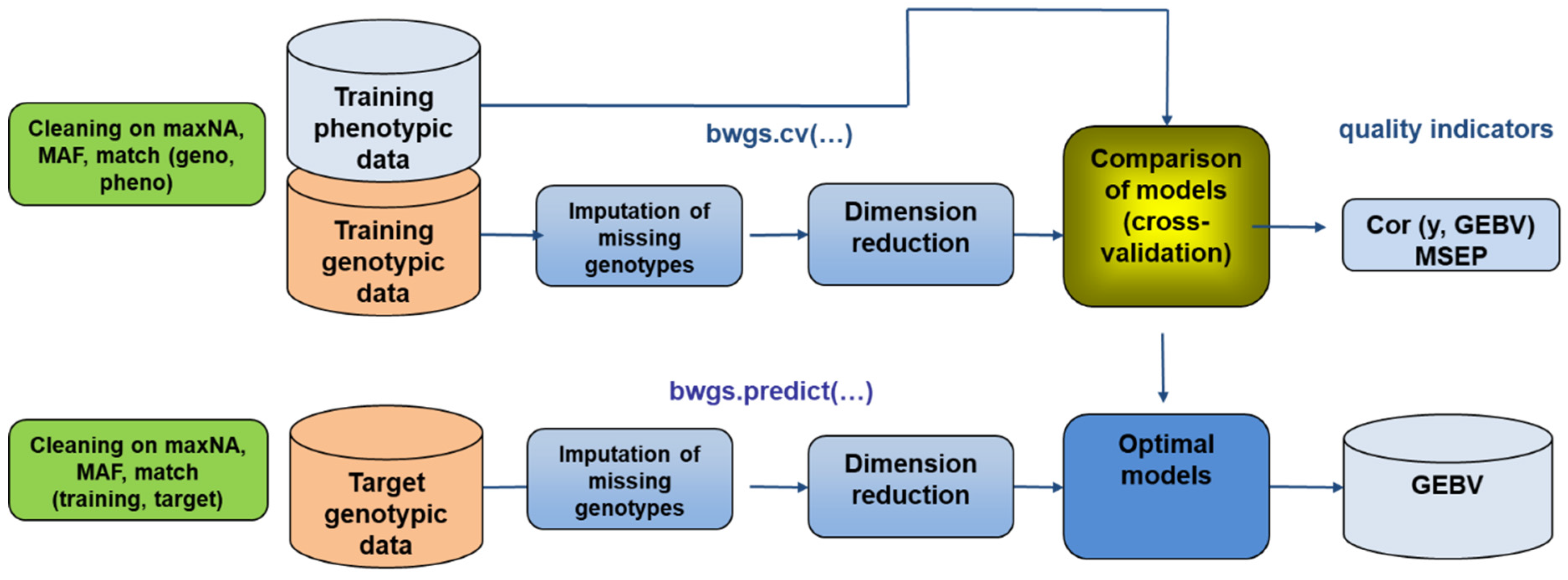
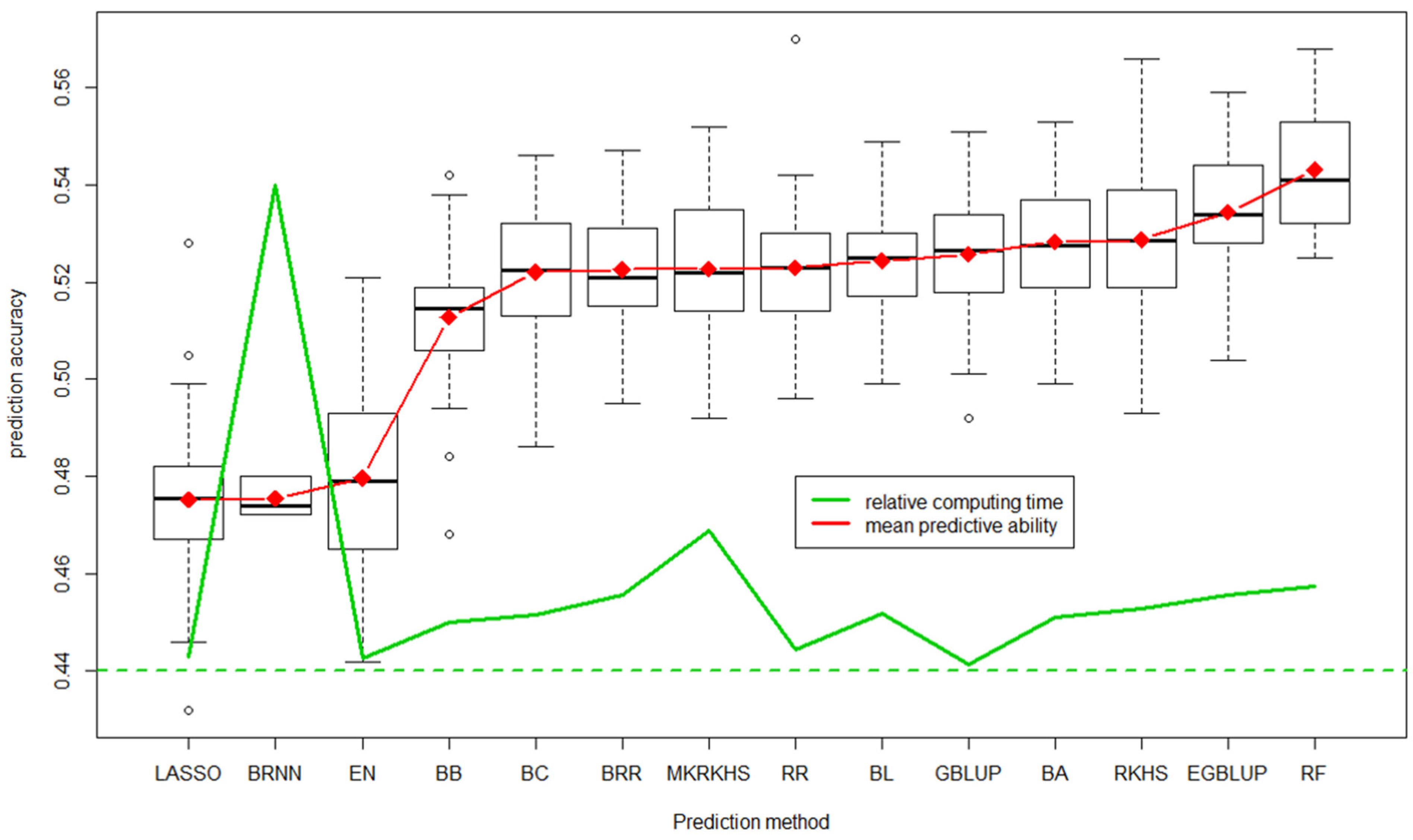
6.1. Genomic Selection
6.2. Phenomic Selection
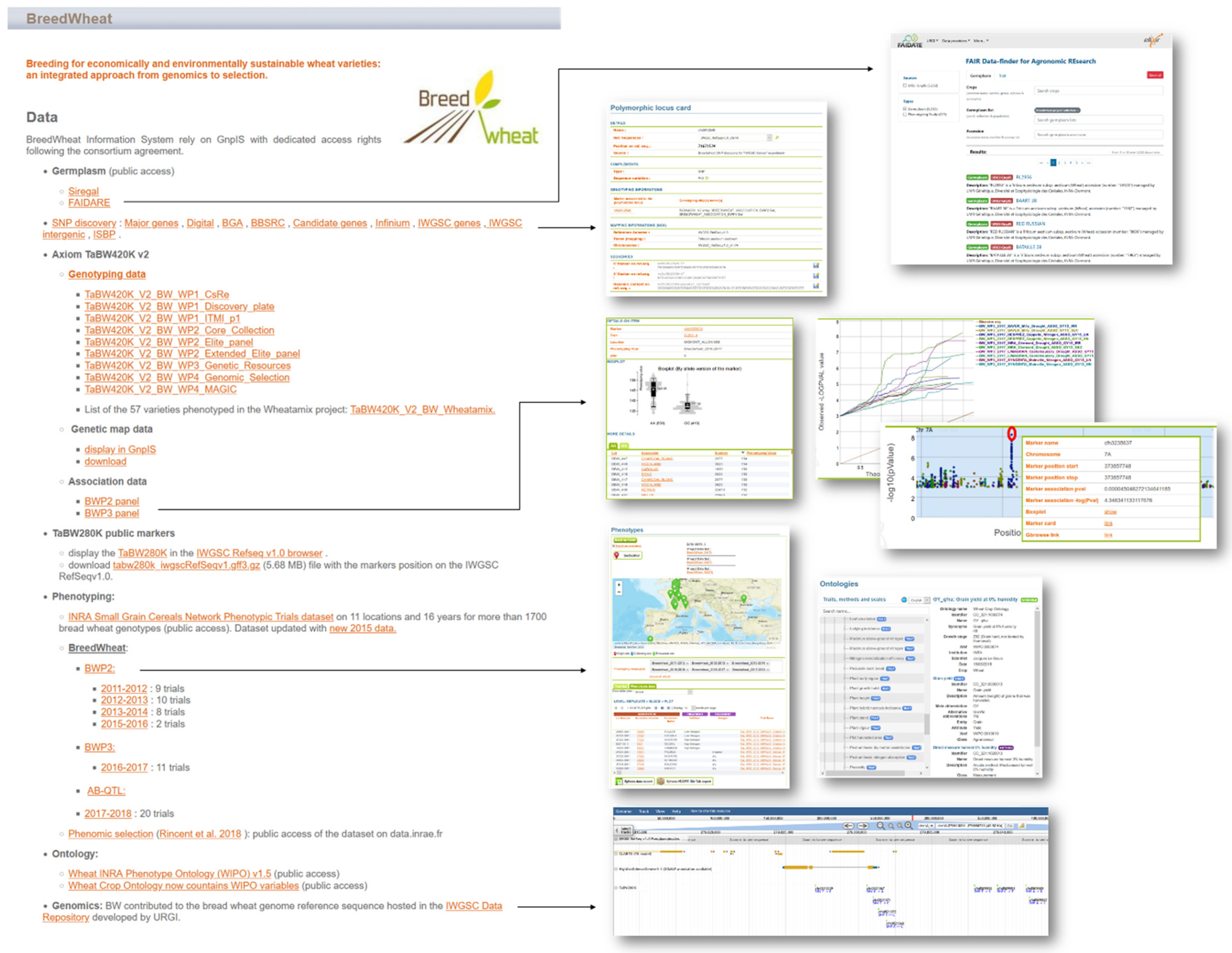
7. Data Integration into an Information System following the FAIR Principles
- (1)
- Long-term storage of the data as GnpIS has been available since 2000 and benefits from perennial funding by INRAE (Plant Biology and Breeding division);
- (2)
- Implementation of a data management plan, which includes a data access mechanism with credentials following the consortium agreement;
- (3)
- Integration of all the project data in a common information system to link the data from genomics to phenomics [132];
- (4)
- To allow the researchers and breeders to query the data through the GnpIS web interfaces (FAIDARE, JBrowse, GnpIS core-DB, detailed below); and
- (5)
- To insure data quality and compliance to the FAIR principles (Findable Accessible Interoperable Reusable) [133].
7.1. Data Quality and FAIRness
- Findability: a DOI (digital object identifier) was generated for each accession; all data are searchable using web interfaces; public BREEDWHEAT data are findable by the whole community via the WheatIS data discovery tool (https://urgi.versailles.inrae.fr/wheatis (accessed on 16 December 2021)).
- Accessibility: phenotyping data are accessible through Breeding API (BrAPI) web services [134].
- Interoperability: phenotyping data followed an ontology developed in the frame of the project and merged with the international wheat crop ontology (CO_321) [135].
- Reusability: a data management timeline defines when each kind of data will be opened; all the GnpIS tools have general terms of use and license.
7.2. Genetic Resources Data Integration
7.3. Genomics Data Integration
7.4. Genotyping, Phenotyping, and GWAS Data Integration
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef]
- Anonymous. Wheat lag. Nature 2014, 507, 399–400. [Google Scholar] [CrossRef][Green Version]
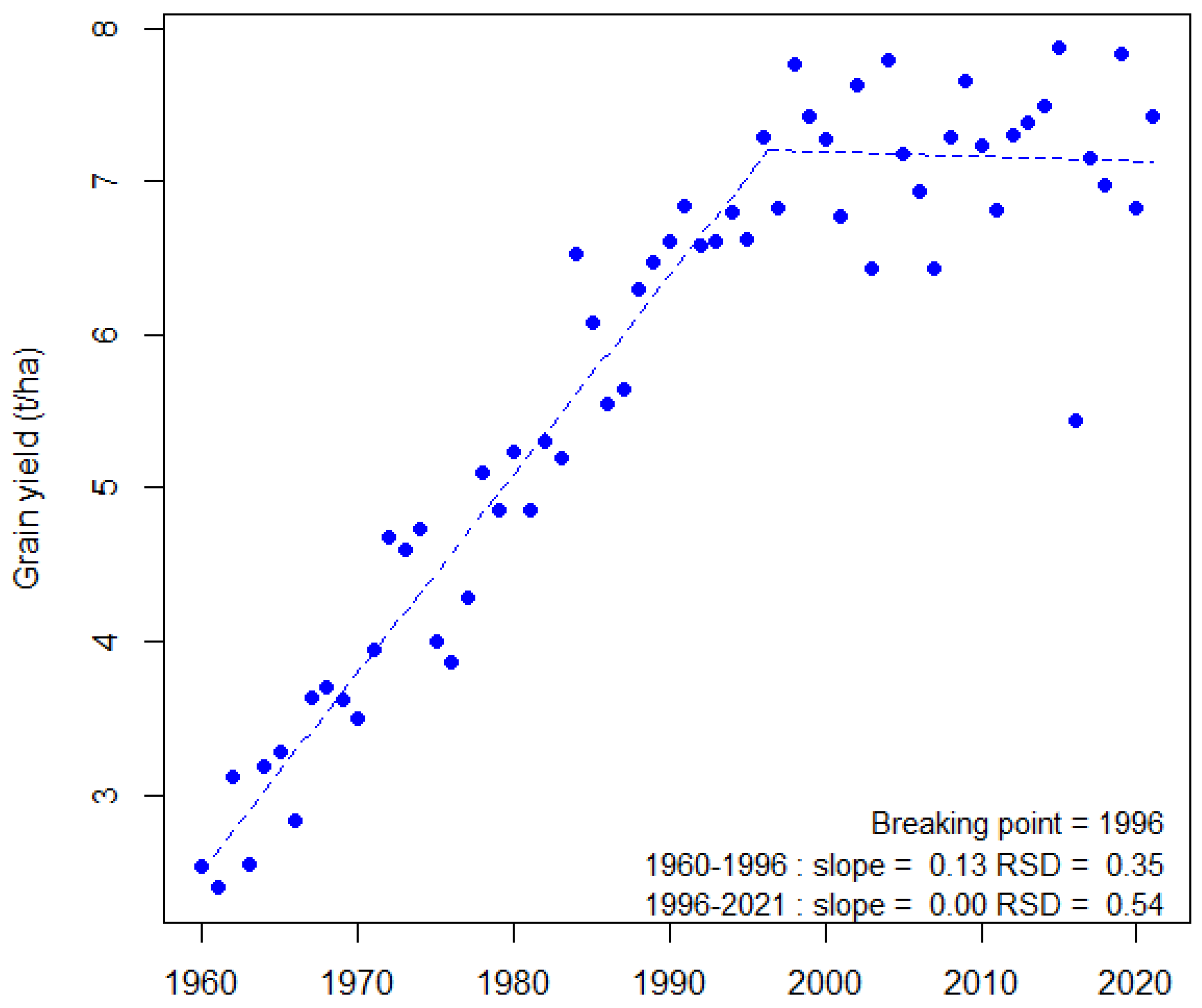
- Brisson, N.; Gate, G.; Gouache, D.; Charmet, G.; Oury, F.-X.; Huard, F. Why are wheat yields stagnating in Europe? A comprehensive data analysis for France. Field Crops Res. 2010, 119, 201–212. [Google Scholar] [CrossRef]
- Porter, J.R.; Semenov, M.A. Crop responses to climatic variation. Philos. Trans. R. Soc. B-Biol. Sci. 2005, 360, 2021–2035. [Google Scholar] [CrossRef]
- Tester, M.; Langridge, P. Breeding technologies to increase crop production in a changing world. Science 2010, 327, 818–822. [Google Scholar] [CrossRef]
- Le Gouis, J.; Oury, F.-X.; Charmet, G. How changes in climate and agricultural practices influence wheat production in Western Europe. J. Cereal Sci. 2020, 93, 102960. [Google Scholar] [CrossRef]
- Muggeo, V.M.R. Estimating regression models with unknown break-points. Stat. Med. 2003, 22, 3055–3071. [Google Scholar] [CrossRef] [PubMed]
- Paux, E.; Sourdille, P.; Salse, J.; Saintenac, C.; Choulet, F.; Leroy, P.; Korol, A.; Michalak, M.; Kianian, S.; Spielmeyer, W.; et al. A physical map of the 1-Gigabase bread wheat chromosome 3B. Science 2008, 322, 101–104. [Google Scholar] [CrossRef]
- Haudry, A.; Cenci, A.; Ravel, C.; Bataillon, T.; Brunel, D.; Poncet, C.; Hochu, I.; Poirier, S.; Santoni, S.; Glemin, S.; et al. Grinding up wheat: A massive loss of nucleotide diversity since domestication. Mol. Biol. Evol. 2007, 24, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Reif, J.C.; Zhang, P.; Dreisigacker, S.; Warburton, M.L.; van Ginkel, M.; Hoisington, D.; Bohn, M.; Melchinger, A.E. Wheat genetic diversity trends during domestication and breeding. Theor. Appl. Genet. 2005, 110, 859–864. [Google Scholar] [CrossRef]
- Feuillet, C.; Langridge, P.; Waugh, R. Cereal breeding takes a walk on the wild side. Trends Genet. 2008, 24, 24–32. [Google Scholar] [CrossRef]
- Tilman, D. Global environmental impacts of agricultural expansion: The need for sustainable and efficient practices. Proc. Natl. Acad. Sci. USA 1999, 96, 5995–6000. [Google Scholar] [CrossRef]
- Verdier, J.; Thompson, R.D. Transcriptional regulation of storage protein synthesis during dicotyledon seed filling. Plant Cell Physiol. 2008, 49, 1263–1271. [Google Scholar] [CrossRef]
- Rimbert, H.; Darrier, B.; Navarro, J.; Cubizolles, N.; Kitt, J.; Choulet, F.; Leveugle, M.; Duarte, J.; Rivière, N.; Eversole, K.; et al. High throughput SNP discovery and genotyping in hexaploid wheat. PLoS ONE 2018, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kitt, J.; Danguy des Désert, A.; Bouchet, S.; Servin, B.; Rimbert, H.; de Oliveira, R.; Choulet, F.; Balfourier, F.; Sourdille, P.; Paux, E. Genotyping of 4506 bread wheat accessions with the TaBW410K SNP array. Zenodo 2021, 13, evab152. [Google Scholar] [CrossRef]
- Balfourier, F.; Bouchet, S.; Robert, S.; De Oliveira, R.; Rimbert, H.; Kitt, J.; Choulet, F.; Appels, R.; Feuillet, C.; Keller, B.; et al. Worldwide phylogeography and history of wheat genetic diversity. Sci. Adv. 2019, 5, eaav053. [Google Scholar] [CrossRef] [PubMed]
- Béral, A.; Rincent, R.; Le Gouis, J.; Girousse, C.; Allard, V. Wheat individual grain-size variance originates from crop development and from specific genetic determinism. PLoS ONE 2020, 15, e0230689. [Google Scholar] [CrossRef]
- Rincent, R.; Charpentier, J.-P.; Faivre-Rampant, P.; Paux, E.; Le Gouis, J.; Bastien, C.; Segura, V. Phenomic selection: A low-cost and high-throughput method based on indirect predictions. Proof of concept on wheat and poplar. G3 2018, 8, 3961–3972. [Google Scholar] [CrossRef]
- Robert, P.; Le Gouis, J.; Rincent, R.; BreedWheat, C. Combining crop growth modeling with trait-assisted prediction improved the prediction of genotype by environment interactions. Front. Plant Sci. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Touzy, G.; Lafarge, S.; Redondo, E.; Lievin, V.; Decoopman, X.; Le Gouis, J.; Praud, S. Genome-wide identification of QTL affecting terminal heat stress responses in bread wheat. Theor. Appl. Genet. 2022. [Google Scholar] [CrossRef]
- Touzy, G.; Rincent, R.; Bogard, M.; Lafarge, S.; Dubreuil, P.; Mini, A.; Deswarte, J.C.; Beauchene, K.; Le Gouis, J.; Praud, S. Using environmental clustering to identify specific drought tolerance QTLs in bread wheat (T. aestivum L.). Theor. Appl. Genet. 2019, 132, 2859–2880. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 661. [Google Scholar] [CrossRef]
- Juery, C.; Concia, L.; De Oliveira, R.; Papon, N.; Ramirez-Gonzalez, R.; Benhamed, M.; Uauy, C.; Choulet, F.; Paux, E. New insights into homoeologous copy number variations in the hexaploid wheat genome. Plant Genome 2021, 14, e20069. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Gonzalez, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Gundlach, H.; Spannagl, M.; Uauy, C.; Borrill, P.; Ramirez-Gonzalez, R.H.; De Oliveira, R.; Mayer, K.F.X.; Paux, E.; Choulet, F.; et al. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biol. 2018, 19, 103. [Google Scholar] [CrossRef]
- Langlands-Perry, C.; Cuenin, M.; Bergez, C.; Krima, S.B.; Gélisse, S.; Sourdille, P.; Valade, R.; Marcel, T.C. Resistance of the wheat cultivar ‘Renan’ to Septoria leaf blotch explained by a combination of strain specific and strain non-specific QTL mapped on an ultra-dense genetic map. Genes 2022, 13, 100. [Google Scholar] [CrossRef]
- Rasheed, A.; Hao, Y.F.; Xia, X.C.; Khan, A.; Xu, Y.B.; Varshney, R.K.; He, Z.H. Crop breeding chips and genotyping platforms: Progress, challenges, and perspectives. Mol. Plant 2017, 10, 1047–1064. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Yang, X.P.; Peng, Z.; Xu, L.P.; Wang, J.P. Development and applications of a high throughput genotyping tool for polyploid crops: Single Nucleotide Polymorphism (SNP) Array. Front. Plant Sci. 2018, 9, 104. [Google Scholar] [CrossRef]
- Winfield, M.O.; Allen, A.M.; Burridge, A.J.; Barker, G.L.A.; Benbow, H.R.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; Scopes, G.; et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 2016, 14, 1195–1206. [Google Scholar] [CrossRef]
- Cavanagh, C.R.; Chao, S.M.; Wang, S.C.; Huang, B.E.; Stephen, S.; Kiani, S.; Forrest, K.; Saintenac, C.; Brown-Guedira, G.L.; Akhunova, A.; et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl. Acad. Sci. USA 2013, 110, 8057–8062. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.X.; Rogers, J.; Dolezel, J.; Pozniak, C.; Eversole, K.; Feuillet, C.; Gill, B.; Friebe, B.; Lukaszewski, A.J.; Sourdille, P.; et al. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Cubizolles, N.; Rey, E.; Choulet, F.; Rimbert, H.; Laugier, C.; Balfourier, F.; Bordes, J.; Poncet, C.; Jack, P.; James, C.; et al. Exploiting the repetitive fraction of the wheat genome for high-throughput single-nucleotide polymorphism discovery and genotyping. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Wong, D.B.; Forrest, K.; Allen, A.; Chao, S.M.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90 000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef]
- Allen, A.M.; Winfield, M.O.; Burridge, A.J.; Downie, R.C.; Benbow, H.R.; Barker, G.L.A.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; et al. Characterization of a wheat breeders’ array suitable for high-throughput SNP genotyping of global accessions of hexaploid bread wheat (Triticum aestivum). Plant Biotechnol. J. 2017, 15, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Doerge, R.W. Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 2002, 3, 43–52. [Google Scholar] [CrossRef]
- Choulet, F.; Alberti, A.; Theil, S.; Glover, N.; Barbe, V.; Daron, J.; Pingault, L.; Sourdille, P.; Couloux, A.; Paux, E.; et al. Structural and functional partitioning of bread wheat chromosome 3B. Science 2014, 345, 1249721. [Google Scholar] [CrossRef]
- McVean, G.; Awadalla, P.; Fearnhead, P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 2002, 160, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Darrier, B.; Rimbert, H.; Balfourier, F.; Pingault, L.; Josselin, A.A.; Servin, B.; Navarro, J.; Choulet, F.; Paux, E.; Sourdille, P. High-resolution mapping of crossover events in the hexaploid wheat genome suggests a universal recombination mechanism. Genetics 2017, 206, 1373–1388. [Google Scholar] [CrossRef]
- Danguy des Désert, A.; Bouchet, S.; Sourdille, P.; Servin, B. Evolution of recombination landscapes in diverging populations of bread wheat. Genome Biol. Evol. 2021, 13, evab152. [Google Scholar] [CrossRef]
- Jordan, K.W.; Wang, S.C.; He, F.; Chao, S.A.M.; Lun, Y.N.; Paux, E.; Sourdille, P.; Sherman, J.; Akhunova, A.; Blake, N.K.; et al. The genetic architecture of genome-wide recombination rate variation in allopolyploid wheat revealed by nested association mapping. Plant J. 2018, 95, 1039–1054. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Lozada, D.N.; Zhang, Z.W.; Pumphrey, M.O.; Carter, A.H. Deep learning for predicting complex traits in spring wheat breeding program. Front. Plant Sci. 2021, 11, 613325. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, K.S.; Mihalyov, P.D.; Lewien, M.J.; Pumphrey, M.O.; Carter, A.H. Combining genomic and phenomic information for predicting grain protein content and grain yield in spring wheat. Front. Plant Sci. 2021, 12, 613300. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, K.S.; Mihalyov, P.D.; Lewien, M.J.; Pumphrey, M.O.; Carter, A.H. Genomic selection and genome-wide association studies for grain protein content stability in a nested association mapping population of wheat. Agronomy 2021, 11, 2528. [Google Scholar] [CrossRef]
- Svacina, R.; Karafiatova, M.; Malurova, M.; Serra, H.; Vitek, D.; Endo, T.R.; Sourdille, P.; Bartos, J. Development of deletion lines for chromosome 3D of bread wheat. Front. Plant Sci. 2020, 10, 1756. [Google Scholar] [CrossRef]
- Serra, H.; Svacina, R.; Baumann, U.; Whitford, R.; Sutton, T.; Bartos, J.; Sourdille, P. Ph2 encodes the mismatch repair protein MSH7-3D that inhibits wheat homoeologous recombination. Nat. Commun. 2021, 12, 803. [Google Scholar] [CrossRef]
- Guan, J.T.; Garcia, D.F.; Zhou, Y.; Appels, R.; Li, A.L.; Mao, L. The battle to sequence the bread wheat genome: A tale of the three kingdoms. Genom. Proteom. Bioinform. 2020, 18, 221–229. [Google Scholar] [CrossRef]
- Feuillet, C.; Stein, N.; Rossini, L.; Praud, S.; Mayer, K.; Schulman, A.; Eversole, K.; Appels, R. Integrating cereal genomics to support innovation in the Triticeae. Funct. Integr. Genom. 2012, 12, 573–583. [Google Scholar] [CrossRef]
- Philippe, R.; Paux, E.; Bertin, I.; Sourdille, P.; Choulet, F.; Laugier, C.; Simkova, H.; Safar, J.; Bellec, A.; Vautrin, S.; et al. A high density physical map of chromosome 1BL supports evolutionary studies, map-based cloning and sequencing in wheat. Genome Biol. 2013, 14, R64. [Google Scholar] [CrossRef]
- Raats, D.; Frenkel, Z.; Krugman, T.; Dodek, I.; Sela, H.; Simkova, H.; Magni, F.; Cattonaro, F.; Vautrin, S.; Berges, H.; et al. The physical map of wheat chromosome 1BS provides insights into its gene space organization and evolution. Genome Biol. 2013, 14, R138. [Google Scholar] [CrossRef]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Chaisson, M.J.; Pevzner, P.A. Short read fragment assembly of bacterial genomes. Genome Res. 2008, 18, 324–330. [Google Scholar] [CrossRef]
- Tulpova, Z.; Luo, M.C.; Toegelova, H.; Visendi, P.; Hayashi, S.; Vojta, P.; Paux, E.; Kilian, A.; Abrouk, M.; Bartos, J.; et al. Integrated physical map of bread wheat chromosome arm 7DS to facilitate gene cloning and comparative studies. N. Biotechnol. 2019, 48, 12–19. [Google Scholar] [CrossRef]
- Dubcovsky, J.; Dvorak, J. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 2007, 316, 1862–1866. [Google Scholar] [CrossRef] [PubMed]
- Marcussen, T.; Sandve, S.R.; Heier, L.; Spannagl, M.; Pfeifer, M.; Jakobsen, K.S.; Wulff, B.B.H.; Steuernagel, B.; Mayer, K.F.X.; Olsen, O.A.; et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science 2014, 345, 1250092. [Google Scholar] [CrossRef]
- Mirzaghaderi, G.; Mason, A.S. Broadening the bread wheat D genome. Theor. Appl. Genet. 2019, 132, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M.; Schreiber, M.; Scholz, U.; Graner, A.; Reif, J.C.; Stein, N. Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet. 2019, 51, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Balfourier, F.; Roussel, V.; Strelchenko, P.; Exbrayat-Vinson, F.; Sourdille, P.; Boutet, G.; Koenig, J.; Ravel, C.; Mitrofanova, O.; Beckert, M.; et al. A worldwide bread wheat core collection arrayed in a 384-well plate. Theor. Appl. Genet. 2007, 114, 1265–1275. [Google Scholar] [CrossRef]
- Bordes, J.; Branlard, G.; Oury, F.X.; Charmet, G.; Balfourier, F. Agronomic characteristics, grain quality and flour rheology of 372 bread wheats in a worldwide core collection. J. Cereal Sci. 2008, 48, 569–579. [Google Scholar] [CrossRef]
- Bordes, J.; Ravel, C.; Le Gouis, J.; Lapierre, A.; Charmet, G.; Balfourier, F. Use of a global wheat core collection for association analysis of flour and dough quality traits. J. Cereal Sci. 2011, 54, 137–147. [Google Scholar] [CrossRef]
- Horvath, A.; Didier, A.; Koenig, J.; Exbrayat, F.; Charmet, G.; Balfourier, F. Analysis of diversity and linkage disequilibrium along chromosome 3B of bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2009, 119, 1523–1537. [Google Scholar] [CrossRef]
- Rincent, R.; Malosetti, M.; Ababaei, B.; Touzy, G.; Mini, A.; Bogard, M.; Martre, P.; Le Gouis, J.; van Eeuwijk, F. Using crop growth model stress covariates and AMMI decomposition to better predict genotype-by-environment interactions. Theor. Appl. Genet. 2019, 132, 3399–3411. [Google Scholar] [CrossRef] [PubMed]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. 2006. Available online: http://darwin.cirad.fr/ (accessed on 16 December 2021).
- Kimball, B.A. Crop responses to elevated CO2 and interactions with H2O, N, and temperature. Curr. Opin. Plant Biol. 2016, 31, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Gammans, M.; Merel, P.; Ortiz-Bobea, A. Negative impacts of climate change on cereal yields: Statistical evidence from France. Environ. Res. Lett. 2017, 12. [Google Scholar] [CrossRef]
- Waldhoff, S.T.; Wing, I.S.; Edmonds, J.; Leng, G.Y.; Zhang, X.S. Future climate impacts on global agricultural yields over the 21st century. Environ. Res. Lett. 2020, 15. [Google Scholar] [CrossRef]
- Zampieri, M.; Ceglar, A.; Dentener, F.; Toreti, A. Wheat yield loss attributable to heat waves, drought and water excess at the global, national and subnational scales. Environ. Res. Lett. 2017, 12, 064008. [Google Scholar] [CrossRef]
- Hossard, L.; Philibert, A.; Bertrand, M.; Colnenne-David, C.; Debaeke, P.; Munier-Jolain, N.; Jeuffroy, M.H.; Richard, G.; Makowski, D. Effects of halving pesticide use on wheat production. Sci. Rep. 2014, 4, 4405. [Google Scholar] [CrossRef]
- Raffan, S.; Oddy, J.; Halford, N.G. The sulphur response in wheat grain and its implications for acrylamide formation and food safety. Int. J. Mol. Sci. 2020, 21, 3876. [Google Scholar] [CrossRef]
- Yu, Z.T.; Juhasz, A.; Islam, S.; Diepeveen, D.; Zhang, J.J.; Wang, P.H.; Ma, W.J. Impact of mid-season sulphur deficiency on wheat nitrogen metabolism and biosynthesis of grain protein. Sci. Rep. 2018, 8, 2499. [Google Scholar] [CrossRef]
- Bonnot, T.; Bancel, E.; Alvarez, D.; Davanture, M.; Boudet, J.; Pailloux, M.; Zivy, M.; Ravel, C.; Martre, P. Grain subproteome responses to nitrogen and sulfur supply in diploid wheat Triticum monococcum ssp monococcum. Plant J. 2017, 91, 894–910. [Google Scholar] [CrossRef]
- Bancel, E.; Bonnot, T.; Davanture, M.; Branlard, G.; Zivy, M.; Martre, P. Proteomic approach to identify nuclear proteins in wheat grain. J. Proteome Res. 2015, 14, 4432–4439. [Google Scholar] [CrossRef]
- Vincent, J.; Martre, P.; Gouriou, B.; Ravel, C.; Dai, Z.; Petit, J.-M.; Pailloux, M. RulNet: A web-oriented platform for regulatory network inference, application to wheat –omics data. PLoS ONE 2015, 10, e0127127. [Google Scholar] [CrossRef]
- Bancel, E.; Bonnot, T.; Davanture, M.; Alvarez, D.; Zivy, M.; Martre, P.; Dejean, S.; Ravel, C. Proteomic data integration highlights central actors involved in einkorn (Triticum monococcum ssp. monococcum) grain filling in relation to grain storage protein composition. Front. Plant Sci. 2019, 10, 832. [Google Scholar] [CrossRef]
- Bonnot, T.; Martre, P.; Hatte, V.; Dardevet, M.; Leroy, P.; Benard, C.; Falagan, N.; Martin-Magniette, M.L.; Deborde, C.; Moing, A.; et al. Omics data reveal putative regulators of einkorn grain protein composition under sulfur deficiency. Plant Physiol. 2020, 183, 501–516. [Google Scholar] [CrossRef]
- Boudet, J.; Merlino, M.; Plessis, A.; Gaudin, J.-C.; Dardevet, M.; Perrochon, S.; Alvarez, D.; Risacher, T.; Martre, P.; Ravel, C. The bZIP transcription factor SPA heterodimenzing protein represses glutenin synthesis in Triticum aestivum. Plant J. 2019, 97, 858–871. [Google Scholar] [CrossRef]
- Calderini, D.F.; Slafer, G.A. Has yield stability changed with genetic improvement of wheat yield? Euphytica 1999, 107, 51–59. [Google Scholar] [CrossRef]
- Tashiro, T.; Wardlaw, I.F. A comparison of the effect of high temperature on grain development in wheat and rice. Ann. Bot. 1989, 64, 59–65. [Google Scholar] [CrossRef]
- Girousse, C.; Roche, J.; Guérin, C.; Le Gouis, J.; Balzègue, S.; Mouzeyar, S.; Bouzidi, F. Coexpression network and phenotypic analysis identify metabolic pathways associated with the effect of warming on grain yield components in wheat. PLoS ONE 2018, 13, e0199434. [Google Scholar] [CrossRef]
- Tardieu, F. Any trait or trait-related allele can confer drought tolerance: Just design the right drought scenario. J. Exp. Bot. 2012, 63, 25–31. [Google Scholar] [CrossRef]
- Farooq, M.; Hussain, M.; Siddique, K.H.M. Drought stress in wheat during flowering and grain-filling periods. Crit. Rev. Plant Sci. 2014, 33, 331–349. [Google Scholar] [CrossRef]
- Gupta, P.K.; Balyan, H.S.; Gahlaut, V. QTL Analysis for drought tolerance in wheat: Present status and future possibilities. Agronomy 2017, 7, 5. [Google Scholar] [CrossRef]
- Tricker, P.J.; ElHabti, A.; Schmidt, J.; Fleury, D. The physiological and genetic basis of combined drought and heat tolerance in wheat. J. Exp. Bot. 2018, 69, 3195–3210. [Google Scholar] [CrossRef]
- Cormier, F.; Foulkes, J.; Hirel, B.; Gouache, D.; Moenne-Locco, Y.; Le Gouis, J. Breeding for increased nitrogen-use efficiency: A review for wheat (T. aestivum L.). Plant Breed. 2016, 135, 255–278. [Google Scholar] [CrossRef]
- Le Gouis, J.; Hawkesford, M. Improving the uptake and assimilation of nitrogen in wheat plants. In Achieving Sustainable Wheat Cultivation; Langridge, P., Ed.; BDS Publishing: Cambridge, UK, 2017; pp. 77–99. [Google Scholar]
- de los Campos, G.; Perez-Rodriguez, P.; Bogard, M.; Gouache, D.; Crossa, J. A data-driven simulation platform to predict cultivars’ performances under uncertain weather conditions. Nat. Commun. 2020, 11, 4876. [Google Scholar] [CrossRef]
- Gouache, D.; Bogard, M.; Pegard, M.; Thepot, S.; Garcia, C.; Hourcade, D.; Paux, E.; Oury, F.X.; Rousset, M.; Deswarte, J.C.; et al. Bridging the gap between ideotype and genotype: Challenges and prospects for modelling as exemplified by the case of adapting wheat (Triticum aestivum L.) phenology to climate change in France. Field Crops Res. 2017, 202, 108–121. [Google Scholar] [CrossRef]
- Bogard, M.; Hourcade, D.; Piquemal, B.; Gouache, D.; Deswarte, J.-C.; Throude, M.; Cohan, J.-P. Marker-based crop model-assisted ideotype design to improve avoidance of abiotic stress in bread wheat. J. Exp. Bot. 2021, 72, 1085–1103. [Google Scholar] [CrossRef]
- Barillot, R.; Chambon, C.; Andrieu, B. CN-Wheat, a functional-structural model of carbon and nitrogen distribution in wheat culms after anthesis. I. Model description. Ann. Bot. 2016, 118, 997–1013. [Google Scholar] [CrossRef]
- Barillot, R.; Chambon, C.; Andrieu, B. CN-Wheat, a functional-structural model of carbon and nitrogen distribution in wheat culms after anthesis. II. Model evaluation. Ann. Bot. 2016, 118, 1015–1031. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barillot, R.; Chambon, C.; Fournier, C.; Combes, D.; Pradal, C.; Andrieu, B. Investigation of complex canopies with a functional-structural plant model as exemplified by leaf inclination effect on the functioning of pure and mixed stands of wheat during grain filling. Ann. Bot. 2019, 123, 727–742. [Google Scholar] [CrossRef]
- Gauthier, M.; Barillot, R.; Schneider, A.; Chambon, C.; Fournier, C.; Pradal, C.; Robert, C.; Andrieu, B. A functional structural model of grass development based on metabolic regulation and coordination rules. J. Exp. Bot. 2020, 71, 5454–5468. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.; Barillot, R.; Andrieu, B. Simulating grass phenotypic plasticity as an emergent property of growth zone responses to carbon and nitrogen metabolites. Silico Plants 2021, 3, diab034. [Google Scholar] [CrossRef]
- Furbank, R.T.; Tester, M. Phenomics-technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 2011, 16, 635–644. [Google Scholar] [CrossRef]
- Araus, J.L.; Kefauver, S.C.; Zaman-Allah, M.; Olsen, M.S.; Cairns, J.E. Translating high-throughput phenotyping into genetic gain. Trends Plant Sci. 2018, 23, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Beauchene, K.; Leroy, F.; Fournier, A.; Huet, C.; Bonnefoy, M.; Lorgeou, J.; de Solan, B.; Piquemal, B.; Thomas, S.; Cohan, J.P. Management and characterization of abiotic stress via PhenoFieldR (R), a high-throughput field phenotyping platform. Front. Plant Sci. 2019, 10, 904. [Google Scholar] [CrossRef] [PubMed]
- Comar, A.; Burger, P.; de Solan, B.; Baret, F.; Daumard, F.; Hanocq, J.F. A semi-automatic system for high throughput phenotyping wheat cultivars in-field conditions: Description and first results. Funct. Plant Biol. 2012, 39, 914–924. [Google Scholar] [CrossRef]
- Madec, S.; Baret, F.; de Solan, B.; Thomas, S.; Dutartre, D.; Jezequel, S.; Hemerlé, M.; Colombeau, G.; Comar, A. High-throughput phenotyping of plant height: Comparing unmanned aerial vehicles and ground LiDAR estimates. Front. Plant Sci. 2017, 8, 2002. [Google Scholar] [CrossRef]
- Baret, F.; Clevers, J.G.P.W.; Steven, M.D. The robustness of canopy gap fraction estimates from red and near-infrared reflectances—A comparison of approaches. Remote Sens. Environ. 1995, 54, 141–151. [Google Scholar] [CrossRef]
- Baret, F.; de Solan, B.; Lopez-Lozano, R.; Ma, K.; Weiss, M. GAI estimates of row crops from downward looking digital photos taken perpendicular to rows at 57.5 degrees zenith angle: Theoretical considerations based on 3D architecture models and application to wheat crops. Agric. For. Meteorol. 2010, 150, 1393–1401. [Google Scholar] [CrossRef]
- Verger, A.; Vigneau, N.; Cheron, C.; Gilliot, J.M.; Comar, A.; Baret, F. Green area index from an unmanned aerial system over wheat and rapeseed crops. Remote Sens. Environ. 2014, 152, 654–664. [Google Scholar] [CrossRef]
- Liu, S.Y.; Baret, F.; Abichou, M.; Boudon, F.; Thomas, S.; Zhao, K.G.; Fournier, C.; Andrieu, B.; Irfan, K.; Hemmerle, M.; et al. Estimating wheat green area index from ground-based LiDAR measurement using a 3D canopy structure model. Agric. For. Meteorol. 2017, 247, 12–20. [Google Scholar] [CrossRef]
- Jiang, J.; Baret, F.; Weiss, M.; Liu, S. The Impact of Canopy Structure Assumption on the Retrieval of GAI (Green Area Index) and FIPAR (Fraction of Intercepted Radiation). In Proceedings of the 5th International Symposium on Recent Advances in Quantitative Remote Sensing (RAQRS’V), Torrent, Spain, 18–22 September 2017. [Google Scholar]
- Liu, S.Y.; Martre, P.; Buis, S.; Abichou, M.; Andrieu, B.; Baret, F. Estimation of plant and canopy architectural traits using the digital plant phenotyping platform. Plant Physiol. 2019, 181, 881–890. [Google Scholar] [CrossRef]
- Jiang, J.; Weiss, M.; Liu, S.; Baret, F.; IEEE. The Impact of Canopy Structure Assumption on the Retrieval of GAI and Leaf Chlorophyll Content for Wheat and Maize Crops. In Proceedings of the IEEE International Geoscience and Remote Sensing Symposium (IGARSS), Yokohama, Japan, 28 July–2 August 2019; pp. 7216–7219. [Google Scholar]
- Lu, N.; Wang, W.H.; Zhang, Q.F.; Li, D.; Yao, X.; Tian, Y.C.; Zhu, Y.; Cao, W.X.; Baret, R.; Liu, S.Y.; et al. Estimation of nitrogen nutrition status in winter wheat from unmanned aerial vehicle based multi-angular multispectral imagery. Front. Plant Sci. 2019, 10, 1601. [Google Scholar] [CrossRef]
- Jay, S.; Maupas, F.; Bendoula, R.; Gorretta, N. Retrieving LAI, chlorophyll and nitrogen contents in sugar beet crops from multi-angular optical remote sensing: Comparison of vegetation indices and PROSAIL inversion for field phenotyping. Field Crops Res. 2017, 210, 33–46. [Google Scholar] [CrossRef]
- Liu, S.; Baret, F.; Allard, D.; Jin, X.; Andrieu, B.; Burger, P.; Hemmerlé, M.; Comar, A. A method to estimate plant density and plant spacing heterogeneity: Application to wheat crops. Plant Methods 2017, 13, 38. [Google Scholar] [CrossRef]
- Jin, X.L.; Madec, S.; Dutartre, D.; de Solan, B.; Comar, A.; Baret, F. High-throughput measurements of stem characteristics to estimate ear density and above-ground biomass. Plant Phenomics 2019, 2019, 4820305. [Google Scholar] [CrossRef]
- Madec, S.; Jin, X.; Lu, H.; De Solan, B.; Liu, S.; Duyme, F.; Heritier, E.; Baret, F. Ear density estimation from high resolution RGB imagery using deep learning technique. Agric. For. Meteorol. 2019, 264, 225–234. [Google Scholar] [CrossRef]
- Velumani, K.; Madec, S.; de Solan, B.; Lopez-Lozano, R.; Gillet, J.; Labrosse, J.; Jezequel, S.; Comar, A.; Baret, F. An automatic method based on daily in situ images and deep learning to date wheat heading stage. Field Crops Res. 2020, 252, 107793. [Google Scholar] [CrossRef]
- Liu, S.; Baret, F.; Andrieu, B.; Abichou, M.; Allard, D.; de Solan, B.; Burger, P. Modeling the spatial distribution of plants on the row for wheat crops: Consequences on the green fraction at the canopy level. Comput. Electron. Agric. 2017, 136, 147–156. [Google Scholar] [CrossRef]
- Liu, S.; Baret, F.; Andrieu, B.; Burger, P.; Hemmerlé, M. Estimation of wheat plant density at early stages using high resolution imagery. Front. Plant Sci. 2017, 8, 739. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R.; Yu, J.M. Prospects for genomewide selection for quantitative traits in maize. Crop Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef]
- Crossa, J.; de los Campos, G.; Perez, P.; Gianola, D.; Burgueno, J.; Araus, J.L.; Makumbi, D.; Singh, R.P.; Dreisigacker, S.; Yan, J.B.; et al. Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 2010, 186, 713–724. [Google Scholar] [CrossRef]
- Heffner, E.L.; Lorenz, A.J.; Jannink, J.L.; Sorrells, M.E. Plant breeding with genomic selection: Gain per unit time and cost. Crop Sci. 2010, 50, 1681–1690. [Google Scholar] [CrossRef]
- Todorovska, E.; Christov, N.; Slavov, S.; Christova, P.; Vassilev, D. Biotic stress resistance in wheat—breeding and genomic selection implications. Biotechnol. Biotechnol. Equip. 2009, 23, 1417–1426. [Google Scholar] [CrossRef]
- Heslot, N.; Akdemir, D.; Sorrells, M.E.; Jannink, J.L. Integrating environmental covariates and crop modeling into the genomic selection framework to predict genotype by environment interactions. Theor. Appl. Genet. 2014, 127, 463–480. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Charmet, G.; Tran, L.G.; Auzanneau, J.; Rincent, R.; Bouchet, S. BWGS: A R package for genomic selection and its application to a wheat breeding programme. PLoS ONE 2020, 15, e0222733. [Google Scholar] [CrossRef]
- Rincent, R.; Laloe, D.; Nicolas, S.; Altmann, T.; Brunel, D.; Revilla, P.; Rodriguez, V.M.; Moreno-Gonzalez, J.; Melchinger, A.; Bauer, E.; et al. Maximizing the reliability of genomic selection by optimizing the calibration set of reference individuals: Comparison of methods in two diverse groups of maize inbreds (Zea mays L.). Genetics 2012, 192, 715. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sadoun, S.; Rincent, R.; Auzanneau, J.; Oury, F.X.; Rolland, B.; Heumez, E.; Ravel, C.; Charmet, G.; Bouchet, S. Economical optimization of a breeding scheme by selective phenotyping of the calibration set in a multi-trait context: Application to bread making quality. Theor. Appl. Genet. 2020, 133, 2197–2212. [Google Scholar] [CrossRef]
- Ben Sadoun, S.; Fugeray-Scarbel, A.; Oury, F.-X.; Heumez, E.; Rolland, B.; Auzanneau, J.; Charmet, G.; Lemarié, S.; Bouchet, S. Integration of genomic selection into bread wheat breeding schemes: A simulation pipeline including economic constraints. Crop Breed. Genet. Genom. 2021, 3, e210008. [Google Scholar]
- Fugeray-Scarbel, A.; Lemarié, S.; Bouchet, S.; Ben Sadoun, S. Analyzing the economic effectiveness of genomic selection relative to conventional breeding approaches. In Genomic Prediction of Complex Traits; Ahmadi, N., Bartholomé, J., Eds.; Methods in Molecular Biology; Springer Nature: New York, NY, USA, 2022; in press. [Google Scholar]
- Jarquin, D.; Crossa, J.; Lacaze, X.; Du Cheyron, P.; Daucourt, J.; Lorgeou, J.; Piraux, F.; Guerreiro, L.; Perez, P.; Calus, M.; et al. A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor. Appl. Genet. 2014, 127, 595–607. [Google Scholar] [CrossRef]
- Ly, D.; Huet, S.; Gauffreteau, A.; Rincent, R.; Touzy, G.; Mini, A.; Jannink, J.-L.; Cormier, F.; Paux, E.; Lafarge, S.; et al. Whole-genome prediction of reaction norms to environmental stress in bread wheat (Triticum aestivum L.) by genomic random regression. Field Crops Res. 2018, 216, 32–41. [Google Scholar] [CrossRef]
- Ly, D.; Chenu, K.; Gauffreteau, A.; Rincent, R.; Huet, S.; Gouache, D.; Martre, P.; Bordes, J.; Charmet, G. Nitrogen nutrition index predicted by a crop model improves the genomic prediction of grain number for a bread wheat core collection. Field Crops Res. 2017, 214, 331–340. [Google Scholar] [CrossRef]
- Rincent, R.; Kuhn, E.; Monod, H.; Oury, F.-X.; Rousset, M.; Allard, V.; Le Gouis, J. Optimization of multi-environment trials for genomic selection based on crop models. Theor. Appl. Genet. 2017, 130, 1735–1752. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.C.; Stone, E.A.; Ayroles, J.F. The genetics of quantitative traits: Challenges and prospects. Nat. Rev. Genet. 2009, 10, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, O.; Urrutia, M.; Bernillon, S.; Giauffret, C.; Tardieu, F.; Le Gouis, J.; Langlade, N.; Charcosset, A.; Moing, A.; Gibon, Y. Fortune telling: Metabolic markers of plant performance. Metabolomics 2016, 12, 158. [Google Scholar] [CrossRef] [PubMed]
- Robert, P.; Brault, C.; Rincent, R.; Segura, V. Phenomic selection: A new and efficient alternative to genomic selection. Methods in Molecular Biology 2021, 8, 3961–3972. [Google Scholar]
- Alaux, M.; Rogers, J.; Letellier, T.; Flores, R.; Alfama, F.; Pommier, C.; Mohellibi, N.; Durand, S.; Kimmel, E.; Michotey, C.; et al. Linking the international wheat genome sequencing consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol. 2018, 19, 111. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; Santos, L.B.D.; Bourne, P.E.; et al. Comment: The FAIR Guiding principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]
- Selby, P.; Abbeloos, R.; Backlund, J.E.; Salido, M.B.; Bauchet, G.; Benites-Alfaro, O.E.; Birkett, C.; Calaminos, V.C.; Carceller, P.; Cornut, G.; et al. BrAPI-an application programming interface for plant breeding applications. Bioinformatics 2019, 35, 4147–4155. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Matteis, L.; Skofic, M.; Portugal, A.; McLaren, G.; Hyman, G.; Arnaud, E. Bridging the phenotypic and genetic data useful for integrated breeding through a data annotation using the crop ontology developed by the crop communities of practice. Front. Physiol. 2012, 3, 326. [Google Scholar] [CrossRef]
- Pommier, C.; Michotey, C.; Cornut, G.; Roumet, P.; Duchêne, E.; Flores, R.; Lebreton, A.; Alaux, M.; Durand, S.; Kimmel, E.; et al. Applying FAIR principles to plant phenotypic data management in GnpIS. Plant Phenom. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- IPCC. Summary for policymakers. In Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S.L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M.I., et al., Eds.; Cambridge University Press: Cambridge, UK, 2021; pp. 1–31. [Google Scholar]
- Ben-Ari, T.; Boe, J.; Ciais, P.; Lecerf, R.; Van der Velde, M.; Makowski, D. Causes and implications of the unforeseen 2016 extreme yield loss in the breadbasket of France. Nat. Commun. 2018, 9, 1627. [Google Scholar] [CrossRef] [PubMed]
- HLPE. Agroecological and other Innovative Approaches for Sustainable Agriculture and Food Systems that Enhance Food Security and Nutrition. A Report by the High Level Panel of Experts on Food Security and Nutrition of the Committee on World Food Security; HLPE: Rome, Italy, 2019; pp. 1–162. [Google Scholar]
- Krishnappa, G.; Savadi, S.; Tyagi, B.S.; Singh, S.K.; Mamrutha, H.M.; Kumar, S.; Mishra, C.N.; Khan, H.; Gangadhara, K.; Uday, G.; et al. Integrated genomic selection for rapid improvement of crops. Genomics 2021, 113, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Saini, D.K.; Chopra, Y.; Singh, J.; Sandhu, K.S.; Kumar, A.; Bazzer, S.; Srivastava, P. Comprehensive evaluation of mapping complex traits in wheat using genome-wide association studies. Mol. Breed. 2022, 42, 1. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Size | Uses | Publications |
|---|---|---|---|
| Axiom SNP arrays | 409,685 SNPs | Phylogeny, mapping, GWAS, GS | [14,15,16,17,18,19,20,21] |
| 34,746 SNPs | GWAS, GS | ||
| Chinese Spring (CS) × Renan Genetic map | 146,602 SNPs | 21 CS pseudomolecules assembly, analysis of the recombination landscape, QTL detection | [22,23,24,25,26] |
| Chromosome 1B sequence | 10,395 BACs 13,277 scaffolds 920 Mb | Analysis of the transcriptional landscape, the impact of transposable elements on genome structure and evolution, etc. | [22,23,24,25] |
| Level | Trait | Method | Sensor | Configuration | Vector | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| RGB | Multispectral | LiDAR | View Direction | Ground Sampling Distance (cm) | UAV | Phenomobile | ||||
| Canopy | Vegetation Index (VI) | Band combination | 0° | 20 | [96] | |||||
| Plant height | Structure from motion | 0° | 1 | [97] | ||||||
| Distribution of height | ±35° | 0.5 | [97] | |||||||
| Vegetation Fraction (VF) | DL segmentation | 0° | 0.05 | Madec et al. (pers.com) | ||||||
| Height threshold | ±35° | 0.5 | Lopez-Lozano et al. (pers.com) | |||||||
| Green Fraction (GF) | SVM/random forest | 0°-45° | 0.05 | Serouart et al. (pers.com) | ||||||
| DL segmentation | 0°–45° | 0.05 | Madec et al. (pers.com) | |||||||
| 1D RTM inversion | 0° | 20 | [98] | |||||||
| Green Area Index (GAI) | Green fraction turbid | 0°–45° | 0.05 | [99] | ||||||
| 1D RTM inversion | 0°–45° | 20 | [100] | |||||||
| 3D RTM inversion | ±35° | 0.05–0.5 | [101] | |||||||
| Plant Area Index (PAI) | 1D turbid | ±35° | 0.5 | Lopez-Lozano et al. (pers.com) | ||||||
| Fraction of Intercepted Radiation (FIPAR) | 1D RTM inversion | 0° | 20 | [102] | ||||||
| Green Fraction turbid | 0°–45° | 0.05 | [103] | |||||||
| 1D turbid | ±35° | 0.5 | Lopez-Lozano et al. (pers.com) | |||||||
| Average Inclination Angle (AIA) | 1D RTM inversion | 0° | 20 | [102] | ||||||
| 1D turbid | 0°–45° | 0.05 | Liu et al. (pers.com) | |||||||
| 1D turbid | ±35° | 0.5 | Lopez-Lozano et al. (pers.com) | |||||||
| 3D inversion | ±35° | 0.05–0.5 | [103] | |||||||
| Canopy Chlorophyll Content (CCC) | 1D RTM inversion | 0°–45° | 20 | [104] | ||||||
| VI empirical | 0° | 20 | [105] | |||||||
| VI empirical | 0° | 0.05 | [106] | |||||||
| 3D Distribution of Area | 1D turbid | ±35° | 0.5 | Lopez-Lozano et al. (pers.com) | ||||||
| Organ | Plant density | DL at emergence | 45° | 0.05 | [107] | |||||
| Stem density | DL at harvest | 0° | 0.02 | [108] | ||||||
| Stem diameter | DL at harvest | 0° | 0.02 | [108] | ||||||
| Ear density | DL at reproductive stage | 0° | 0.05 | [109] | ||||||
| Leaf Chlorophyll Content | 1D RTM inversion | 0° | 20 | [105] | ||||||
| VI empirical | 0° | 0.05 | Jay et al. (pers.com) | |||||||
| VI empirical | 0° | 0.05 | Jay et al. (pers.com) | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paux, E.; Lafarge, S.; Balfourier, F.; Derory, J.; Charmet, G.; Alaux, M.; Perchet, G.; Bondoux, M.; Baret, F.; Barillot, R.; et al. Breeding for Economically and Environmentally Sustainable Wheat Varieties: An Integrated Approach from Genomics to Selection. Biology 2022, 11, 149. https://doi.org/10.3390/biology11010149
Paux E, Lafarge S, Balfourier F, Derory J, Charmet G, Alaux M, Perchet G, Bondoux M, Baret F, Barillot R, et al. Breeding for Economically and Environmentally Sustainable Wheat Varieties: An Integrated Approach from Genomics to Selection. Biology. 2022; 11(1):149. https://doi.org/10.3390/biology11010149
Chicago/Turabian StylePaux, Etienne, Stéphane Lafarge, François Balfourier, Jérémy Derory, Gilles Charmet, Michael Alaux, Geoffrey Perchet, Marion Bondoux, Frédéric Baret, Romain Barillot, and et al. 2022. "Breeding for Economically and Environmentally Sustainable Wheat Varieties: An Integrated Approach from Genomics to Selection" Biology 11, no. 1: 149. https://doi.org/10.3390/biology11010149
APA StylePaux, E., Lafarge, S., Balfourier, F., Derory, J., Charmet, G., Alaux, M., Perchet, G., Bondoux, M., Baret, F., Barillot, R., Ravel, C., Sourdille, P., Le Gouis, J., & on behalf of the BREEDWHEAT Consortium. (2022). Breeding for Economically and Environmentally Sustainable Wheat Varieties: An Integrated Approach from Genomics to Selection. Biology, 11(1), 149. https://doi.org/10.3390/biology11010149