
The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation


Abstract
Simple Summary
Abstract
1. Introduction
2. Type I IFNs: A Complex Signaling Network
2.1. Upstream Triggers of Type I IFNs
2.2. Downstream Effectors of Type I IFNs
3. Type I IFN-Induced Genetic and Epigenetic Signatures
4. Type I IFNs and Cancer: A Troubled Relationship
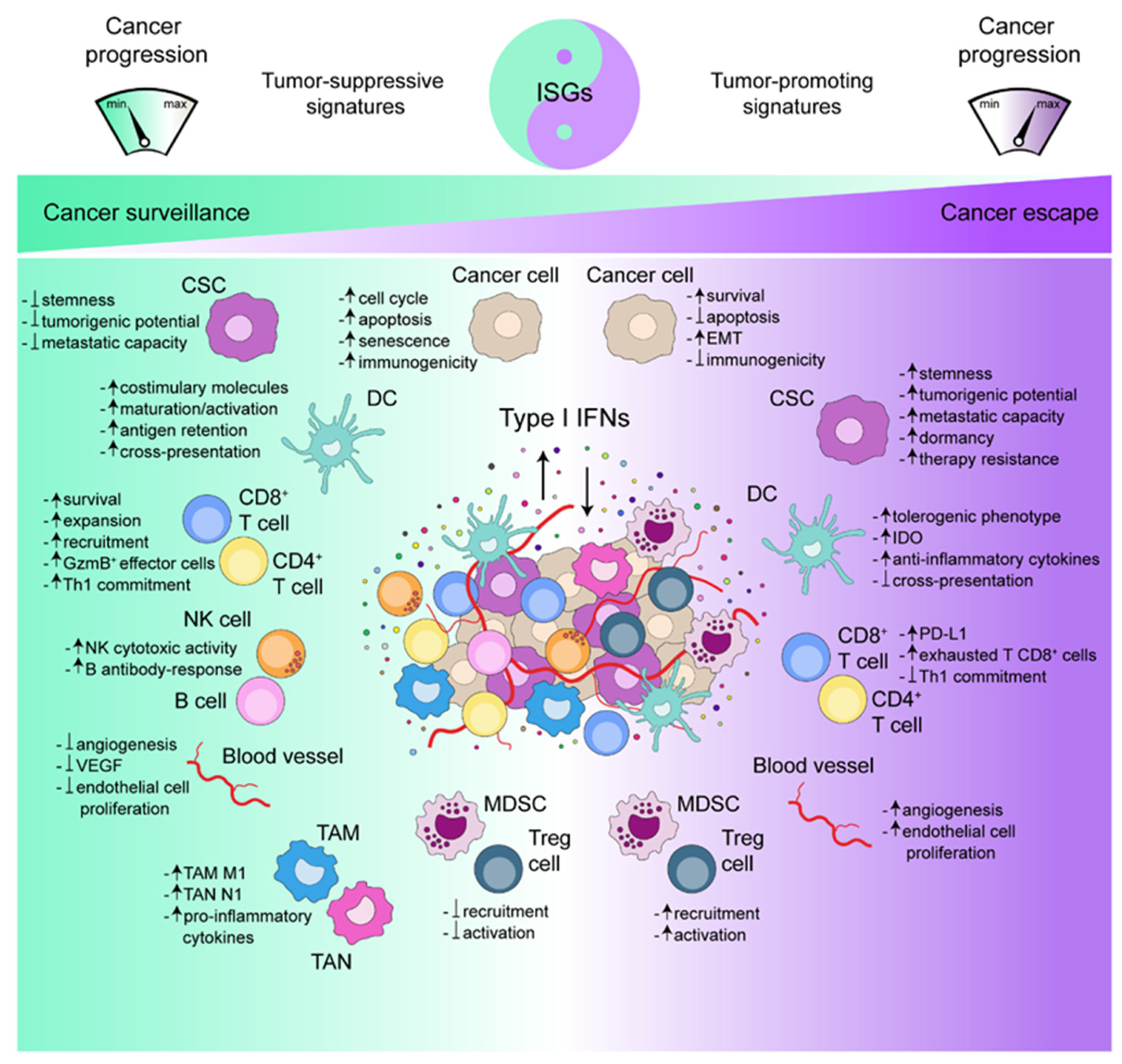
4.1. Type I IFNs and Cancer Immunosurveillance
4.2. Type I IFNs and Cancer Immunoescape
5. Type I IFNs and Cancer Therapy: From Response to Resistance
5.1. Type I IFNs and Cytotoxic Therapies
5.2. Type I IFN Monotherapies
5.3. Type I IFNs and Immunotherapies
5.4. Type I IFNs and Therapy Resistance
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.; Remédios, C.; et al. Cancer cell—Autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma Emerg. Surg. 2019, 46, 751–775. [Google Scholar] [CrossRef]
- Alick, I.; Lindenmann, J. Virus interference: I. The interferon. Proc. R. Soc. Lond. B 1957, 147, 258–267, Republished in CA Cancer J. Clin. 1988, 38, 280–290. [Google Scholar]
- Fuertes, M.B.; Woo, S.-R.; Burnett, B.; Fu, Y.-X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2012, 34, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. Type I interferons: Diversity of sources, production pathways and effects on immune responses. Curr. Opin. Virol. 2011, 1, 463–475. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-F.; Gong, M.-J.; Zhao, F.-R.; Shao, J.-J.; Xie, Y.-L.; Zhang, Y.-G.; Chang, H.-Y. Type I interferons: Distinct biological activities and current applications for viral infection. Cell. Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2013, 14, 36–49. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Baldridge, M.T. Interferon-lambda: A potent regulator of intestinal viral infections. Front. Immunol. 2017, 8, 749. [Google Scholar] [CrossRef]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Krause, C.D.; Pestka, S. Cut, copy, move, delete: The study of human interferon genes reveal multiple mechanisms underlying their evolution in amniotes. Cytokine 2015, 76, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Björck, P.; Leong, H.X.; Engleman, E.G. Plasmacytoid dendritic cell dichotomy: Identification of IFN-α producing cells as a phenotypically and functionally distinct subset. J. Immunol. 2010, 186, 1477–1485. [Google Scholar] [CrossRef]
- Gough, D.J.; Messina, N.L.; Clarke, C.J.; Johnstone, R.W.; Levy, D.E. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity 2012, 36, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Leviyang, S.; Strawn, N.; Griva, I. Regulation of interferon stimulated gene expression levels at homeostasis. Cytokine 2020, 126, 154870. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 663–671. [Google Scholar] [CrossRef]
- Garg, A.D.; Agostinis, P. Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol. Rev. 2017, 280, 126–148. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017, 6, e1314424. [Google Scholar] [CrossRef] [PubMed]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of endogenous nucleic acids by the innate immune system. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Detection of microbial infections through innate immune sensing of nucleic acids. Annu. Rev. Microbiol. 2018, 72, 447–478. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Perkins, D.J.; Vogel, S.N. Space and time: New considerations about the relationship between toll-like receptors (TLRs) and type I interferons (IFNs). Cytokine 2015, 74, 171–174. [Google Scholar] [CrossRef]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Sousa, C.R. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, X.; Wu, L.; Wang, X.; Liu, Z. The anticancer functions of RIG-I-like receptors, RIG-I and MDA5, and their applications in cancer therapy. Transl. Res. 2017, 190, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.X.; Wan, H.; Nie, L.; Shao, T.; Xiang, L.X.; Shao, J.Z. RIG-I: A multifunctional protein beyond a pattern recognition receptor. Protein Cell 2018, 9, 246–253. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Tsolis, R.M. NOD1 and NOD2: Beyond peptidoglycan sensing. Trends Immunol. 2017, 38, 758–767. [Google Scholar] [CrossRef]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Wang, Z.; Choi, M.K.; Ban, T.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 5477–5482. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Chen, R.H.; Du, Y.; Han, P.; Wang, H.B.; Liang, F.Y.; Feng, G.K.; Zhou, A.J.; Cai, M.Y.; Zhong, Q.; Zeng, M.S.; et al. ISG15 predicts poor prognosis and promotes cancer stem cell phenotype in nasopharyngeal carcinoma. Oncotarget 2016, 7, 16910–16922. [Google Scholar] [CrossRef]
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef]
- Di Micco, A.; Frera, G.; Lugrin, J.; Jamilloux, Y.; Hsu, E.T.; Tardivel, A.; De Gassart, A.; Zaffalon, L.; Bujisic, B.; Siegert, S.; et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl. Acad. Sci. USA 2016, 113, E4671–E4680. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, S.A.; Morrone, S.R.; van Oijen, A.M.; Sohn, J. The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. eLife 2015, 4, e11721. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Kim, T.; Pazhoor, S.; Bao, M.; Zhang, Z.; Hanabuchi, S.; Facchinetti, V.; Bover, L.; Plumas, J.; Chaperot, L.; Qin, J.; et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15181–15186. [Google Scholar] [CrossRef]
- Meng, J.; Liu, X.; Cao, X. A new cytosolic DNA-recognition pathway for DNA-induced inflammatory responses. Cell. Mol. Immunol. 2014, 11, 506–509. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kobayashi, J.; Saitoh, T.; Maruyama, K.; Ishii, K.J.; Barber, G.N.; Komatsu, K.; Akira, S.; Kawai, T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA 2013, 110, 2969–2974. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Demaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA sensing in organismal tumor control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Iwanaszko, M.; Kimmel, M. NF-kappaB and IRF pathways: Cross-regulation on target genes promoter level. BMC Genom. 2015, 16, 307. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Negishi, H.; Taniguchi, T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef]
- Savan, R. Post-transcriptional regulation of interferons and their signaling pathways. J. Interf. Cytokine Res. 2014, 34, 318–329. [Google Scholar] [CrossRef]
- Khabar, K.S.; Young, H.A. Post-transcriptional control of the interferon system. Biochimie 2007, 89, 761–769. [Google Scholar] [CrossRef]
- Venkatesh, D.; Ernandez, T.; Rosetti, F.; Batal, I.; Cullere, X.; Luscinskas, F.W.; Zhang, Y.; Stavrakis, G.; Garcia-Cardena, G.; Horwitz, B.H.; et al. Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-beta autocrine signaling to promote monocyte recruitment. Immunity 2013, 38, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Yarilina, A.; Park-Min, K.H.; Antoniv, T.; Hu, X.; Ivashkiv, L.B. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol. 2008, 9, 378–387. [Google Scholar] [CrossRef]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I interferon (IFN)-regulated activation of canonical and non-canonical signaling pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Moraga, I.; Levin, D.; Krutzik, P.O.; Podoplelova, Y.; Trejo, A.; Lee, C.; Yarden, G.; Vleck, S.E.; Glenn, J.S.; et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 2011, 146, 621–632. [Google Scholar] [CrossRef]
- Schreiber, G. The molecular basis for differential type I interferon signaling. J. Biol. Chem. 2017, 292, 7285–7294. [Google Scholar] [CrossRef]
- Lavoie, T.B.; Kalie, E.; Crisafulli-Cabatu, S.; Abramovich, R.; DiGioia, G.; Moolchan, K.; Pestka, S.; Schreiber, G. Binding and activity of all human alpha interferon subtypes. Cytokine 2011, 56, 282–289. [Google Scholar] [CrossRef]
- Uze, G.; Schreiber, G.; Piehler, J.; Pellegrini, S. The receptor of the type I interferon family. Curr. Top. Microbiol. Immunol. 2007, 316, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Wallweber, H.J.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E.J. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.C.; Nogusa, S.; Thapa, R.J.; Basagoudanavar, S.H.; Gill, A.L.; Merali, S.; Barrero, C.A.; Balachandran, S.; Gamero, A.M. Identification of STAT2 serine 287 as a novel regulatory phosphorylation site in type I interferon-induced cellular responses. J. Biol. Chem. 2013, 288, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Lukhele, S.; Boukhaled, G.M.; Brooks, D.G. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol. 2019, 43, 101277. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Cheon, H.; Wang, Y. Responses to cytokines and interferons that depend upon JAKs and STATs. Cold Spring Harb. Perspect. Biol. 2018, 10, a028555. [Google Scholar] [CrossRef]
- Cheon, H.; Borden, E.C.; Stark, G.R. Interferons and their stimulated genes in the tumor microenvironment. Semin. Oncol. 2014, 41, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoite, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.Y.; Feng, T.; et al. Parsing the interferon transcriptional network and its disease associations. Cell 2016, 164, 564–578. [Google Scholar] [CrossRef]
- Cheon, H.; Stark, G.R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Grandvaux, N. STAT2 and IRF9: Beyond ISGF3. JAK-STAT 2013, 2, e27521. [Google Scholar] [CrossRef]
- Blaszczyk, K.; Nowicka, H.; Kostyrko, K.; Antonczyk, A.; Wesoly, J.; Bluyssen, H.A. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev. 2016, 29, 71–81. [Google Scholar] [CrossRef]
- Platanitis, E.; Demiroz, D.; Schneller, A.; Fischer, K.; Capelle, C.; Hartl, M.; Gossenreiter, T.; Muller, M.; Novatchkova, M.; Decker, T. A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat. Commun. 2019, 10, 2921. [Google Scholar] [CrossRef]
- Wang, W.B.; Levy, D.E.; Lee, C.K. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 2011, 187, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; He, S.F.; Wang, W.; Ren, H.; Qi, Z.T. Interferon alpha antagonizes STAT3 and SOCS3 signaling triggered by hepatitis C virus. Cytokine 2016, 80, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Muller, M.; Decker, T. The regulation of inflammation by interferons and their STATs. JAK-STAT 2013, 2, e23820. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jiang, M.; Pernis, A.B. IFN-alpha activates Stat6 and leads to the formation of Stat2:Stat6 complexes in B cells. J. Immunol. 1999, 163, 3834–3841. [Google Scholar]
- Nguyen, K.B.; Watford, W.T.; Salomon, R.; Hofmann, S.R.; Pien, G.C.; Morinobu, A.; Gadina, M.; O’Shea, J.J.; Biron, C.A. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 2002, 297, 2063–2066. [Google Scholar] [CrossRef]
- Uddin, S.; Lekmine, F.; Sassano, A.; Rui, H.; Fish, E.N.; Platanias, L.C. Role of Stat5 in type I interferon-signaling and transcriptional regulation. Biochem. Biophys. Res. Commun. 2003, 308, 325–330. [Google Scholar] [CrossRef]
- Hsu, Y.A.; Huang, C.C.; Kung, Y.J.; Lin, H.J.; Chang, C.Y.; Lee, K.R.; Wan, L. The anti-proliferative effects of type I IFN involve STAT6-mediated regulation of SP1 and BCL6. Cancer Lett. 2016, 375, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.N.; Uddin, S.; Korkmaz, M.; Majchrzak, B.; Druker, B.J.; Platanias, L.C. Activation of a CrkL-stat5 signaling complex by type I interferons. J. Biol. Chem. 1999, 274, 571–573. [Google Scholar] [CrossRef]
- Katsoulidis, E.; Li, Y.; Mears, H.; Platanias, L.C. The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J. Interf. Cytokine Res. 2005, 25, 749–756. [Google Scholar] [CrossRef]
- Li, Y.; Sassano, A.; Majchrzak, B.; Deb, D.K.; Levy, D.E.; Gaestel, M.; Nebreda, A.R.; Fish, E.N.; Platanias, L.C. Role of p38alpha map kinase in type I interferon signaling. J. Biol. Chem. 2004, 279, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Sassano, A.; Joseph, A.M.; Majchrzak-Kita, B.; Eklund, E.A.; Verma, A.; Brachmann, S.M.; Fish, E.N.; Platanias, L.C. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J. Immunol. 2008, 181, 7316–7323. [Google Scholar] [CrossRef] [PubMed]
- Saleiro, D.; Mehrotra, S.; Kroczynska, B.; Beauchamp, E.M.; Lisowski, P.; Majchrzak-Kita, B.; Bhagat, T.D.; Stein, B.L.; McMahon, B.; Altman, J.K.; et al. Central role of ULK1 in type I interferon signaling. Cell Rep. 2015, 11, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Saleiro, D.; Platanias, L.C. Interferon signaling in cancer. Non-canonical pathways and control of intracellular immune checkpoints. Semin. Immunol. 2019, 43, 101299. [Google Scholar] [CrossRef]
- Mavrommatis, E.; Fish, E.N.; Platanias, L.C. The schlafen family of proteins and their regulation by interferons. J. Interf. Cytokine Res. 2013, 33, 206–210. [Google Scholar] [CrossRef]
- Kosciuczuk, E.M.; Mehrotra, S.; Saleiro, D.; Kroczynska, B.; Majchrzak-Kita, B.; Lisowski, P.; Driehaus, C.; Rogalska, A.; Turner, A.; Lienhoop, T.; et al. Sirtuin 2-mediated deacetylation of cyclin-dependent kinase 9 promotes STAT1 signaling in type I interferon responses. J. Biol. Chem. 2019, 294, 827–837. [Google Scholar] [CrossRef]
- Arslan, A.D.; Sassano, A.; Saleiro, D.; Lisowski, P.; Kosciuczuk, E.M.; Fischietti, M.; Eckerdt, F.; Fish, E.N.; Platanias, L.C. Human SLFN5 is a transcriptional co-repressor of STAT1-mediated interferon responses and promotes the malignant phenotype in glioblastoma. Oncogene 2017, 36, 6006–6019. [Google Scholar] [CrossRef]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Targeting of type I interferon in systemic autoimmune diseases. Transl. Res. 2015, 165, 296–305. [Google Scholar] [CrossRef]
- Khoo, J.J.; Forster, S.; Mansell, A. Toll-like receptors as interferon-regulated genes and their role in disease. J. Interf. Cytokine Res. 2011, 31, 13–25. [Google Scholar] [CrossRef]
- Kumar, K.G.; Barriere, H.; Carbone, C.J.; Liu, J.; Swaminathan, G.; Xu, P.; Li, Y.; Baker, D.P.; Peng, J.; Lukacs, G.L.; et al. Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J. Cell Biol. 2007, 179, 935–950. [Google Scholar] [CrossRef]
- Kumar, K.G.; Varghese, B.; Banerjee, A.; Baker, D.P.; Constantinescu, S.N.; Pellegrini, S.; Fuchs, S.Y. Basal ubiquitin-independent internalization of interferon alpha receptor is prevented by Tyk2-mediated masking of a linear endocytic motif. J. Biol. Chem. 2008, 283, 18566–18572. [Google Scholar] [CrossRef]
- Liu, J.; Plotnikov, A.; Banerjee, A.; Suresh Kumar, K.G.; Ragimbeau, J.; Marijanovic, Z.; Baker, D.P.; Pellegrini, S.; Fuchs, S.Y. Ligand-independent pathway that controls stability of interferon alpha receptor. Biochem. Biophys. Res. Commun. 2008, 367, 388–393. [Google Scholar] [CrossRef][Green Version]
- Wilmes, S.; Beutel, O.; Li, Z.; Francois-Newton, V.; Richter, C.P.; Janning, D.; Kroll, C.; Hanhart, P.; Hotte, K.; You, C.; et al. Receptor dimerization dynamics as a regulatory valve for plasticity of type I interferon signaling. J. Cell Biol. 2015, 209, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef]
- Arimoto, K.I.; Lochte, S.; Stoner, S.A.; Burkart, C.; Zhang, Y.; Miyauchi, S.; Wilmes, S.; Fan, J.B.; Heinisch, J.J.; Li, Z.; et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat. Struct. Mol. Biol. 2017, 24, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Francois-Newton, V.; Magno de Freitas, G.A.; Payelle-Brogard, B.; Monneron, D.; Pichard-Garcia, L.; Piehler, J.; Pellegrini, S.; Uze, G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS ONE 2011, 6, e22200. [Google Scholar] [CrossRef]
- Francois-Newton, V.; Livingstone, M.; Payelle-Brogard, B.; Uze, G.; Pellegrini, S. USP18 establishes the transcriptional and anti-proliferative interferon alpha/beta differential. Biochem. J. 2012, 446, 509–516. [Google Scholar] [CrossRef]
- Goldmann, T.; Zeller, N.; Raasch, J.; Kierdorf, K.; Frenzel, K.; Ketscher, L.; Basters, A.; Staszewski, O.; Brendecke, S.M.; Spiess, A.; et al. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015, 34, 1612–1629. [Google Scholar] [CrossRef]
- Speer, S.D.; Li, Z.; Buta, S.; Payelle-Brogard, B.; Qian, L.; Vigant, F.; Rubino, E.; Gardner, T.J.; Wedeking, T.; Hermann, M.; et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat. Commun. 2016, 7, 11496. [Google Scholar] [CrossRef] [PubMed]
- Linossi, E.M.; Nicholson, S.E. Kinase inhibition, competitive binding and proteasomal degradation: Resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol. Rev. 2015, 266, 123–133. [Google Scholar] [CrossRef]
- Piganis, R.A.; De Weerd, N.A.; Gould, J.A.; Schindler, C.W.; Mansell, A.; Nicholson, S.E.; Hertzog, P.J. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon alpha receptor (IFNAR1)-associated tyrosine kinase Tyk2. J. Biol. Chem. 2011, 286, 33811–33818. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-stimulated genes: What do they all do? Annu. Rev. 2019, 6, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, P.J.; Williams, B.R. Fine tuning type I interferon responses. Cytokine Growth Factor Rev. 2013, 24, 217–225. [Google Scholar] [CrossRef]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of type I interferon-mediated responses in cancer therapy: From anti-tumor immunity to resistance. Front. Oncol. 2018, 8, 322. [Google Scholar] [CrossRef] [PubMed]
- Platanitis, E.; Decker, T. Regulatory networks involving STATs, IRFs, and NFkappaB in inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef]
- Josset, L.; Tchitchek, N.; Gralinski, L.E.; Ferris, M.T.; Eisfeld, A.J.; Green, R.R.; Thomas, M.J.; Tisoncik-Go, J.; Schroth, G.P.; Kawaoka, Y.; et al. Annotation of long non-coding RNAs expressed in collaborative cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biol. 2014, 11, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.C.; Tate, M.D.; Hertzog, P.J. MicroRNA as type I interferon-regulated transcripts and modulators of the innate immune response. Front. Immunol. 2015, 6, 334. [Google Scholar] [CrossRef] [PubMed]
- Trilling, M.; Bellora, N.; Rutkowski, A.J.; de Graaf, M.; Dickinson, P.; Robertson, K.; da Costa, O.P.; Ghazal, P.; Friedel, C.C.; Alba, M.M.; et al. Deciphering the modulation of gene expression by type I and II interferons combining 4sU-tagging, translational arrest and in silico promoter analysis. Nucleic Acids Res. 2013, 41, 8107–8125. [Google Scholar] [CrossRef]
- Megger, D.A.; Philipp, J.; Le-Trilling, V.T.K.; Sitek, B.; Trilling, M. Deciphering of the human interferon-regulated proteome by mass spectrometry-based quantitative analysis reveals extent and dynamics of protein induction and repression. Front. Immunol. 2017, 8, 1139. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J. Type I interferonopathies: A novel set of inborn errors of immunity. Ann. N. Y. Acad. Sci. 2011, 1238, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhao, M.; Chang, C.; Wu, H.; Lu, Q. Type I interferons in the pathogenesis and treatment of autoimmune diseases. Clin. Rev. Allergy Immunol. 2020, 59, 248–272. [Google Scholar] [CrossRef]
- Ascierto, M.L.; Kmieciak, M.; Idowu, M.O.; Manjili, R.; Zhao, Y.; Grimes, M.; Dumur, C.; Wang, E.; Ramakrishnan, V.; Wang, X.Y.; et al. A signature of immune function genes associated with recurrence-free survival in breast cancer patients. Breast Cancer Res. Treat. 2012, 131, 871–880. [Google Scholar] [CrossRef]
- Linsley, P.S.; Speake, C.; Whalen, E.; Chaussabel, D. Copy number loss of the interferon gene cluster in melanomas is linked to reduced T cell infiltrate and poor patient prognosis. PLoS ONE 2014, 9, e109760. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Duarte, C.W.; Willey, C.D.; Zhi, D.; Cui, X.; Harris, J.J.; Vaughan, L.K.; Mehta, T.; McCubrey, R.O.; Khodarev, N.N.; Weichselbaum, R.R.; et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS ONE 2012, 7, e29653. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef]
- Smale, S.T.; Tarakhovsky, A.; Natoli, G. Chromatin contributions to the regulation of innate immunity. Annu. Rev. Immunol. 2014, 32, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, R.; Piccolo, V.; Barozzi, I.; Polletti, S.; Termanini, A.; Bonifacio, S.; Curina, A.; Prosperini, E.; Ghisletti, S.; Natoli, G. Latent enhancers activated by stimulation in differentiated cells. Cell 2013, 152, 157–171. [Google Scholar] [CrossRef]
- Kamada, R.; Yang, W.; Zhang, Y.; Patel, M.C.; Yang, Y.; Ouda, R.; Dey, A.; Wakabayashi, Y.; Sakaguchi, K.; Fujita, T.; et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl. Acad. Sci. USA 2018, 115, E9162–E9171. [Google Scholar] [CrossRef]
- Park, S.H.; Kang, K.; Giannopoulou, E.; Qiao, Y.; Kang, K.; Kim, G.; Park-Min, K.H.; Ivashkiv, L.B. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 2017, 18, 1104–1116. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, S.; Natoli, G. Short-term memory of danger signals and environmental stimuli in immune cells. Nat. Immunol. 2013, 14, 777–784. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Musella, M.; Corradi, F.; Sistigu, A.; Vitale, S.; Abdel Rehim, S.S.; Mattiello, L.; Malacaria, E.; Galassi, C.; Signore, M.; et al. Control of replication stress and mitosis in colorectal cancer stem cells through the interplay of PARP1, MRE11 and RAD51. Cell Death Differ. 2021, 28, 2060–2082. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, S.; Turdo, A.; Todaro, M.; Stassi, G. Role of type I and II interferons in colorectal cancer and melanoma. Front. Immunol. 2017, 8, 878. [Google Scholar] [CrossRef]
- Hobeika, A.C.; Subramaniam, P.S.; Johnson, H.M. IFNalpha induces the expression of the cyclin-dependent kinase inhibitor p21 in human prostate cancer cells. Oncogene 1997, 14, 1165–1170. [Google Scholar] [CrossRef]
- Balkwill, F.; Watling, D.; Taylor-Papadimitriou, J. Inhibition by lymphoblastoid interferon of growth of cells derived from the human breast. Int. J. Cancer 1978, 22, 258–265. [Google Scholar] [CrossRef]
- Sangfelt, O.; Erickson, S.; Grander, D. Mechanisms of interferon-induced cell cycle arrest. Front. Biosci. 2000, 5, D479–D487. [Google Scholar] [CrossRef]
- Matsuoka, M.; Tani, K.; Asano, S. Interferon-alpha-induced G1 phase arrest through up-regulated expression of CDK inhibitors, p19Ink4D and p21Cip1 in mouse macrophages. Oncogene 1998, 16, 2075–2086. [Google Scholar] [CrossRef]
- Katayama, T.; Nakanishi, K.; Nishihara, H.; Kamiyama, N.; Nakagawa, T.; Kamiyama, T.; Iseki, K.; Tanaka, S.; Todo, S. Type I interferon prolongs cell cycle progression via p21WAF1/CIP1 induction in human colon cancer cells. Int. J. Oncol. 2007, 31, 613–620. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, W.; Li, Y.; Berenzon, D.; Wang, X.; Wang, J.; Mascarenhas, J.; Xu, M.; Hoffman, R. Interferon-alpha targets JAK2V617F-positive hematopoietic progenitor cells and acts through the p38 MAPK pathway. Exp. Hematol. 2010, 38, 472–480. [Google Scholar] [CrossRef]
- Platanias, L.C.; Uddin, S.; Bruno, E.; Korkmaz, M.; Ahmad, S.; Alsayed, Y.; Van Den Berg, D.; Druker, B.J.; Wickrema, A.; Hoffman, R. CrkL and CrkII participate in the generation of the growth inhibitory effects of interferons on primary hematopoietic progenitors. Exp. Hematol. 1999, 27, 1315–1321. [Google Scholar] [CrossRef]
- Thyrell, L.; Erickson, S.; Zhivotovsky, B.; Pokrovskaja, K.; Sangfelt, O.; Castro, J.; Einhorn, S.; Grander, D. Mechanisms of interferon-alpha induced apoptosis in malignant cells. Oncogene 2002, 21, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.A.; Lei, H.; Maron, D.J.; Wilson, J.M.; Barsoum, J.; Fraker, D.L.; El-Deiry, W.S.; Spitz, F.R. Stat1-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand and the cell-surface death signaling pathway by interferon beta in human cancer cells. Cancer Res. 2003, 63, 5299–5307. [Google Scholar] [PubMed]
- Bernardo, A.R.; Cosgaya, J.M.; Aranda, A.; Jimenez-Lara, A.M. Synergy between RA and TLR3 promotes type I IFN-dependent apoptosis through upregulation of TRAIL pathway in breast cancer cells. Cell Death Dis. 2013, 4, e479. [Google Scholar] [CrossRef]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: Role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Frank, T.; Tuppi, M.; Hugle, M.; Dotsch, V.; van Wijk, S.J.L.; Fulda, S. Cell cycle arrest in mitosis promotes interferon-induced necroptosis. Cell Death Differ. 2019, 26, 2046–2060. [Google Scholar] [CrossRef] [PubMed]
- Katlinskaya, Y.V.; Katlinski, K.V.; Yu, Q.; Ortiz, A.; Beiting, D.P.; Brice, A.; Davar, D.; Sanders, C.; Kirkwood, J.M.; Rui, H.; et al. Suppression of type I interferon signaling overcomes oncogene-induced senescence and mediates melanoma development and progression. Cell Rep. 2016, 15, 171–180. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed]
- Gluck, S.; Ablasser, A. Innate immunosensing of DNA in cellular senescence. Curr. Opin. Immunol. 2019, 56, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Gresser, I.; Maury, C.; Brouty-Boye, D. Mechanism of the antitumour effect of interferon in mice. Nature 1972, 239, 167–168. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Ferrantini, M.; Proietti, E.; Santodonato, L.; Gabriele, L.; Peretti, M.; Plavec, I.; Meyer, F.; Kaido, T.; Gresser, I.; Belardelli, F. Alpha 1-interferon gene transfer into metastatic friend leukemia cells abrogated tumorigenicity in immunocompetent mice: Antitumor therapy by means of interferon-producing cells. Cancer Res. 1993, 53, 1107–1112. [Google Scholar]
- Ferrantini, M.; Giovarelli, M.; Modesti, A.; Musiani, P.; Modica, A.; Venditti, M.; Peretti, E.; Lollini, P.L.; Nanni, P.; Forni, G.; et al. IFN-alpha 1 gene expression into a metastatic murine adenocarcinoma (TS/A) results in CD8+ T cell-mediated tumor rejection and development of antitumor immunity. Comparative studies with IFN-gamma-producing TS/A cells. J. Immunol. 1994, 153, 4604–4615. [Google Scholar] [PubMed]
- Dunn, G.P.; Sheehan, K.C.; Old, L.J.; Schreiber, R.D. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005, 65, 3447–3453. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Boyer, C.M.; Dawson, D.V.; Neal, S.E.; Winchell, L.F.; Leslie, D.S.; Ring, D.; Bast, R.C.J. Differential induction by interferons of major histocompatibility complex-encoded and non-major histocompatibility complex-encoded antigens in human breast and ovarian carcinoma cell lines. Cancer Res. 1989, 49, 2928–2934. [Google Scholar]
- Greiner, J.W.; Hand, P.H.; Noguchi, P.; Fisher, P.B.; Pestka, S.; Schlom, J. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte alpha-interferon treatment. Cancer Res. 1984, 44, 3208–3214. [Google Scholar]
- Giacomini, P.; Fraioli, R.; Nistico, P.; Tecce, R.; Nicotra, M.R.; Di Filippo, F.; Fisher, P.B.; Natali, P.G. Modulation of the antigenic phenotype of early-passage human melanoma cells derived from multiple autologous metastases by recombinant human leukocyte, fibroblast and immune interferon. Int. J. Cancer 1990, 46, 539–545. [Google Scholar] [CrossRef]
- Dunn, I.S.; Haggerty, T.J.; Kono, M.; Durda, P.J.; Butera, D.; Macdonald, D.B.; Benson, E.M.; Rose, L.B.; Kurnick, J.T. Enhancement of human melanoma antigen expression by IFN-beta. J. Immunol. 2007, 179, 2134–2142. [Google Scholar] [CrossRef]
- Dolei, A.; Capobianchi, M.R.; Ameglio, F. Human interferon-gamma enhances the expression of class I and class II major histocompatibility complex products in neoplastic cells more effectively than interferon-alpha and interferon-beta. Infect. Immun. 1983, 40, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Lanza, L.; Peirano, L.; Bosco, O.; Contini, P.; Filaci, G.; Setti, M.; Puppo, F.; Indiveri, F.; Scudeletti, M. Interferons up-regulate with different potency HLA class I antigen expression in M14 human melanoma cell line. Possible interaction with glucocorticoid hormones. Cancer Immunol. Immunother. 1995, 41, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Blanpain, C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer stem cells: The architects of the tumor ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Castiello, L.; Sestili, P.; Schiavoni, G.; Dattilo, R.; Monque, D.M.; Ciaffoni, F.; Iezzi, M.; Lamolinara, A.; Sistigu, A.; Moschella, F.; et al. Disruption of IFN-I signaling promotes HER2/Neu tumor progression and breast cancer stem cells. Cancer Immunol. Res. 2018, 6, 658–670. [Google Scholar] [CrossRef]
- Doherty, M.R.; Cheon, H.; Junk, D.J.; Vinayak, S.; Varadan, V.; Telli, M.L.; Ford, J.M.; Stark, G.R.; Jackson, M.W. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13792–13797. [Google Scholar] [CrossRef]
- Yuki, K.; Natsume, A.; Yokoyama, H.; Kondo, Y.; Ohno, M.; Kato, T.; Chansakul, P.; Ito, M.; Kim, S.U.; Wakabayashi, T. Induction of oligodendrogenesis in glioblastoma-initiating cells by IFN-mediated activation of STAT3 signaling. Cancer Lett. 2009, 284, 71–79. [Google Scholar] [CrossRef]
- Du, Z.; Cai, C.; Sims, M.; Boop, F.A.; Davidoff, A.M.; Pfeffer, L.M. The effects of type I interferon on glioblastoma cancer stem cells. Biochem. Biophys. Res. Commun. 2017, 491, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Guo, S.; Li, Y.; Ran, H.; Huang, H.; Mi, L.; Wu, J.; Wang, X.; Xiao, D.; Chen, L.; et al. Glioma stem-like cells evade interferon suppression through MBD3/NuRD complex-mediated STAT1 downregulation. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Ota, M.; Okada, Y. Isolation of cancer stem cells by side population method. Methods Mol. Biol. 2018, 1692, 49–59. [Google Scholar] [CrossRef]
- Liang, Y.; Hannan, R.; Fu, Y.X. Type I IFN activating type I dendritic cells for antitumor immunity. Clin. Cancer Res. 2021, 27, 3818–3824. [Google Scholar] [CrossRef]
- Lorenzi, S.; Mattei, F.; Sistigu, A.; Bracci, L.; Spadaro, F.; Sanchez, M.; Spada, M.; Belardelli, F.; Gabriele, L.; Schiavoni, G. Type I IFNs control antigen retention and survival of CD8alpha(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J. Immunol. 2011, 186, 5142–5150. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Valenzuela, J.O.; Agarwal, P.; Lins, D.; Mescher, M.F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005, 174, 4465–4469. [Google Scholar] [CrossRef]
- Tough, D.F. Modulation of T-cell function by type I interferon. Immunol. Cell Biol. 2012, 90, 492–497. [Google Scholar] [CrossRef]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef]
- Lim, J.Y.; Gerber, S.A.; Murphy, S.P.; Lord, E.M. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol. Immunother. 2014, 63, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Medrano, R.F.V.; Hunger, A.; Mendonca, S.A.; Barbuto, J.A.M.; Strauss, B.E. Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 2017, 8, 71249–71284. [Google Scholar] [CrossRef]
- Le Bon, A.; Durand, V.; Kamphuis, E.; Thompson, C.; Bulfone-Paus, S.; Rossmann, C.; Kalinke, U.; Tough, D.F. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 2006, 176, 4682–4689. [Google Scholar] [CrossRef]
- Richer, M.J.; Nolz, J.C.; Harty, J.T. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity 2013, 38, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Klement, J.D.; Ibrahim, M.L.; Xiao, W.; Redd, P.S.; Nayak-Kapoor, A.; Zhou, G.; Liu, K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer 2019, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019, 50, 195–211.e110. [Google Scholar] [CrossRef]
- Snell, L.M.; MacLeod, B.L.; Law, J.C.; Osokine, I.; Elsaesser, H.J.; Hezaveh, K.; Dickson, R.J.; Gavin, M.A.; Guidos, C.J.; McGaha, T.L.; et al. CD8(+) T cell priming in established chronic viral infection preferentially directs differentiation of memory-like cells for sustained immunity. Immunity 2018, 49, 678–694.e675. [Google Scholar] [CrossRef] [PubMed]
- Miron, M.; Kumar, B.V.; Meng, W.; Granot, T.; Carpenter, D.J.; Senda, T.; Chen, D.; Rosenfeld, A.M.; Zhang, B.; Lerner, H.; et al. Human lymph nodes maintain TCF-1(hi) memory T cells with high functional potential and clonal diversity throughout life. J. Immunol. 2018, 201, 2132–2140. [Google Scholar] [CrossRef]
- Huber, J.P.; Farrar, J.D. Regulation of effector and memory T-cell functions by type I interferon. Immunology 2011, 132, 466–474. [Google Scholar] [CrossRef]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Hashimoto, H.; Ueda, R.; Narumi, K.; Heike, Y.; Yoshida, T.; Aoki, K. Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther. 2014, 21, 532–541. [Google Scholar] [CrossRef]
- Hirata, A.; Hashimoto, H.; Shibasaki, C.; Narumi, K.; Aoki, K. Intratumoral IFN-alpha gene delivery reduces tumor-infiltrating regulatory T cells through the downregulation of tumor CCL17 expression. Cancer Gene Ther. 2019, 26, 334–343. [Google Scholar] [CrossRef]
- Pace, L.; Vitale, S.; Dettori, B.; Palombi, C.; La Sorsa, V.; Belardelli, F.; Proietti, E.; Doria, G. APC activation by IFN-alpha decreases regulatory T cell and enhances Th cell functions. J. Immunol. 2010, 184, 5969–5979. [Google Scholar] [CrossRef]
- Mizutani, T.; Neugebauer, N.; Putz, E.M.; Moritz, N.; Simma, O.; Zebedin-Brandl, E.; Gotthardt, D.; Warsch, W.; Eckelhart, E.; Kantner, H.P.; et al. Conditional IFNAR1 ablation reveals distinct requirements of type I IFN signaling for NK cell maturation and tumor surveillance. Oncoimmunology 2012, 1, 1027–1037. [Google Scholar] [CrossRef]
- Rautela, J.; Baschuk, N.; Slaney, C.Y.; Jayatilleke, K.M.; Xiao, K.; Bidwell, B.N.; Lucas, E.C.; Hawkins, E.D.; Lock, P.; Wong, C.S.; et al. Loss of host type-I IFN signaling accelerates metastasis and impairs NK-cell antitumor function in multiple models of breast cancer. Cancer Immunol. Res. 2015, 3, 1207–1217. [Google Scholar] [CrossRef]
- Zanker, D.J.; Owen, K.L.; Baschuk, N.; Spurling, A.J.; Parker, B.S. Loss of type I IFN responsiveness impairs natural killer cell antitumor activity in breast cancer. Cancer Immunol. Immunother. 2021, 70, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Zoglmeier, C.; Bauer, H.; Noerenberg, D.; Wedekind, G.; Bittner, P.; Sandholzer, N.; Rapp, M.; Anz, D.; Endres, S.; Bourquin, C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 2011, 17, 1765–1775. [Google Scholar] [CrossRef]
- Muller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Oynebraten, I.; Corthay, A. Both type I and type II interferons can activate antitumor M1 macrophages when combined with TLR stimulation. Front. Immunol. 2018, 9, 2520. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, B.; Zaritskaya, L.; Semenuk, M.; Cho, Y.H.; LaFleur, D.W.; Shah, D.; Ullrich, S.; Girolomoni, G.; Albanesi, C.; Moore, P.A. Regulatory effect of IFN-kappa, a novel type I IFN, on cytokine production by cells of the innate immune system. J. Immunol. 2002, 169, 4822–4830. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, J.; Yang, Y.; Liu, L.; Zhu, G.; Wang, X.; Wang, S.; Guo, W.; Yue, Q.; Zhao, T.; et al. Impact of interferon-alpha1b (IFN-alpha1b) on antitumor immune response: An interpretation of the promising therapeutic effect of IFN-alpha1b on melanoma. Med Sci. Monit. 2020, 26, e922790. [Google Scholar] [CrossRef]
- Villanueva, A.I.; Haeryfar, S.M.; Mallard, B.A.; Kulkarni, R.R.; Sharif, S. Functions of invariant NK T cells are modulated by TLR ligands and IFN-alpha. Innate Immun. 2015, 21, 275–288. [Google Scholar] [CrossRef]
- Watanabe, N.; Narita, M.; Yokoyama, A.; Sekiguchi, A.; Saito, A.; Tochiki, N.; Furukawa, T.; Toba, K.; Aizawa, Y.; Takahashi, M. Type I IFN-mediated enhancement of anti-leukemic cytotoxicity of gammadelta T cells expanded from peripheral blood cells by stimulation with zoledronate. Cytotherapy 2006, 8, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Thompson, C.; Kamphuis, E.; Durand, V.; Rossmann, C.; Kalinke, U.; Tough, D.F. Cutting edge: Enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 2006, 176, 2074–2078. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.L.; Wilson, T.J.; Strauch, P.; Colonna, M.; Pelanda, R.; Torres, R.M. Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response. J. Exp. Med. 2010, 207, 1485–1500. [Google Scholar] [CrossRef] [PubMed]
- Boukhaled, G.M.; Harding, S.; Brooks, D.G. Opposing roles of type I interferons in cancer immunity. Annu. Rev. Pathol. 2021, 16, 167–198. [Google Scholar] [CrossRef] [PubMed]
- Indraccolo, S. Interferon-alpha as angiogenesis inhibitor: Learning from tumor models. Autoimmunity 2010, 43, 244–247. [Google Scholar] [CrossRef]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Investig. 2019, 129, 4350–4364. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.H.; Liu, Z.; et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef]
- Pantel, A.; Teixeira, A.; Haddad, E.; Wood, E.G.; Steinman, R.M.; Longhi, M.P. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 2014, 12, e1001759. [Google Scholar] [CrossRef]
- York, A.G.; Williams, K.J.; Argus, J.P.; Zhou, Q.D.; Brar, G.; Vergnes, L.; Gray, E.E.; Zhen, A.; Wu, N.C.; Yamada, D.H.; et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 2015, 163, 1716–1729. [Google Scholar] [CrossRef]
- Pfeffer, L.M. The role of nuclear factor kappaB in the interferon response. J. Interf. Cytokine Res. 2011, 31, 553–559. [Google Scholar] [CrossRef]
- Yang, C.H.; Murti, A.; Pfeffer, S.R.; Basu, L.; Kim, J.G.; Pfeffer, L.M. IFNalpha/beta promotes cell survival by activating NF-kappa B. Proc. Natl. Acad. Sci. USA 2000, 97, 13631–13636. [Google Scholar] [CrossRef]
- Cheriyath, V.; Kuhns, M.A.; Jacobs, B.S.; Evangelista, P.; Elson, P.; Downs-Kelly, E.; Tubbs, R.; Borden, E.C. G1P3, an interferon- and estrogen-induced survival protein contributes to hyperplasia, tamoxifen resistance and poor outcomes in breast cancer. Oncogene 2012, 31, 2222–2236. [Google Scholar] [CrossRef]
- Puthier, D.; Thabard, W.; Rapp, M.; Etrillard, M.; Harousseau, J.; Bataille, R.; Amiot, M. Interferon alpha extends the survival of human myeloma cells through an upregulation of the Mcl-1 anti-apoptotic molecule. Br. J. Haematol. 2001, 112, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Hatano, H.; Kudo, Y.; Ogawa, I.; Tsunematsu, T.; Kikuchi, A.; Abiko, Y.; Takata, T. IFN-induced transmembrane protein 1 promotes invasion at early stage of head and neck cancer progression. Clin. Cancer Res. 2008, 14, 6097–6105. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Jiang, Y.; Lu, J.; Wu, J.; Zhang, M. Inhibiting of proliferation, migration, and invasion in lung cancer induced by silencing interferon-induced transmembrane protein 1 (IFITM1). BioMed Res. Int. 2019, 2019, 9085435. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liang, X.; Peterson, A.J.; Munn, D.H.; Blazar, B.R. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J. Immunol. 2008, 181, 5396–5404. [Google Scholar] [CrossRef]
- Cunningham, C.R.; Champhekar, A.; Tullius, M.V.; Dillon, B.J.; Zhen, A.; de la Fuente, J.R.; Herskovitz, J.; Elsaesser, H.; Snell, L.M.; Wilson, E.B.; et al. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. 2016, 12, e1005356. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878. [Google Scholar] [CrossRef]
- Baban, B.; Chandler, P.R.; Sharma, M.D.; Pihkala, J.; Koni, P.A.; Munn, D.H.; Mellor, A.L. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J. Immunol. 2009, 183, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 2019, 178, 933–948.e914. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef]
- Bald, T.; Landsberg, J.; Lopez-Ramos, D.; Renn, M.; Glodde, N.; Jansen, P.; Gaffal, E.; Steitz, J.; Tolba, R.; Kalinke, U.; et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014, 4, 674–687. [Google Scholar] [CrossRef]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vetizou, M.; Daillere, R.; et al. Sustained type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Li, S.; Xie, Y.; Zhang, W.; Gao, J.; Wang, M.; Zheng, G.; Yin, X.; Xia, H.; Tao, X. Interferon alpha-inducible protein 27 promotes epithelial-mesenchymal transition and induces ovarian tumorigenicity and stemness. J. Surg. Res. 2015, 193, 255–264. [Google Scholar] [CrossRef]
- Ma, H.; Jin, S.; Yang, W.; Tian, Z.; Liu, S.; Wang, Y.; Zhou, G.; Zhao, M.; Gvetadze, S.; Zhang, Z.; et al. Interferon-alpha promotes the expression of cancer stem cell markers in oral squamous cell carcinoma. J. Cancer 2017, 8, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Karakhanova, S.; Huang, X.; Deng, S.P.; Werner, J.; Bazhin, A.V. Influence of interferon-alpha on the expression of the cancer stem cell markers in pancreatic carcinoma cells. Exp. Cell Res. 2014, 324, 146–156. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Martin, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef] [PubMed]
- Qadir, A.S.; Ceppi, P.; Brockway, S.; Law, C.; Mu, L.; Khodarev, N.N.; Kim, J.; Zhao, J.C.; Putzbach, W.; Murmann, A.E.; et al. CD95/Fas increases stemness in cancer cells by inducing a STAT1-dependent type I interferon response. Cell Rep. 2017, 18, 2373–2386. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Chanda, P.; Thandavarayan, R.A.; Cooke, J.P. Transflammation: Innate immune signaling in nuclear reprogramming. Adv. Drug Deliv. Rev. 2017, 120, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sayed, N.; Hunter, A.; Au, K.F.; Wong, W.H.; Mocarski, E.S.; Pera, R.R.; Yakubov, E.; Cooke, J.P. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012, 151, 547–558. [Google Scholar] [CrossRef]
- Sayed, N.; Ospino, F.; Himmati, F.; Lee, J.; Chanda, P.; Mocarski, E.S.; Cooke, J.P. Retinoic acid inducible gene 1 protein (RIG1)-like receptor pathway is required for efficient nuclear reprogramming. Stem Cells 2017, 35, 1197–1207. [Google Scholar] [CrossRef]
- Jia, D.; Yang, W.; Li, L.; Liu, H.; Tan, Y.; Ooi, S.; Chi, L.; Filion, L.G.; Figeys, D.; Wang, L. Beta-Catenin and NF-kappaB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ. 2015, 22, 298–310. [Google Scholar] [CrossRef]
- Lan, Q.; Peyvandi, S.; Duffey, N.; Huang, Y.T.; Barras, D.; Held, W.; Richard, F.; Delorenzi, M.; Sotiriou, C.; Desmedt, C.; et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene 2019, 38, 2814–2829. [Google Scholar] [CrossRef]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning cancer fate: Tumor microenvironment’s role in cancer stem cell quiescence and reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Sistigu, A.; Valentini, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sanchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011, 71, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Pitt, J.M.; Daillere, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Swann, J.B.; Vesely, M.D.; Silva, A.; Sharkey, J.; Akira, S.; Schreiber, R.D.; Smyth, M.J. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 652–656. [Google Scholar] [CrossRef]
- Heyer, J.; Kwong, L.N.; Lowe, S.W.; Chin, L. Non-germline genetically engineered mouse models for translational cancer research. Nat. Rev. Cancer 2010, 10, 470–480. [Google Scholar] [CrossRef]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized mouse models of clinical disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Galassi, C.; Manic, G.; Musella, M.; Sistigu, A.; Vitale, I. Assessment of IFN-gamma and granzyme-B production by in sitro technology. Methods Enzymol. 2020, 631, 391–414. [Google Scholar] [CrossRef]
- De Ninno, A.; Bertani, F.R.; Gerardino, A.; Schiavoni, G.; Musella, M.; Galassi, C.; Mattei, F.; Sistigu, A.; Businaro, L. Microfluidic co-culture models for dissecting the immune response in in vitro tumor microenvironments. J. Vis. Exp. 2021, 170. [Google Scholar] [CrossRef]
- Vacchelli, E.; Sistigu, A.; Yamazaki, T.; Vitale, I.; Zitvogel, L.; Kroemer, G. Autocrine signaling of type 1 interferons in successful anticancer chemotherapy. Oncoimmunology 2015, 4, e988042. [Google Scholar] [CrossRef]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.F.; Duret, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 7142–7147. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Fu, M.L.; Weichselbaum, R.R.; Gajewski, T.F.; Guo, Y.; Fu, Y.X. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Sistigu, A.; Proietti, E.; Moschella, F. The added value of type I interferons to cytotoxic treatments of cancer. Cytokine Growth Factor Rev. 2017, 36, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Gresser, I.; Bourali, C. Exogenous interferon and inducers of interferon in the treatment Balb-c mice inoculated with RC19 tumour cells. Nature 1969, 223, 844–845. [Google Scholar] [CrossRef] [PubMed]
- Arico, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I interferons and cancer: An evolving story demanding novel clinical applications. Cancers 2019, 11, 1943. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Suciu, S.; Testori, A.; Santinami, M.; Kruit, W.H.; Marsden, J.; Punt, C.J.; Sales, F.; Dummer, R.; Robert, C.; et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J. Clin. Oncol. 2012, 30, 3810–3818. [Google Scholar] [CrossRef]
- Moschos, S.J.; Edington, H.D.; Land, S.R.; Rao, U.N.; Jukic, D.; Shipe-Spotloe, J.; Kirkwood, J.M. Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J. Clin. Oncol. 2006, 24, 3164–3171. [Google Scholar] [CrossRef]
- Burchert, A.; Muller, M.C.; Kostrewa, P.; Erben, P.; Bostel, T.; Liebler, S.; Hehlmann, R.; Neubauer, A.; Hochhaus, A. Sustained molecular response with interferon alfa maintenance after induction therapy with imatinib plus interferon alfa in patients with chronic myeloid leukemia. J. Clin. Oncol. 2010, 28, 1429–1435. [Google Scholar] [CrossRef]
- Preudhomme, C.; Guilhot, J.; Nicolini, F.E.; Guerci-Bresler, A.; Rigal-Huguet, F.; Maloisel, F.; Coiteux, V.; Gardembas, M.; Berthou, C.; Vekhoff, A.; et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N. Engl. J. Med. 2010, 363, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Riley, C.H.; Hansen, M.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M.; Jensen, M.K. Expansion of circulating CD56bright natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-alpha. Eur. J. Haematol. 2015, 94, 227–234. [Google Scholar] [CrossRef]
- Simonsson, B.; Gedde-Dahl, T.; Markevarn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Bjoreman, M.; Flogegard, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-alpha2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011, 118, 3228–3235. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.G.; Liu, X.; et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sceneay, J.; Goreczny, G.J.; Wilson, K.; Morrow, S.; DeCristo, M.J.; Ubellacker, J.M.; Qin, Y.; Laszewski, T.; Stover, D.G.; Barrera, V.; et al. Interferon signaling is diminished with age and is associated with immune checkpoint blockade efficacy in triple-negative breast cancer. Cancer Discov. 2019, 9, 1208–1227. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Cherian, J.; Moschos, S.J.; Tawbi, H.A.; Shuai, Y.; Gooding, W.E.; Sander, C.; Kirkwood, J.M. Safety and efficacy of combination immunotherapy with interferon alfa-2b and tremelimumab in patients with stage IV melanoma. J. Clin. Oncol. 2012, 30, 322–328. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Robb, R.J.; Kreijveld, E.; Kuns, R.D.; Wilson, Y.A.; Olver, S.D.; Don, A.L.; Raffelt, N.C.; De Weerd, N.A.; Lineburg, K.E.; Varelias, A.; et al. Type I-IFNs control GVHD and GVL responses after transplantation. Blood 2011, 118, 3399–3409. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra252. [Google Scholar] [CrossRef] [PubMed]
- Miquilena-Colina, M.E.; Lozano-Rodriguez, T.; Garcia-Pozo, L.; Saez, A.; Rizza, P.; Capone, I.; Rapicetta, M.; Chionne, P.; Capobianchi, M.; Selleri, M.; et al. Recombinant interferon-alpha2b improves immune response to hepatitis B vaccination in haemodialysis patients: Results of a randomised clinical trial. Vaccine 2009, 27, 5654–5660. [Google Scholar] [CrossRef]
- Urbani, F.; Ferraresi, V.; Capone, I.; Macchia, I.; Palermo, B.; Nuzzo, C.; Torsello, A.; Pezzotti, P.; Giannarelli, D.; Pozzi, A.F.; et al. Clinical and immunological outcomes in high-risk resected melanoma patients receiving peptide-based vaccination and interferon alpha, with or without dacarbazine preconditioning: A phase II study. Front. Oncol. 2020, 10, 202. [Google Scholar] [CrossRef]
- Geoffroy, K.; Bourgeois-Daigneault, M.C. The pros and cons of interferons for oncolytic virotherapy. Cytokine Growth Factor Rev. 2020, 56, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Quetglas, J.I.; Reboredo, M.; Dubrot, J.; Rodriguez-Madoz, J.R.; Mancheno, U.; Casales, E.; Riezu-Boj, J.I.; Ruiz-Guillen, M.; Ochoa, M.C.; et al. Strict requirement for vector-induced type I interferon in efficacious antitumor responses to virally encoded IL12. Cancer Res. 2015, 75, 497–507. [Google Scholar] [CrossRef]
- Bhattacharya, S.; HuangFu, W.C.; Dong, G.; Qian, J.; Baker, D.P.; Karar, J.; Koumenis, C.; Diehl, J.A.; Fuchs, S.Y. Anti-tumorigenic effects of type 1 interferon are subdued by integrated stress responses. Oncogene 2013, 32, 4214–4221. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, W.C.; Qian, J.; Liu, C.; Liu, J.; Lokshin, A.E.; Baker, D.P.; Rui, H.; Fuchs, S.Y. Inflammatory signaling compromises cell responses to interferon alpha. Oncogene 2012, 31, 161–172. [Google Scholar] [CrossRef]
- Zheng, H.; Qian, J.; Carbone, C.J.; Leu, N.A.; Baker, D.P.; Fuchs, S.Y. Vascular endothelial growth factor-induced elimination of the type 1 interferon receptor is required for efficient angiogenesis. Blood 2011, 118, 4003–4006. [Google Scholar] [CrossRef]
- Tomita, S.; Ishibashi, K.; Hashimoto, K.; Sugino, T.; Yanagida, T.; Kushida, N.; Shishido, K.; Aikawa, K.; Sato, Y.; Suzutani, T.; et al. Suppression of SOCS3 increases susceptibility of renal cell carcinoma to interferon-alpha. Cancer Sci. 2011, 102, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Landolfo, S.; Guarini, A.; Riera, L.; Gariglio, M.; Gribaudo, G.; Cignetti, A.; Cordone, I.; Montefusco, E.; Mandelli, F.; Foa, R. Chronic myeloid leukemia cells resistant to interferon-alpha lack STAT1 expression. Hematol. J. 2000, 1, 7–14. [Google Scholar] [CrossRef]
- Romero-Weaver, A.L.; Wang, H.W.; Steen, H.C.; Scarzello, A.J.; Hall, V.L.; Sheikh, F.; Donnelly, R.P.; Gamero, A.M. Resistance to IFN-alpha-induced apoptosis is linked to a loss of STAT2. Mol. Cancer Res. 2010, 8, 80–92. [Google Scholar] [CrossRef]
- Bi, X.; Hameed, M.; Mirani, N.; Pimenta, E.M.; Anari, J.; Barnes, B.J. Loss of interferon regulatory factor 5 (IRF5) expression in human ductal carcinoma correlates with disease stage and contributes to metastasis. Breast Cancer Res. 2011, 13, R111. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, W.; Zhu, W.; Meng, H.; Chen, J.; Zhang, J. Overexpression of interferon regulatory factor 7 (IRF7) reduces bone metastasis of prostate cancer cells in mice. Oncol. Res. 2017, 25, 511–522. [Google Scholar] [CrossRef]
- Minn, A.J. Interferons and the immunogenic effects of cancer therapy. Trends Immunol. 2015, 36, 725–737. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Beckett, M.; Labay, E.; Darga, T.; Roizman, B.; Weichselbaum, R.R. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.H.; Cook, J.A.; Chandramouli, G.V.; DeGraff, W.; Yan, H.; Zhao, S.; Coleman, C.N.; Mitchell, J.B.; Chuang, E.Y. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res. 2007, 67, 3845–3852. [Google Scholar] [CrossRef] [PubMed]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint, C.V.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef]
- Gaston, J.; Cheradame, L.; Yvonnet, V.; Deas, O.; Poupon, M.F.; Judde, J.G.; Cairo, S.; Goffin, V. Intracellular STING inactivation sensitizes breast cancer cells to genotoxic agents. Oncotarget 2016, 7, 77205–77224. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Name | Company | Type I IFN Subtype | Indication(s) |
|---|---|---|---|
| Alfaferone | Alfa Wassermann | Natural leukocyte interferon alpha | Hairy cell leukemia, multiple myeloma, non-Hodgkin lymphoma, follicular lymphoma, chronic myelogenous leukemia, malignant melanoma, AIDS-related Kaposi’s sarcoma |
| Belerofon | Nautilus Biotechnology | Interferon alpha | / |
| HeberFERON | Center for Genetic Engineering and Biotechnology | Interferon alpha-2b/interferon gamma | Basal cell cancer |
| IFN alfa-2b XL | Flamel Technologies | Interferon alpha-2b | / |
| Infergen | Amgen | Interferon alfacon-1 | Non-Hodgkin’s lymphoma, ovarian cancer |
| Intron A | Biogen Idec | Interferon alpha-2b | Chronic myeloid leukemia, follicular lymphoma, hairy cell leukemia, malignant melanoma, multiple myeloma, non-Hodgkin’s lymphoma, AIDS-related Kaposi’s sarcoma |
| Joulferon | Human Genome Sciences | Albinterferon alpha-2b | / |
| Locteron | Biolex | Interferon alpha-2b | / |
| Multiferon | Viragen | HuIFN-alpha | Chronic myeloid leukemia, hairy cell leukemia, malignant melanoma |
| Novaferon | Genova Biotech Company | Interferon alpha | Colorectal cancer, neuroendocrine tumors, pancreatic cancer |
| Pegasys | Hoffmann-La Roche | Peginterferon alpha-2a | Malignant melanoma, renal cell carcinoma |
| PegIntron | Enzon Pharmaceuticals | Peginterferon alpha-2b | Malignant melanoma, cholangiocarcinoma, chronic myeloid leukemia, solid tumors |
| Reiferon Retard | Rhein Minapharm Biogenetics | Peginterferon alpha-2a | / |
| Roferon A | Hoffmann-La Roche | Interferon alpha-2a | Chronic myeloid leukemia, cutaneous T-cell lymphoma, hairy cell leukemia, Kaposi’s sarcoma, malignant melanoma, non-Hodgkin’s lymphoma, renal cell carcinoma |
| Sylatron | Merck | Peginterferon alpha-2b | Melanoma |
| Wellferon | Glaxo Wellcome SA | Interferon alpha-n1 | Chronic myeloid leukemia, Hairy cell leukemia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musella, M.; Galassi, C.; Manduca, N.; Sistigu, A. The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation. Biology 2021, 10, 856. https://doi.org/10.3390/biology10090856
Musella M, Galassi C, Manduca N, Sistigu A. The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation. Biology. 2021; 10(9):856. https://doi.org/10.3390/biology10090856
Chicago/Turabian StyleMusella, Martina, Claudia Galassi, Nicoletta Manduca, and Antonella Sistigu. 2021. "The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation" Biology 10, no. 9: 856. https://doi.org/10.3390/biology10090856
APA StyleMusella, M., Galassi, C., Manduca, N., & Sistigu, A. (2021). The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation. Biology, 10(9), 856. https://doi.org/10.3390/biology10090856