
Actin Cytoskeleton Dynamics and Type I IFN-Mediated Immune Response: A Dangerous Liaison in Cancer?


Abstract
Simple Summary
Abstract
1. Introduction
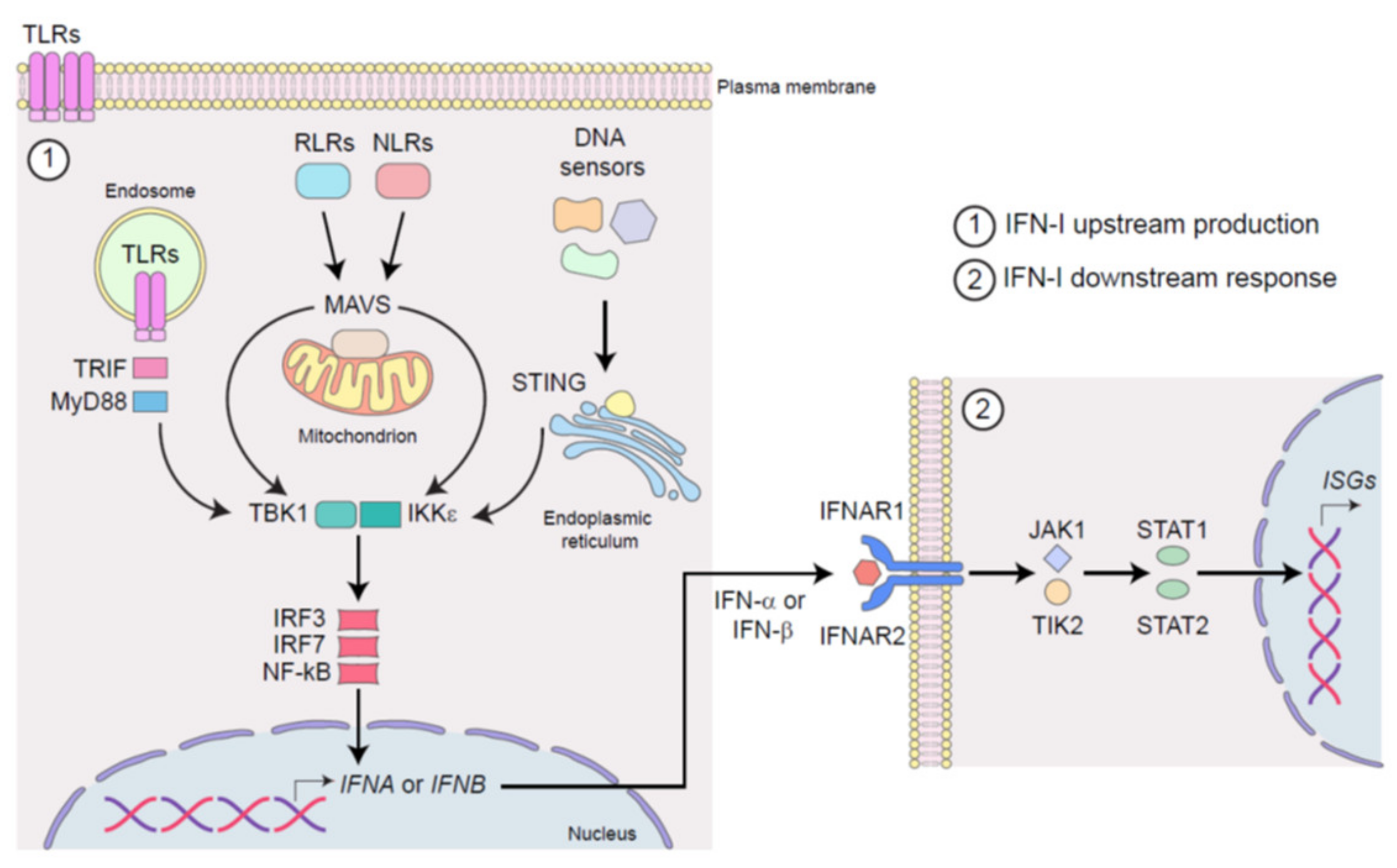
2. Mechanisms of IFN-I Signaling in Infection and Cancer
3. Actin Cytoskeleton: Components, Dynamics, and Emerging Role in Innate Immunity
4. Cytoskeleton Interactions with Components of Innate Immunity
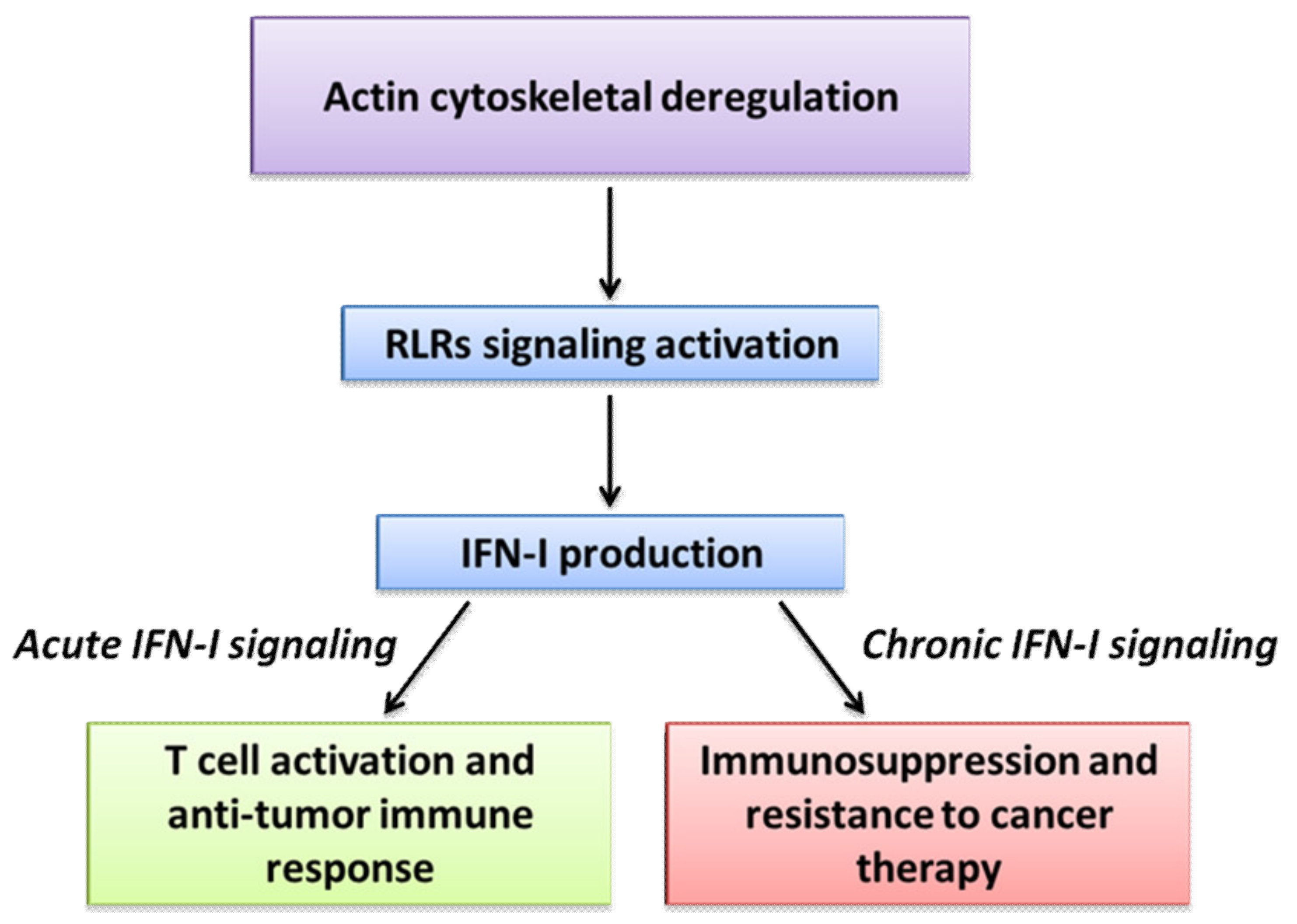
5. Opposing Effects of IFN-Is in Disease Progression and Resistance to Therapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meizlish, M.L.; Franklin, R.A.; Zhou, X.; Medzhitov, R. Tissue Homeostasis and Inflammation. Annu. Rev. Immunol. 2021, 39, 557–581. [Google Scholar] [CrossRef]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Coyne, C.B. The actin cytoskeleton as a barrier to virus infection of polarized epithelial cells. Viruses 2011, 3, 2462. [Google Scholar] [CrossRef]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 2011, 9, 427–439. [Google Scholar] [CrossRef]
- Datta, A.; Deng, S.; Gopal, V.; Yap, K.C.; Halim, C.E.; Lye, M.L.; Ong, M.S.; Tan, T.Z.; Sethi, G.; Hooi, S.C.; et al. Cytoskeletal Dynamics in Epithelial-Mesenchymal Transition: Insights into Therapeutic Targets for Cancer Metastasis. Cancers 2021, 13, 1882. [Google Scholar] [CrossRef]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Haglund, C.M.; Welch, M.D. Pathogens and polymers: Microbe-host interactions illuminate the cytoskeleton. J. Cell Biol. 2011, 195, 7–17. [Google Scholar] [CrossRef]
- Mostowy, S.; Shenoy, A.R. The cytoskeleton in cell-autonomous immunity: Structural determinants of host defence. Nat. Rev. Immunol. 2015, 15, 559–573. [Google Scholar] [CrossRef]
- Ezelle, H.J.; Malathi, K.; Hassel, B.A. The Roles of RNase-L in Antimicrobial Immunity and the Cytoskeleton-Associated Innate Response. Int. J. Mol. Sci. 2016, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017, 6, e1314424. [Google Scholar] [CrossRef]
- Hardy, M.P.; Owczarek, C.M.; Jermiin, L.S.; Ejdeback, M.; Hertzog, P.J. Characterization of the type I interferon locus and identification of novel genes. Genomics 2004, 84, 331–345. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vetizou, M.; Daillere, R.; et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Lupfer, C.; Kanneganti, T.D. The expanding role of NLRs in antiviral immunity. Immunol. Rev. 2013, 255, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Kienes, I.; Weidl, T.; Mirza, N.; Chamaillard, M.; Kufer, T.A. Role of NLRs in the Regulation of Type I Interferon Signaling, Host Defense and Tolerance to Inflammation. Int. J. Mol. Sci. 2021, 22, 1301. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Hata, N.; Asagiri, M.; Nakaya, T.; Taniguchi, T.; Tanaka, N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998, 441, 106–110. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Van Boxel-Dezaire, A.H.; Rani, M.R.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Sistigu, A.; Proietti, E.; Moschella, F. The added value of type I interferons to cytotoxic treatments of cancer. Cytokine Growth Factor Rev. 2017, 36, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J. Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol. 2015, 36, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Minn, A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef]
- Kloc, M.; Chanana, P.; Vaughn, N.; Uosef, A.; Kubiak, J.Z.; Ghobrial, R.M. New Insights into Cellular Functions of Nuclear Actin. Biology 2021, 10, 304. [Google Scholar] [CrossRef]
- Wei, M.; Fan, X.; Ding, M.; Li, R.; Shao, S.; Hou, Y.; Meng, S.; Tang, F.; Li, C.; Sun, Y. Nuclear actin regulates inducible transcription by enhancing RNA polymerase II clustering. Sci. Adv. 2020, 6, eaay6515. [Google Scholar] [CrossRef]
- Au-Yeung, N.; Horvath, C.M. Transcriptional and chromatin regulation in interferon and innate antiviral gene expression. Cytokine Growth Factor Rev. 2018, 44, 11–17. [Google Scholar] [CrossRef]
- Barone, M.; Müller, M.; Chiha, S.; Ren, J.; Albat, D.; Soicke, A.; Dohmen, S.; Klein, M.; Bruns, J.; van Dinther, M.; et al. Designed nanomolar small-molecule inhibitors of Ena/VASP EVH1 interaction impair invasion and extravasation of breast cancer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 29684–29690. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; LoGrasso, P.V.; Defert, O.; Li, R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef] [PubMed]
- Unbekandt, M.; Croft, D.R.; Crighton, D.; Mezna, M.; McArthur, D.; McConnell, P.; Schüttelkopf, A.W.; Belshaw, S.; Pannifer, A.; Sime, M.; et al. A novel small-molecule MRCK inhibitor blocks cancer cell invasion. Cell Commun. Signal. 2014, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Day, B. Battlefield Cytoskeleton: Turning the Tide on Plant Immunity. Mol. Plant. Microbe Interact. 2019, 32, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Krausgruber, T.; Fortelny, N.; Fife-Gernedl, V.; Senekowitsch, M.; Schuster, L.C.; Lercher, A.; Nemc, A.; Schmidl, C.; Rendeiro, A.F.; Bergthaler, A.; et al. Structural cells are key regulators of organ-specific immune responses. Nature 2020, 583, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Irving, A.T.; Wang, D.; Vasilevski, O.; Latchoumanin, O.; Kozer, N.; Clayton, A.H.; Szczepny, A.; Morimoto, H.; Xu, D.; Williams, B.R.; et al. Regulation of actin dynamics by protein kinase R control of gelsolin enforces basal innate immune defense. Immunity 2012, 36, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef]
- Li, G.H.; Arora, P.D.; Chen, Y.; McCulloch, C.A.; Liu, P. Multifunctional roles of gelsolin in health and diseases. Med. Res. Rev. 2012, 32, 999–1025. [Google Scholar] [CrossRef]
- Lacy-Hulbert, A.; Stuart, L.M. Penetration resistance: PKR’s other talent. Immunity 2012, 36, 695–696. [Google Scholar] [CrossRef][Green Version]
- Razinia, Z.; Makela, T.; Ylanne, J.; Calderwood, D.A. Filamins in mechanosensing and signaling. Annu. Rev. Biophys. 2012, 41, 227–246. [Google Scholar] [CrossRef]
- Malathi, K.; Siddiqui, M.A.; Dayal, S.; Naji, M.; Ezelle, H.J.; Zeng, C.; Zhou, A.; Hassel, B.A. RNase L interacts with Filamin A to regulate actin dynamics and barrier function for viral entry. mBio 2014, 5, e02012-14. [Google Scholar] [CrossRef]
- Bozym, R.A.; Delorme-Axford, E.; Harris, K.; Morosky, S.; Ikizler, M.; Dermody, T.S.; Sarkar, S.N.; Coyne, C.B. Focal adhesion kinase is a component of antiviral RIG-I-like receptor signaling. Cell Host Microbe 2012, 11, 153–166. [Google Scholar] [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Nabergoj, S.; Mlinaric-Rascan, I.; Jakopin, Z. Harnessing the untapped potential of nucleotide-binding oligomerization domain ligands for cancer immunotherapy. Med. Res. Rev. 2019, 39, 1447–1484. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Poels, S.; Kustermans, G.; Bex, F.; Kremmer, E.; Kufer, T.A.; Piette, J. Modulation of Nod2-dependent NF-kappaB signaling by the actin cytoskeleton. J. Cell Sci. 2007, 120, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Machacek, M.; Hodgson, L.; Welch, C.; Elliott, H.; Pertz, O.; Nalbant, P.; Abell, A.; Johnson, G.L.; Hahn, K.M.; Danuser, G. Coordination of Rho GTPase activities during cell protrusion. Nature 2009, 461, 99–103. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Tsolis, R.M. NOD1 and NOD2: Beyond Peptidoglycan Sensing. Trends Immunol. 2017, 38, 758–767. [Google Scholar] [CrossRef]
- Tiku, V.; Tan, M.W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.R.; Pon, L.A. Mitochondria on the move. Trends Cell Biol. 2007, 17, 502–510. [Google Scholar] [CrossRef]
- Kouwaki, T.; Okamoto, M.; Tsukamoto, H.; Fukushima, Y.; Matsumoto, M.; Seya, T.; Oshiumi, H. Zyxin stabilizes RIG-I and MAVS interactions and promotes type I interferon response. Sci. Rep. 2017, 7, 11905. [Google Scholar] [CrossRef]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane crosstalk between the extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2001, 2, 793–805. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Lamb, M.C.; Tootle, T.L. Fascin in Cell Migration: More Than an Actin Bundling Protein. Biology 2020, 9, 403. [Google Scholar] [CrossRef]
- Tan, V.Y.; Lewis, S.J.; Adams, J.C.; Martin, R.M. Association of fascin-1 with mortality, disease progression and metastasis in carcinomas: A systematic review and meta-analysis. BMC Med. 2013, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Hida, S.; Kitazawa, M.; Fujii, C.; Kobayashi, A.; Takeoka, M.; Taniguchi, S.I.; Miyagawa, S.I. Fascin1 suppresses RIG-I-like receptor signaling and interferon-beta production by associating with IkappaB kinase (IKK) in colon cancer. J. Biol. Chem. 2018, 293, 6326–6336. [Google Scholar] [CrossRef]
- Crawford, A.W.; Beckerle, M.C. Purification and characterization of zyxin, an 82,000-dalton component of adherens junctions. J. Biol. Chem. 1991, 266, 5847–5853. [Google Scholar] [CrossRef]
- Hirata, H.; Tatsumi, H.; Sokabe, M. Mechanical forces facilitate actin polymerization at focal adhesions in a zyxin-dependent manner. J. Cell Sci. 2008, 121, 2795–2804. [Google Scholar] [CrossRef]
- Beckerle, M.C. Zyxin: Zinc fingers at sites of cell adhesion. Bioessays 1997, 19, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Ohman, T.; Rintahaka, J.; Kalkkinen, N.; Matikainen, S.; Nyman, T.A. Actin and RIG-I/MAVS signaling components translocate to mitochondria upon influenza A virus infection of human primary macrophages. J. Immunol. 2009, 182, 5682–5692. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Shen, L.; Weber, C.R.; Turner, J.R.; Kim, K.S.; Wang, T.; Coyne, C.B. Retinoic acid-induced gene-1 (RIG-I) associates with the actin cytoskeleton via caspase activation and recruitment domain-dependent interactions. J. Biol. Chem. 2009, 284, 6486–6494. [Google Scholar] [CrossRef] [PubMed]
- Gertler, F.B.; Niebuhr, K.; Reinhard, M.; Wehland, J.; Soriano, P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell 1996, 87, 227–239. [Google Scholar] [CrossRef]
- Reinhard, M.; Halbrugge, M.; Scheer, U.; Wiegand, C.; Jockusch, B.M.; Walter, U. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 1992, 11, 2063–2070. [Google Scholar] [CrossRef]
- Di Modugno, F.; Iapicca, P.; Boudreau, A.; Mottolese, M.; Terrenato, I.; Perracchio, L.; Carstens, R.P.; Santoni, A.; Bissell, M.J.; Nistico, P. Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 19280–19285. [Google Scholar] [CrossRef]
- Di Modugno, F.; Spada, S.; Palermo, B.; Visca, P.; Iapicca, P.; Di Carlo, A.; Antoniani, B.; Sperduti, I.; Di Benedetto, A.; Terrenato, I.; et al. hMENA isoforms impact NSCLC patient outcome through fibronectin/beta1 integrin axis. Oncogene 2018, 37, 5605–5617. [Google Scholar] [CrossRef]
- Trono, P.; Di Modugno, F.; Circo, R.; Spada, S.; Di Benedetto, A.; Melchionna, R.; Palermo, B.; Matteoni, S.; Soddu, S.; Mottolese, M.; et al. hMENA(11a) contributes to HER3-mediated resistance to PI3K inhibitors in HER2-overexpressing breast cancer cells. Oncogene 2016, 35, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Melchionna, R.; Iapicca, P.; Di Modugno, F.; Trono, P.; Sperduti, I.; Fassan, M.; Cataldo, I.; Rusev, B.C.; Lawlor, R.T.; Diodoro, M.G.; et al. The pattern of hMENA isoforms is regulated by TGF-beta1 in pancreatic cancer and may predict patient outcome. Oncoimmunology 2016, 5, e1221556. [Google Scholar] [CrossRef] [PubMed]
- Gertler, F.; Condeelis, J. Metastasis: Tumor cells becoming MENAcing. Trends Cell Biol. 2011, 21, 81–90. [Google Scholar] [CrossRef]
- Di Modugno, F.; DeMonte, L.; Balsamo, M.; Bronzi, G.; Nicotra, M.R.; Alessio, M.; Jager, E.; Condeelis, J.S.; Santoni, A.; Natali, P.G.; et al. Molecular cloning of hMena (ENAH) and its splice variant hMena+11a: Epidermal growth factor increases their expression and stimulates hMena+11a phosphorylation in breast cancer cell lines. Cancer Res. 2007, 67, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, C.C.; Jiang, P.; Amirikian, K.; Dittmar, K.A.; Lu, H.; Shen, S.; Guo, W.; Xing, Y.; Carstens, R.P. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 2010, 29, 3286–3300. [Google Scholar] [CrossRef]
- Bria, E.; Di Modugno, F.; Sperduti, I.; Iapicca, P.; Visca, P.; Alessandrini, G.; Antoniani, B.; Pilotto, S.; Ludovini, V.; Vannucci, J.; et al. Prognostic impact of alternative splicing-derived hMENA isoforms in resected, node-negative, non-small-cell lung cancer. Oncotarget 2014, 5, 11054–11063. [Google Scholar] [CrossRef][Green Version]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Sandler, N.G.; Bosinger, S.E.; Estes, J.D.; Zhu, R.T.R.; Tharp, G.K.; Boritz, E.; Levin, D.; Wijeyesinghe, S.; Makamdop, K.N.; del Prete, G.Q.; et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 2014, 7511, 601–605. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef]
- Wagner, T.C.; Velichko, S.; Chesney, S.K.; Biroc, S.; Harde, D.; Vogel, D.; Croze, E. Interferon receptor expression regulates the antiproliferative effects of interferons on cancer cells and solid tumors. Int. J. Cancer 2004, 111, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Sheehan, K.C.; Old, L.J.; Schreiber, R.D. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005, 65, 3447–3453. [Google Scholar] [CrossRef]
- Romero-Weaver, A.L.; Wang, H.W.; Steen, H.C.; Scarzello, A.J.; Hall, V.L.; Sheikh, F.; Donnelly, R.P.; Gamero, A.M. Resistance to IFN-alpha-induced apoptosis is linked to a loss of STAT2. Mol. Cancer Res. 2010, 8, 80–92. [Google Scholar] [CrossRef]
- Wellbrock, C.; Weisser, C.; Hassel, J.C.; Fischer, P.; Becker, J.; Vetter, C.S.; Behrmann, I.; Kortylewski, M.; Heinrich, P.C.; Schartl, M. STAT5 contributes to interferon resistance of melanoma cells. Curr. Biol. 2005, 15, 1629–1639. [Google Scholar] [CrossRef]
- Zitzmann, K.; Brand, S.; De Toni, E.N.; Baehs, S.; Göke, B.; Meinecke, J.; Spöttl, G.; Meyer, H.H.; Auernhammer, C.J. SOCS1 silencing enhances antitumor activity of type I IFNs by regulating apoptosis in neuroendocrine tumor cells. Cancer Res. 2007, 67, 5025–5032. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of Functional Beta 2 -Microglobulin in Metastatic Melanomas From Five Patients Receiving Immunotherapy. JNCI J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, L.; Wei, T.; Xiao, Y.T.; Sheng, H.; Su, H.; Hollern, D.P.; Zhang, X.; Ma, J.; Wen, S.; et al. FOXA1 overexpression suppresses interferon signaling and immune response in cancer. J. Clin. Investg. 2021, 131, e147025. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Ye, Z.; Dong, H.; Li, Y.; Ma, T.; Huang, H.; Leong, H.S.; Eckel-Passow, J.; Kocher, J.A.; Liang, H.; Wang, L. Prevalent Homozygous Deletions of Type I Interferon and Defensin Genes in Human Cancers Associate with Immunotherapy Resistance. Clin. Cancer Res. 2018, 24, 3299–3308. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef]
- Liu, H.; Golji, J.; Brodeur, L.K.; Chung, F.S.; Chen, J.T.; deBeaumont, R.S.; Bullock, C.P.; Jones, M.D.; Kerr, G.; Li, L.; et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 2019, 25, 95–102. [Google Scholar] [CrossRef]
- Ma, H.; Yang, W.; Zhang, L.; Liu, S.; Zhao, M.; Zhou, G.; Wang, L.; Jin, S.; Zhang, Z.; Hu, J. Interferon-alpha promotes immunosuppression through IFNAR1/STAT1 signalling in head and neck squamous cell carcinoma. Br. J. Cancer 2019, 120, 317–330. [Google Scholar] [CrossRef]
- Donlon, N.E.; Power, R.; Hayes, C.; Reynolds, J.V.; Lysaght, J. Radiotherapy, immunotherapy, and the tumour microenvironment: Turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021, 502, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther. Adv. Med. Oncol. 2018, 10, 1758834017742575. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investg. 2019, 129, 4224–4238. [Google Scholar] [CrossRef]
- Choi, H.J.; Lui, A.; Ogony, J.; Jan, R.; Sims, P.J.; Lewis-Wambi, J. Targeting interferon response genes sensitizes aromatase inhibitor resistant breast cancer cells to estrogen-induced cell death. Breast Cancer Res. 2015, 17, 6. [Google Scholar] [CrossRef]
- De Angelis, C.; Fu, X.; Cataldo, M.L.; Nardone, A.; Pereira, R.; Veeraraghavan, J.; Nanda, S.; Qin, L.; Sethunath, V.; Wang, T.; et al. Activation of the IFN Signaling Pathway is Associated with Resistance to CDK4/6 Inhibitors and Immune Checkpoint Activation in ER-Positive Breast Cancer. Clin. Cancer Res. 2021, 27. [Google Scholar] [CrossRef]
- Dold, C.; Rodriguez Urbiola, C.; Wollmann, G.; Egerer, L.; Muik, A.; Bellmann, L.; Fiegl, H.; Marth, C.; Kimpel, J.; von Laer, D. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV-GP oncolytic viral therapy. Mol. Ther. Oncolytics 2016, 3, 16021. [Google Scholar] [CrossRef]
- Jackson, J.D.; Markert, J.M.; Li, L.; Carroll, S.L.; Cassady, K.A. STAT1 and NF-κB Inhibitors Diminish Basal Interferon-Stimulated Gene Expression and Improve the Productive Infection of Oncolytic HSV in MPNST Cells. Mol. Cancer Res. 2016, 14, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Chawla-Sarkar, M.; Leaman, D.W.; Borden, E.C. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: Correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin. Cancer Res. 2001, 7, 1821–1831. [Google Scholar]
- Mullally, A.; Bruedigam, C.; Poveromo, L.; Heidel, F.H.; Purdon, A.; Vu, T.; Austin, R.; Heckl, D.; Breyfogle, L.J.; Kuhn, C.P.; et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood 2013, 121, 3692–3702. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Liu, D.D.; Choudhury, A.; Hang, X.; Wei, Y.; Zamalloa, J.; Alfaro-Aco, R.; Chakrabarti, R.; Jiang, Y.Z.; Koh, B.I.; et al. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis. Nat. Cell Biol. 2017, 19, 711–723. [Google Scholar] [CrossRef]
- Greiner, J.W.; Hand, P.H.; Noguchi, P.; Fisher, P.B.; Pestka, S.; Schlom, J. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte alpha-interferon treatment. Cancer Res. 1984, 44, 3208–3214. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Acute IFN-I Signaling * | Chronic IFN-I Signaling |
|---|---|
| Apoptotic and antiproliferative effect [111] | Resistance to immune-checkpoint blockade therapy [16,103] |
| Induction of cell differentiation [112,113] | Resistance to radiation therapy [106] |
| Induction of senescence [113] | Resistance to oncolytic viral therapy [109,110] |
| Up regulation of antigen presenting and processing machinery genes [114] | Resistance to aromatase inhibitors [107] |
| Resistance to CDK4/6 inhibitors [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trono, P.; Tocci, A.; Musella, M.; Sistigu, A.; Nisticò, P. Actin Cytoskeleton Dynamics and Type I IFN-Mediated Immune Response: A Dangerous Liaison in Cancer? Biology 2021, 10, 913. https://doi.org/10.3390/biology10090913
Trono P, Tocci A, Musella M, Sistigu A, Nisticò P. Actin Cytoskeleton Dynamics and Type I IFN-Mediated Immune Response: A Dangerous Liaison in Cancer? Biology. 2021; 10(9):913. https://doi.org/10.3390/biology10090913
Chicago/Turabian StyleTrono, Paola, Annalisa Tocci, Martina Musella, Antonella Sistigu, and Paola Nisticò. 2021. "Actin Cytoskeleton Dynamics and Type I IFN-Mediated Immune Response: A Dangerous Liaison in Cancer?" Biology 10, no. 9: 913. https://doi.org/10.3390/biology10090913
APA StyleTrono, P., Tocci, A., Musella, M., Sistigu, A., & Nisticò, P. (2021). Actin Cytoskeleton Dynamics and Type I IFN-Mediated Immune Response: A Dangerous Liaison in Cancer? Biology, 10(9), 913. https://doi.org/10.3390/biology10090913