
Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice

 ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
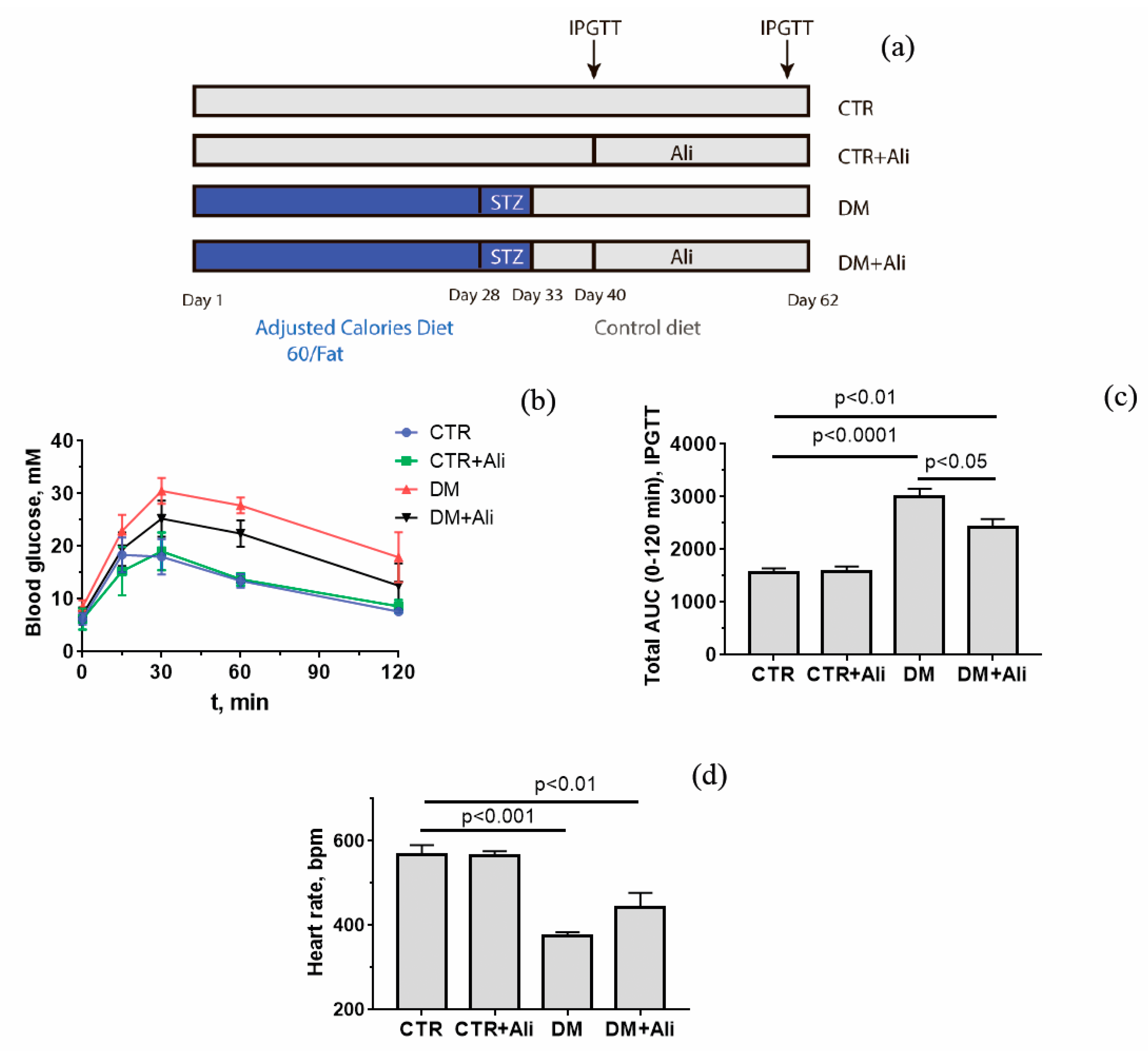
2.1. Animals and the Induction of Diabetes
2.2. Electron Microscopy
2.3. RNA Extraction and Quantitative Real-Time PCR
2.4. Mitochondrial DNA Estimation
2.5. Mitochondria Isolation and Determination of Functional Parameters
2.6. ECG
2.7. Statistical Analysis
2.8. Materials
3. Results
3.1. Somatic and Biochemical Characteristics of Experimental Mice
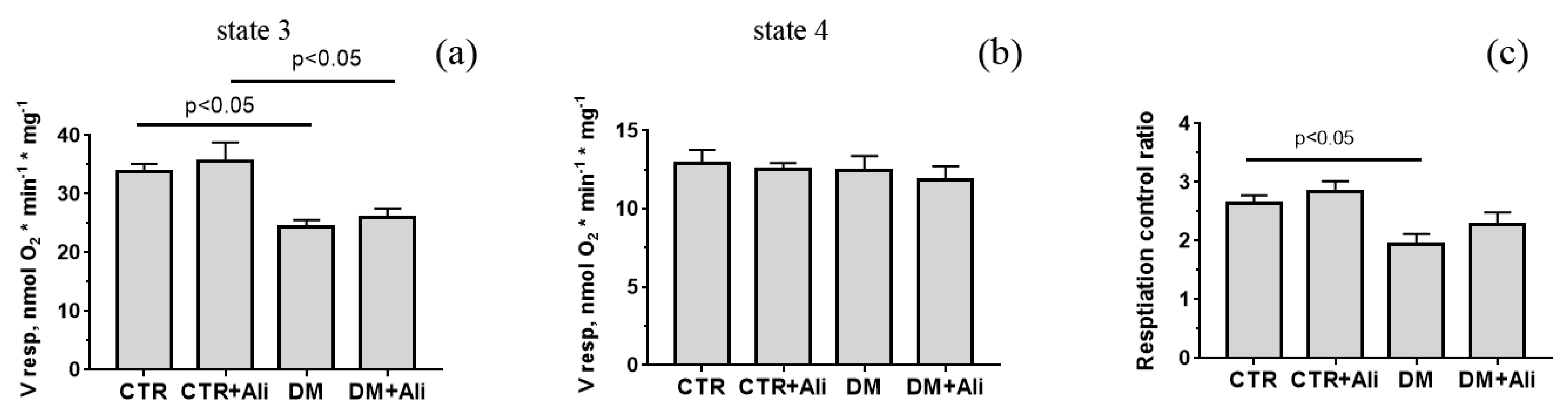
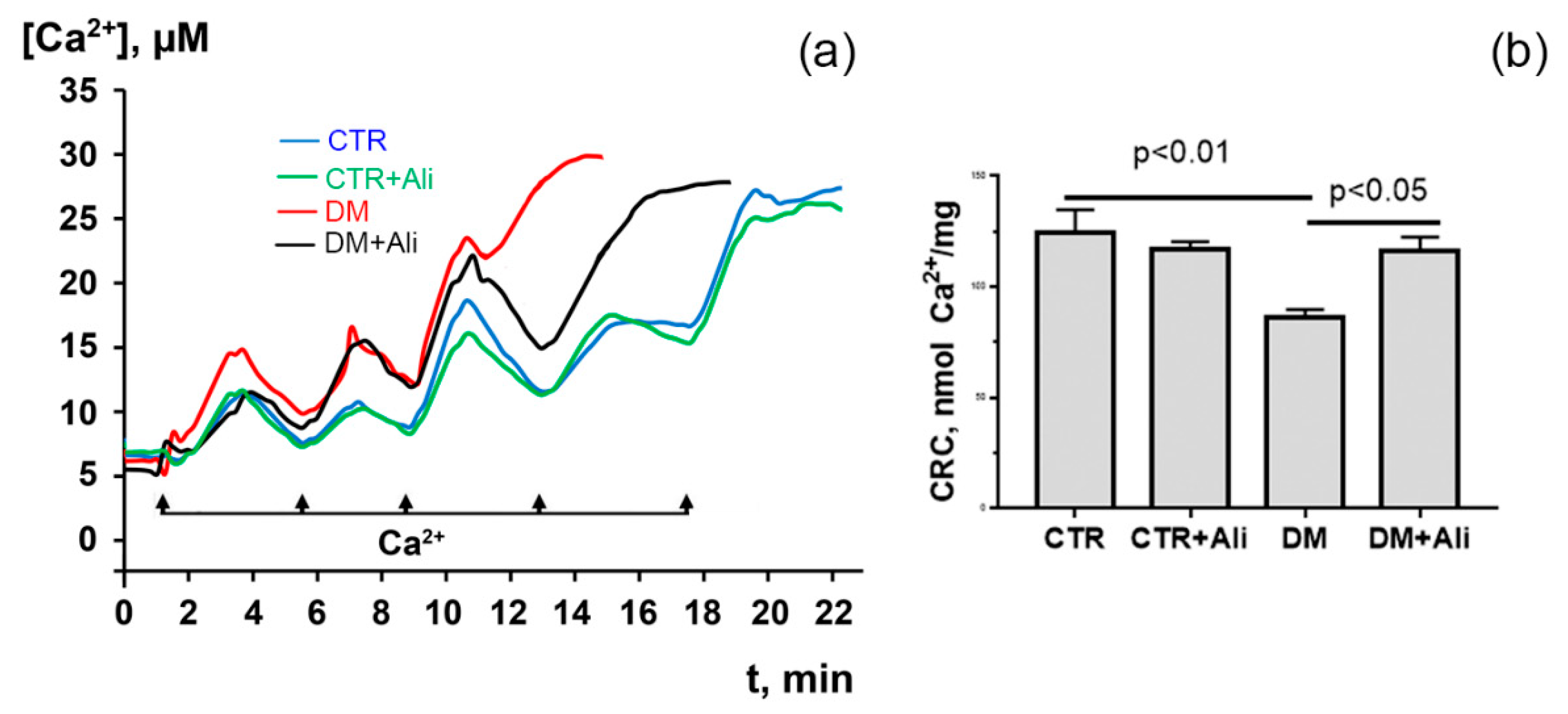
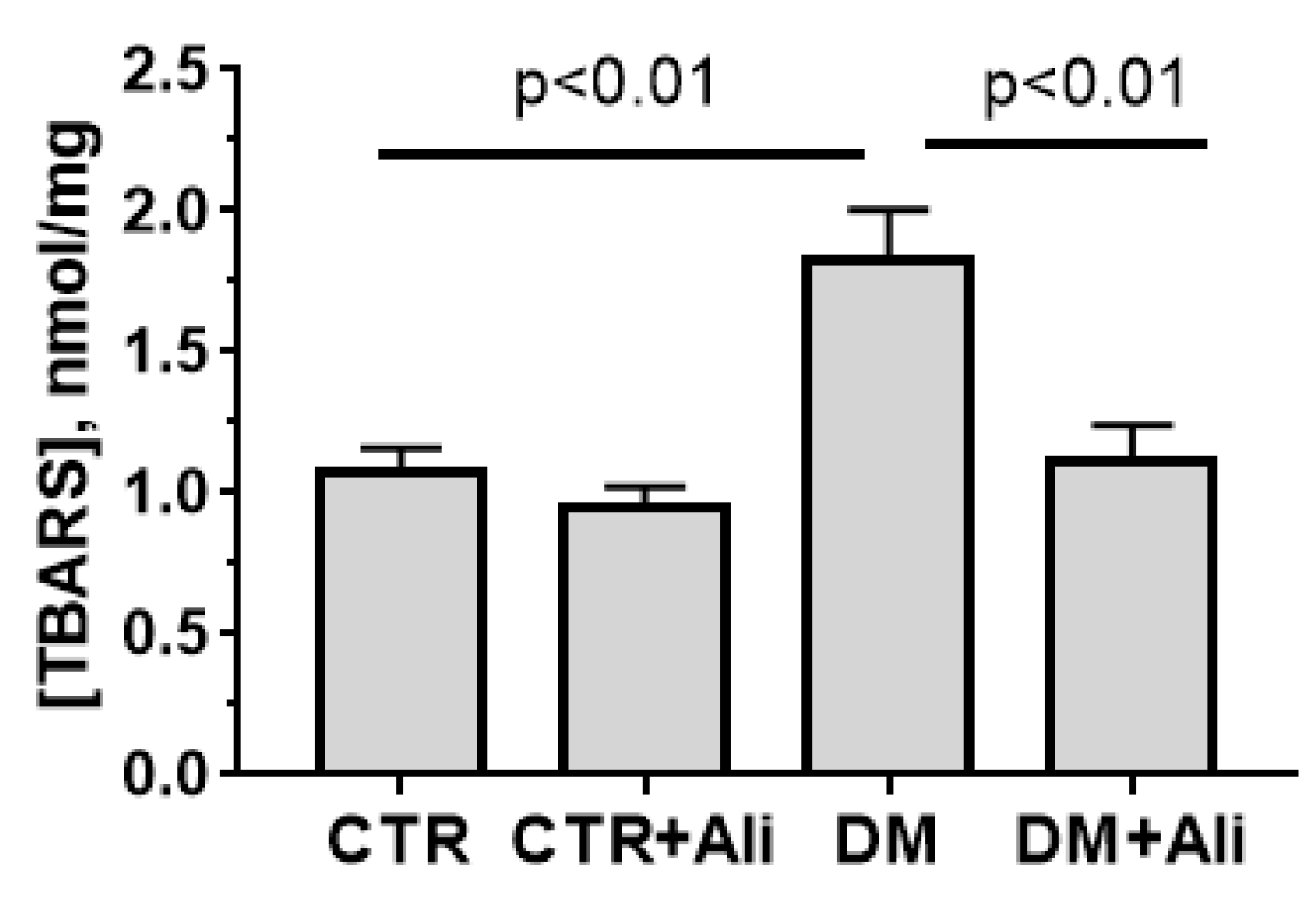
3.2. Effects of Alisporivir on Diabetes-Induced Changes in the Functioning of Heart Mitochondria
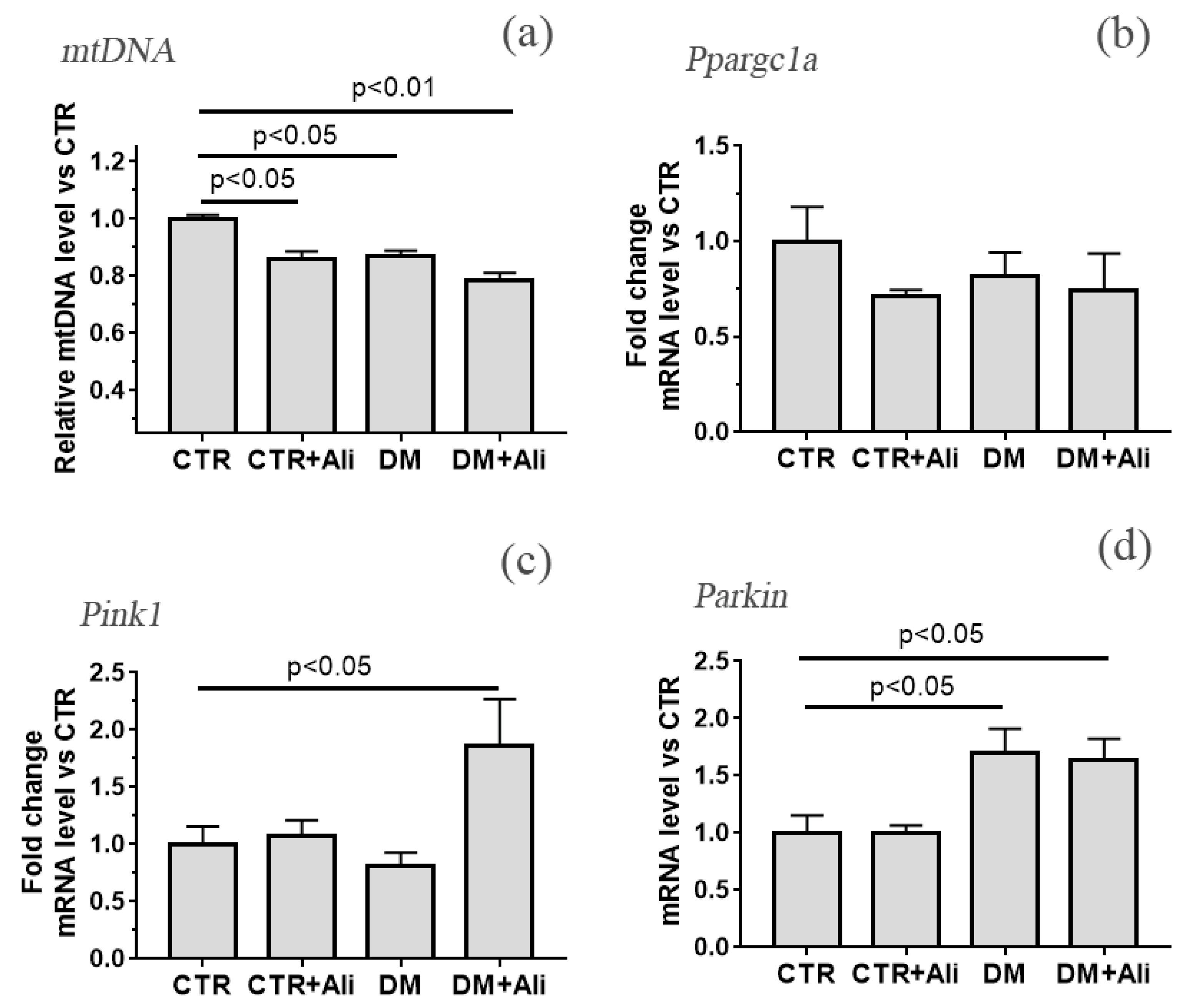
3.3. The Effect of Alisporivir on Ultrastructural Changes in Heart Mitochondria of Mice with Experimental DM and on the Expression of Protein Genes Responsible for Mitochondrial Biogenesis and Mitophagy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boudina, S.; Abel, E.D. Diabetic Cardiomyopathy Revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.K.; Murohara, T. Diabetes-Related Heart Failure. Circ. J. 2014, 78, 576–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef]
- Dillmann, W.H. Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Käämbre, T.; Han, W.H.; De Goede, P.; et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2019, 228, e13430. [Google Scholar] [CrossRef] [Green Version]
- Gollmer, J.; Zirlik, A.; Bugger, H. Mitochondrial Mechanisms in Diabetic Cardiomyopathy. Diabetes Metab. J. 2020, 44, 33–53. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.; Belosludtseva, N.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Fauconnier, J.; Lanner, J.T.; Zhang, S.-J.; Tavi, P.; Bruton, J.D.; Katz, A.; Westerblad, H. Insulin and Inositol 1,4,5-Trisphosphate Trigger Abnormal Cytosolic Ca2+ Transients and Reveal Mitochondrial Ca2+ Handling Defects in Cardiomyocytes of ob/ob Mice. Diabetes 2005, 54, 2375–2381. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, P.J.; Seiça, R.; Coxito, P.M.; Rolo, A.P.; Palmeira, C.; Santos, M.S.; Moreno, A.J. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003, 554, 511–514. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Juarez, J.; Suarez, J.; Cividini, F.; Scott, B.; Diemer, T.; Dai, A.; Dillmann, W.H. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am. J. Physiol. Physiol. 2016, 311, C1005–C1013. [Google Scholar] [CrossRef] [Green Version]
- Suarez, J.; Cividini, F.; Scott, B.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta Rev. Biomembr. 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Pan, M.; Ma, J.; Song, X.; Cao, W.; Zhang, P. Recent progress in the use of mitochondrial membrane permeability transition pore in mitochondrial dysfunction-related disease therapies. Mol. Cell. Biochem. 2021, 476, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Baetz, D.; Ovize, M. Cyclophilin D and myocardial ischemia–reperfusion injury: A fresh perspective. J. Mol. Cell. Cardiol. 2015, 78, 80–89. [Google Scholar] [CrossRef]
- Amanakis, G.; Murphy, E. Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol. 2020, 11, 595. [Google Scholar] [CrossRef]
- Itoh, T.; Kouzu, H.; Miki, T.; Tanno, M.; Kuno, A.; Sato, T.; Sunaga, D.; Murase, H.; Miura, T. Cytoprotective regulation of the mitochondrial permeability transition pore is impaired in type 2 diabetic Goto–Kakizaki rat hearts. J. Mol. Cell. Cardiol. 2012, 53, 870–879. [Google Scholar] [CrossRef]
- Riojas-Hernández, A.; Bernal-Ramírez, J.; Rodríguez-Mier, D.; Marroquin, F.E.M.; Domínguez-Barragán, E.M.; Borja-Villa, C.; Rivera-Álvarez, I.; García-Rivas, G.; Altamirano, J.; García, N. Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from Zucker Fa/fa rats with type 2 diabetes. Life Sci. 2015, 141, 32–43. [Google Scholar] [CrossRef]
- Sloan, R.C.; Moukdar, F.; Frasier, C.R.; Patel, H.D.; Bostian, P.A.; Lust, R.M.; Brown, D.A. Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell. Cardiol. 2012, 52, 1009–1018. [Google Scholar] [CrossRef]
- Najafi, M.; Farajnia, S.; Mohammadi, M.; Badalzadeh, R.; Asl, N.A.; Baradaran, B.; Amani, M. Inhibition of mitochondrial permeability transition pore restores the cardioprotection by postconditioning in diabetic hearts. J. Diabetes Metab. Disord. 2014, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Huhn, R.; Heinen, A.; Hollmann, M.; Schlack, W.; Preckel, B.; Weber, N. Cyclosporine A administered during reperfusion fails to restore cardioprotection in prediabetic Zucker obese rats in vivo. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.; Mattiasson, G.; Månsson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.; Dumont, J.-M.; Besseghir, K.; Elmér, E. The Nonimmunosuppressive Cyclosporin Analogs NIM811 and UNIL025 Display Nanomolar Potencies on Permeability Transition in Brain-Derived Mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Thibault, H.; Gharib, A.; Dumont, J.-M.; Vuagniaux, G.; Scalfaro, P.; Derumeaux, G.; Ovize, M. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am. J. Physiol. Circ. Physiol. 2007, 293, H1654–H1661. [Google Scholar] [CrossRef] [Green Version]
- Wissing, E.R.; Millay, D.P.; Vuagniaux, G.; Molkentin, J.D. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscul. Disord. 2010, 20, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a cyclophilin inhibitor, in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. Br. J. Pharmacol. 2008, 155, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Softic, L.; Brillet, R.; Berry, F.; Ahnou, N.; Nevers, Q.; Morin-Dewaele, M.; Hamadat, S.; Bruscella, P.; Fourati, S.; Pawlotsky, J.-M.; et al. Inhibition of SARS-CoV-2 Infection by the Cyclophilin Inhibitor Alisporivir (Debio 025). Antimicrob. Agents Chemother. 2020, 64, e00876-20. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Norouzi, S.; Ruggiero, A.; Khan, M.; Myers, S.; Kavanagh, K.; Vemuri, R. Type-2 Diabetes as a Risk Factor for Severe COVID-19 Infection. Microorganisms 2021, 9, 1211. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology 2020, 9, 309. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludtsev, K.N.; Dubinin, M.V.; Talanov, E.Y.; Starinets, V.S.; Tenkov, K.S.; Zakharova, N.M.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in the Liver and Heart Mitochondria of Rats with Different Tolerance to Acute Hypoxia. Biomolecules 2020, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Kosareva, E.A.; Talanov, E.Y.; Gudkov, S.V.; Dubinin, M.V. Itaconic acid impairs the mitochondrial function by the inhibition of complexes II and IV and induction of the permeability transition pore opening in rat liver mitochondria. Biochimie 2020, 176, 150–157. [Google Scholar] [CrossRef]
- Astashev, M.E.; Serov, D.A.; Tankanag, A.V. Anesthesia effects on the low frequency blood flow oscillations in mouse skin. Ski. Res. Technol. 2018, 25, 40–46. [Google Scholar] [CrossRef]
- Kawai, S.; Takagi, Y.; Kaneko, S.; Kurosawa, T. Effect of Three Types of Mixed Anesthetic Agents Alternate to Ketamine in Mice. Exp. Anim. 2011, 60, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, A.; Serizawa, K.; Sato, R.; Yamazaki, J.; Inomata, T. Vital signs monitoring during injectable and inhalant anesthesia in mice. Exp. Anim. 2015, 64, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Lindblom, R.S.J.; Higgins, G.C.; Nguyen, T.-V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. 2020, 134, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Taddeo, E.; Laker, R.; Breen, D.; Akhtar, Y.; Kenwood, B.; Liao, J.; Zhang, M.; Fazakerley, D.; Tomsig, J.; Harris, T.; et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 2014, 3, 124–134. [Google Scholar] [CrossRef]
- Qi, R.; Wang, D.; Xing, L.; Wu, Z. Cyclosporin A inhibits mitochondrial biogenesis in Hep G2 cells. Biochem. Biophys. Res. Commun. 2018, 496, 941–946. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Nd4 | ATTATTATTACCCGATGAGGGAACC | ATTAAGATGAGGGCAATTAGCAGT |
| Gapdh | GTGAGGGAGATGCYCAGTGT | CTGGCATTGCTCTCAATGAC |
| Ppargc1a | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Pink1 | TTGCCCCACACCCTAACATC | GCAGGGTACAGGGGTAGTTCT |
| Parkin | AGCCAGAGGTCCAGCAGTTA | GAGGGTTGCTTGTTTGCAGG |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
| CTR | CTR + Ali | DM | DM + Ali | |
|---|---|---|---|---|
| Body weight, g | 31.0 ± 0.8 | 29.7 ± 0.7 | 29.4 ± 0.7 | 29.2 ± 0.8 |
| Heart weight, mg | 152.9 ± 4.5 | 154.1 ± 4.1 | 128.3 ± 5.3 * | 137.7 ± 4.0 |
| BG (fed state), mM | 10.4 ± 0.8 | 10.5 ± 1.1 | 20.5 ± 1.2 * | 16.9 ± 1.3 * |
| Triglycerides, mM | 1.61 ± 0.06 | 1.50 ± 0.07 | 1.96 ± 0.11 * | 1.70 ± 0.11 |
| [TBARS], nmol/mg | 5.3 ± 0.8 | 5.4 ± 0.6 | 10.9 ± 1.4 * | 9.4 ± 1.5 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtseva, N.V.; Starinets, V.S.; Mikheeva, I.B.; Serov, D.A.; Astashev, M.E.; Belosludtsev, M.N.; Dubinin, M.V.; Belosludtsev, K.N. Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice. Biology 2021, 10, 839. https://doi.org/10.3390/biology10090839
Belosludtseva NV, Starinets VS, Mikheeva IB, Serov DA, Astashev ME, Belosludtsev MN, Dubinin MV, Belosludtsev KN. Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice. Biology. 2021; 10(9):839. https://doi.org/10.3390/biology10090839
Chicago/Turabian StyleBelosludtseva, Natalia V., Vlada S. Starinets, Irina B. Mikheeva, Dmitriy A. Serov, Maxim E. Astashev, Maxim N. Belosludtsev, Mikhail V. Dubinin, and Konstantin N. Belosludtsev. 2021. "Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice" Biology 10, no. 9: 839. https://doi.org/10.3390/biology10090839
APA StyleBelosludtseva, N. V., Starinets, V. S., Mikheeva, I. B., Serov, D. A., Astashev, M. E., Belosludtsev, M. N., Dubinin, M. V., & Belosludtsev, K. N. (2021). Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice. Biology, 10(9), 839. https://doi.org/10.3390/biology10090839