Human Papillomavirus in Breast Carcinogenesis: A Passenger, a Cofactor, or a Causal Agent?

 ,
, 
Abstract
Simple Summary
Abstract
1. Breast Cancer as a Global Health Concern
1.1. Breast Cancer Epidemiology
1.2. Breast Cancer Classification
1.3. Breast Cancer Risk Factors
2. Human Papillomavirus (HPV)
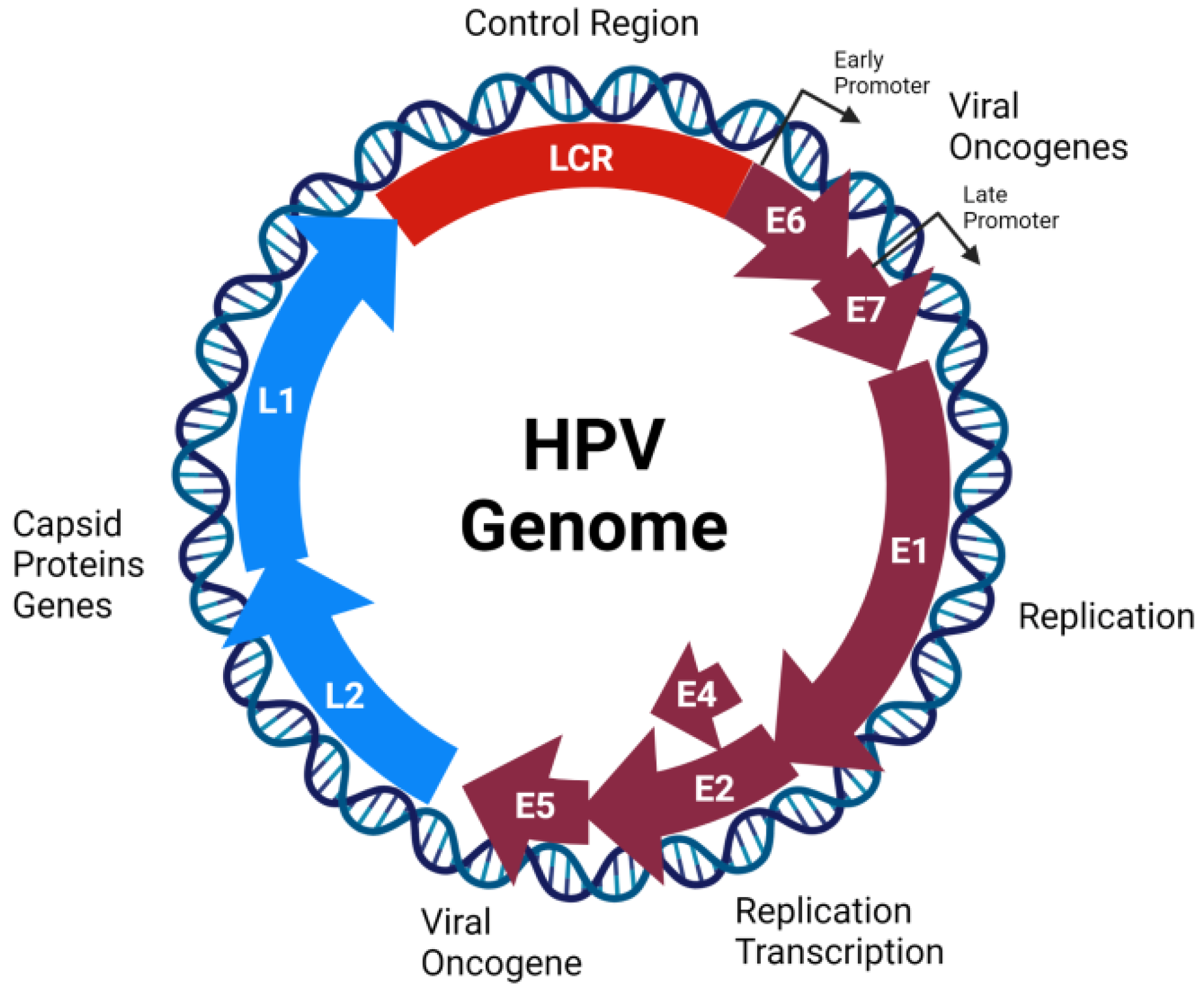
2.1. Genome Organization, Structure, and Replication Cycle
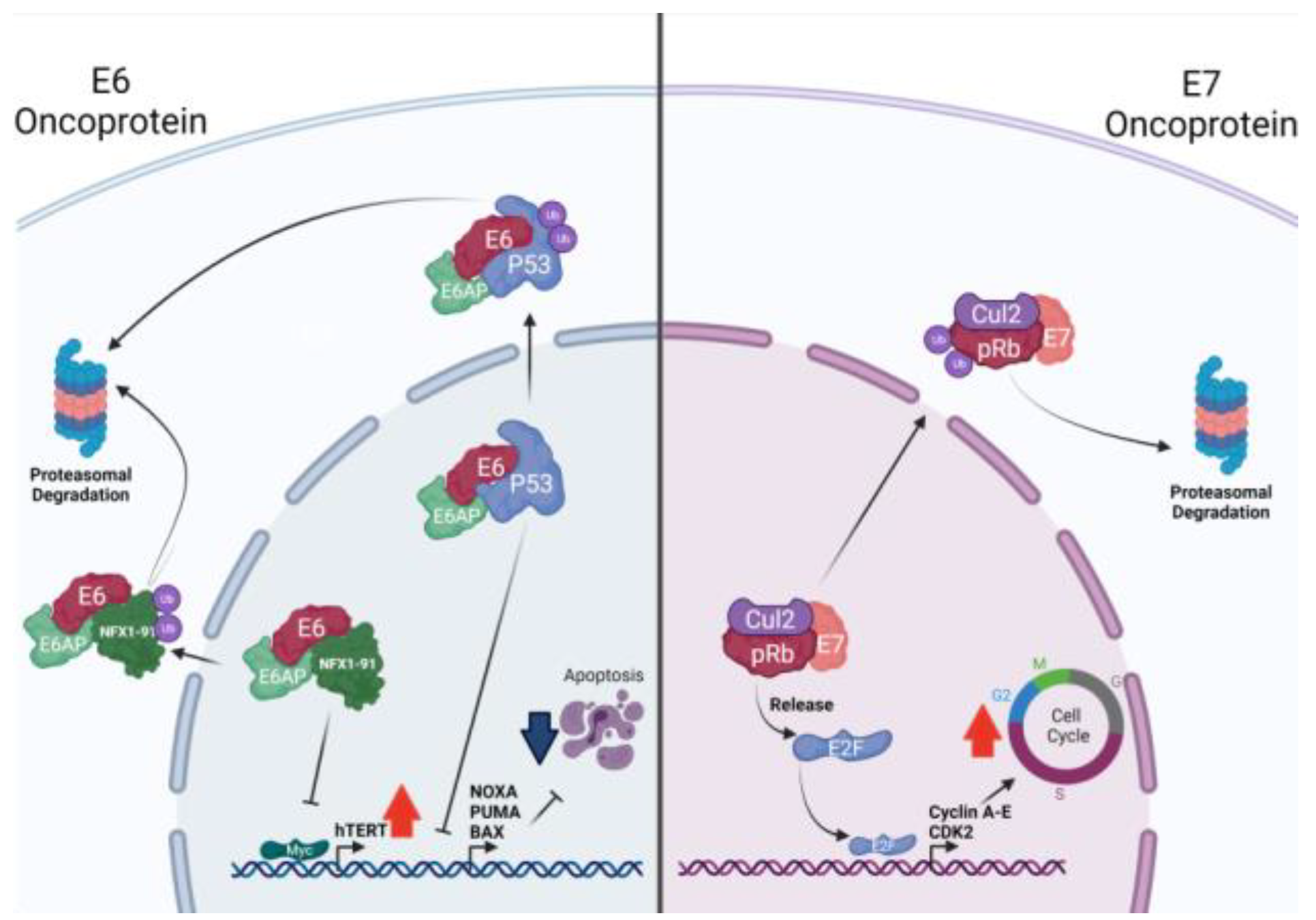
2.2. HPV Oncoproteins
3. HPV Infection and Breast Cancer
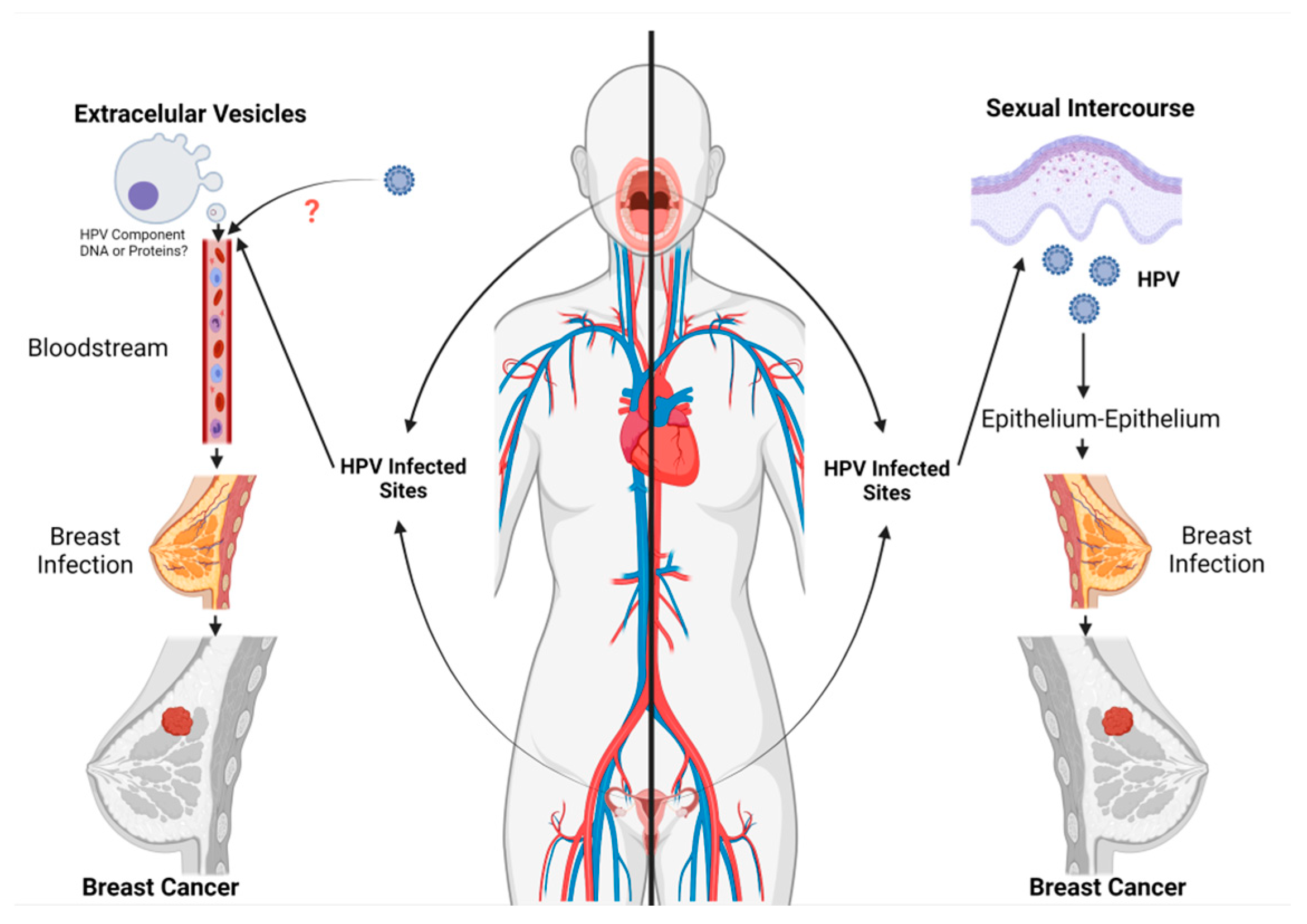
3.1. Routes for HPV Infection in Breast
3.2. Epidemiology of HPV Infection in Breast Cancer
3.3. Role of HPV Infection in Breast Carcinogenesis
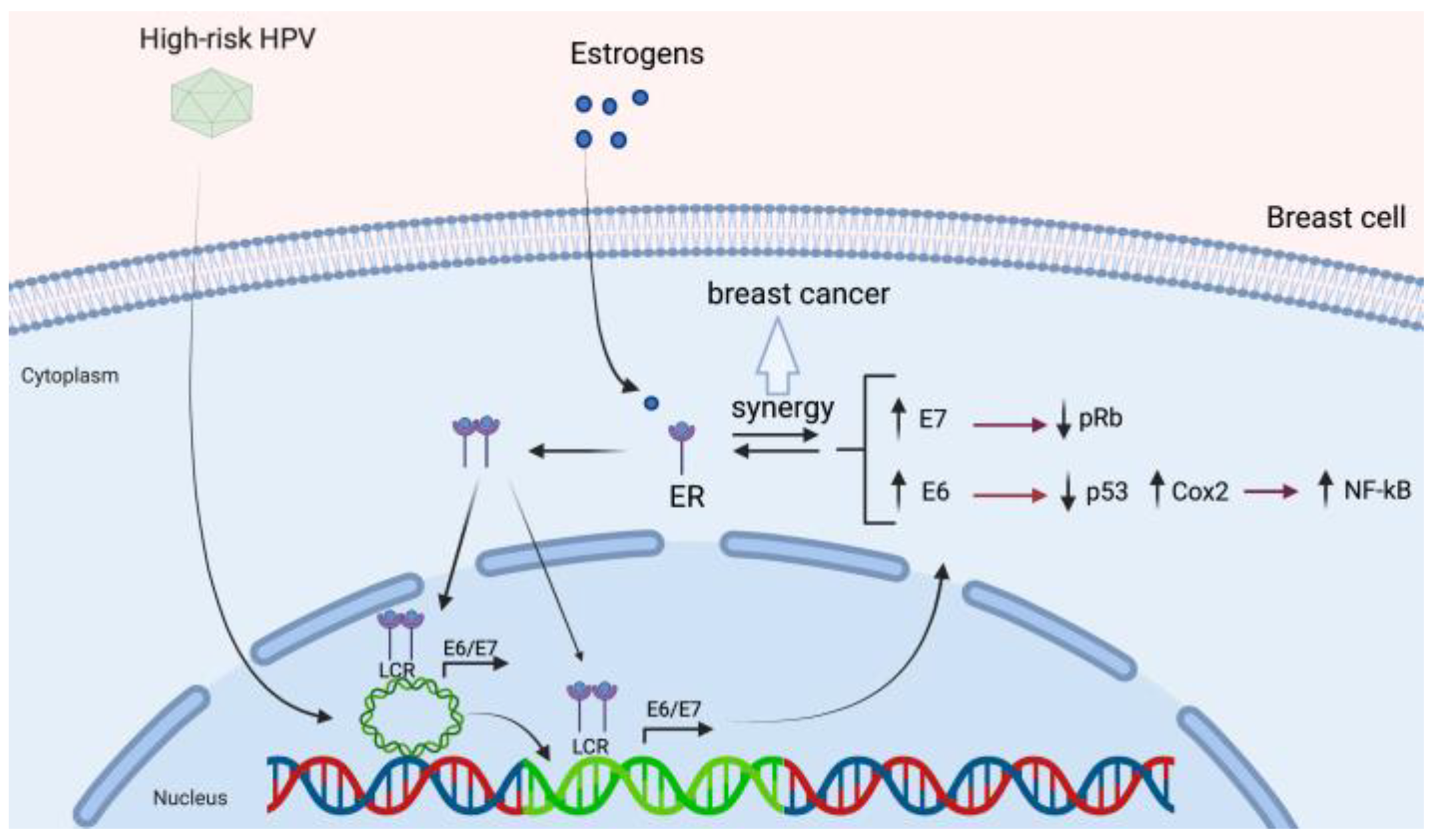
3.4. HPV and Its Association with Estrogen Receptor Signaling
4. Conclusions and Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Francies, F.Z.; Hull, R.; Khanyile, R.; Dlamini, Z. Breast cancer in low-middle income countries: Abnormality in splicing and lack of targeted treatment options. Am. J. Cancer Res. 2020, 10, 1568–1591. [Google Scholar] [PubMed]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Gonçalves, H.; Guerra, M.R.; Duarte Cintra, J.R.; Fayer, V.A.; Brum, I.V.; Bustamante Teixeira, M.T. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Makki, J. Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Liu, Z.; Cheng, R.; Sun, L.; Huang, S.; Fang, Y.; Wang, J. Variation in Breast Cancer Subtype Incidence and Distribution by Race/Ethnicity in the United States From 2010 to 2015. JAMA Netw. Open 2020, 3, e2020303. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [PubMed]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H.J.; members, P. Strategies for subtypes--dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Osako, T.; Okumura, Y.; Tashima, R.; Toyozumi, Y.; Arima, N. Changes in the ER, PgR, HER2, p53 and Ki-67 biological markers between primary and recurrent breast cancer: Discordance rates and prognosis. World J. Surg. Oncol. 2011, 9, 131. [Google Scholar] [CrossRef]
- Kim, C.; Lee, J.; Lee, W.; Kim, A. Changes in intrinsic subtype of breast cancer during tumor progression in the same patient. Int. J. Clin. Exp. Pathol. 2015, 8, 15184–15190. [Google Scholar]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef]
- Changavi, A.A.; Shashikala, A.; Ramji, A.S. Epidermal Growth Factor Receptor Expression in Triple Negative and Nontriple Negative Breast Carcinomas. J. Lab. Phys. 2015, 7, 79–83. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, X.; Zhang, C.; Xue, L.; Yang, L. Expression and clinical significance of MAPK and EGFR in triple-negative breast cancer. Oncol. Lett. 2020, 19, 1842–1848. [Google Scholar] [CrossRef]
- Sun, Y.S.; Zhao, Z.; Yang, Z.N.; Xu, F.; Lu, H.J.; Zhu, Z.Y.; Shi, W.; Jiang, J.; Yao, P.P.; Zhu, H.P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef]
- Brewer, H.R.; Jones, M.E.; Schoemaker, M.J.; Ashworth, A.; Swerdlow, A.J. Family history and risk of breast cancer: An analysis accounting for family structure. Breast Cancer Res. Treat. 2017, 165, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Nickels, S.; Truong, T.; Hein, R.; Stevens, K.; Buck, K.; Behrens, S.; Eilber, U.; Schmidt, M.; Häberle, L.; Vrieling, A.; et al. Evidence of gene-environment interactions between common breast cancer susceptibility loci and established environmental risk factors. PLoS Genet. 2013, 9, e1003284. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.F.; Nathanson, K.L.; Couch, F.J.; Offit, K. Genomic Biomarkers for Breast Cancer Risk. Adv. Exp. Med. Biol. 2016, 882, 1–32. [Google Scholar] [CrossRef]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Campa, D.; Kaaks, R.; Le Marchand, L.; Haiman, C.A.; Travis, R.C.; Berg, C.D.; Buring, J.E.; Chanock, S.J.; Diver, W.R.; Dostal, L.; et al. Interactions between genetic variants and breast cancer risk factors in the breast and prostate cancer cohort consortium. J. Natl. Cancer Inst. 2011, 103, 1252–1263. [Google Scholar] [CrossRef]
- Travis, R.C.; Reeves, G.K.; Green, J.; Bull, D.; Tipper, S.J.; Baker, K.; Beral, V.; Peto, R.; Bell, J.; Zelenika, D.; et al. Gene-environment interactions in 7610 women with breast cancer: Prospective evidence from the Million Women Study. Lancet 2010, 375, 2143–2151. [Google Scholar] [CrossRef]
- Prentice, R.L.; Huang, Y.; Hinds, D.A.; Peters, U.; Pettinger, M.; Cox, D.R.; Beilharz, E.; Chlebowski, R.T.; Rossouw, J.E.; Caan, B.; et al. Variation in the FGFR2 gene and the effects of postmenopausal hormone therapy on invasive breast cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3079–3085. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Gaudet, M.M.; Spurdle, A.B.; Fasching, P.A.; Couch, F.J.; Benítez, J.; Arias Pérez, J.I.; Zamora, M.P.; Malats, N.; Dos Santos Silva, I.; et al. Assessing interactions between the associations of common genetic susceptibility variants, reproductive history and body mass index with breast cancer risk in the breast cancer association consortium: A combined case-control study. Breast Cancer Res. 2010, 12, R110. [Google Scholar] [CrossRef]
- Naccarato, A.G.; Lessi, F.; Zavaglia, K.; Scatena, C.; Al Hamad, M.A.; Aretini, P.; Menicagli, M.; Roncella, M.; Ghilli, M.; Caligo, M.A.; et al. Mouse mammary tumor virus (MMTV)-like exogenous sequences are associated with sporadic but not hereditary human breast carcinoma. Aging 2019, 11, 7236–7241. [Google Scholar] [CrossRef]
- Witt, A.; Hartmann, B.; Marton, E.; Zeillinger, R.; Schreiber, M.; Kubista, E. The mouse mammary tumor virus-like env gene sequence is not detectable in breast cancer tissue of Austrian patients. Oncol. Rep. 2003, 10, 1025–1029. [Google Scholar] [CrossRef]
- Bindra, A.; Muradrasoli, S.; Kisekka, R.; Nordgren, H.; Wärnberg, F.; Blomberg, J. Search for DNA of exogenous mouse mammary tumor virus-related virus in human breast cancer samples. J. Gen. Virol. 2007, 88, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Henle, G.; Henle, W. Immunofluorescence in cells derived from Burkitt’s lymphoma. J. Bacteriol. 1966, 91, 1248–1256. [Google Scholar] [CrossRef]
- Green, M.; Michaels, M.G. Epstein-Barr virus infection and posttransplant lymphoproliferative disorder. Am. J. Transpl. 2013, 13 (Suppl. 3), 41–54. [Google Scholar] [CrossRef]
- Matalka, I.; Al Hamad, M.; Al-Hussaini, M.; Alzoubi, F.Q. The incidence of Epstein-Barr virus in nasopharyngeal carcinoma of Jordanian patients. Eur. Arch. Otorhinolaryngol. 2012, 269, 229–234. [Google Scholar] [CrossRef]
- Teras, L.R.; Rollison, D.E.; Pawlita, M.; Michel, A.; Brozy, J.; de Sanjose, S.; Blase, J.L.; Gapstur, S.M. Epstein-Barr virus and risk of non-Hodgkin lymphoma in the cancer prevention study-II and a meta-analysis of serologic studies. Int. J. Cancer 2015, 136, 108–116. [Google Scholar] [CrossRef]
- Bonnet, M.; Guinebretiere, J.M.; Kremmer, E.; Grunewald, V.; Benhamou, E.; Contesso, G.; Joab, I. Detection of Epstein-Barr virus in invasive breast cancers. J. Natl. Cancer Inst. 1999, 91, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Fina, F.; Romain, S.; Ouafik, L.; Palmari, J.; Ben Ayed, F.; Benharkat, S.; Bonnier, P.; Spyratos, F.; Foekens, J.A.; Rose, C.; et al. Frequency and genome load of Epstein-Barr virus in 509 breast cancers from different geographical areas. Br. J. Cancer 2001, 84, 783–790. [Google Scholar] [CrossRef]
- Pai, T.; Gupta, S.; Gurav, M.; Nag, S.; Shet, T.; Patil, A.; Desai, S. Evidence for the association of Epstein-Barr Virus in breast cancer in Indian patients using in-situ hybridization technique. Breast J. 2018, 24, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wang, T.; Zhu, H.; Guo, J.; Li, K.; Yao, Q.; Lv, Y.; Zhang, J.; He, C.; Chen, J.; et al. Multiplex PCR/mass spectrometry screening of biological carcinogenic agents in human mammary tumors. J. Clin. Virol. 2014, 61, 255–259. [Google Scholar] [CrossRef]
- Marrão, G.; Habib, M.; Paiva, A.; Bicout, D.; Fallecker, C.; Franco, S.; Fafi-Kremer, S.; Simões da Silva, T.; Morand, P.; Freire de Oliveira, C.; et al. Epstein-Barr virus infection and clinical outcome in breast cancer patients correlate with immune cell TNF-α/IFN-γ response. BMC Cancer 2014, 14, 665. [Google Scholar] [CrossRef]
- Mohammadizadeh, F.; Zarean, M.; Abbasi, M. Association of Epstein-Barr virus with invasive breast carcinoma and its impact on well-known clinicopathologic parameters in Iranian women. Adv. Biomed. Res. 2014, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Kadivar, M.; Monabati, A.; Joulaee, A.; Hosseini, N. Epstein-Barr virus and breast cancer: Lack of evidence for an association in Iranian women. Pathol. Oncol. Res. 2011, 17, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Perrigoue, J.G.; den Boon, J.A.; Friedl, A.; Newton, M.A.; Ahlquist, P.; Sugden, B. Lack of association between EBV and breast carcinoma. Cancer Epidemiol. Biomark. Prev. 2005, 14, 809–814. [Google Scholar] [CrossRef]
- Herrmann, K.; Niedobitek, G. Lack of evidence for an association of Epstein-Barr virus infection with breast carcinoma. Breast Cancer Res. 2003, 5, R13–R17. [Google Scholar] [CrossRef] [PubMed]
- Pinidis, P.; Tsikouras, P.; Iatrakis, G.; Zervoudis, S.; Koukouli, Z.; Bothou, A.; Galazios, G.; Vladareanu, S. Human Papilloma Virus’ Life Cycle and Carcinogenesis. Maedica 2016, 11, 48–54. [Google Scholar]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J.L.; the International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of HPV transcription. Clinics 2018, 73, e486s. [Google Scholar] [CrossRef] [PubMed]
- Giroglou, T.; Florin, L.; Schäfer, F.; Streeck, R.E.; Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Abban, C.Y.; Meneses, P.I. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 2010, 403, 1–16. [Google Scholar] [CrossRef]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kühling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef] [PubMed]
- Wasson, C.W.; Morgan, E.L.; Müller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef]
- Adam, J.L.; Briggs, M.W.; McCance, D.J. A mutagenic analysis of the E5 protein of human papillomavirus type 16 reveals that E5 binding to the vacuolar H+-ATPase is not sufficient for biological activity, using mammalian and yeast expression systems. Virology 2000, 272, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.I.; Finbow, M.E.; Alonso, A. Binding of human papillomavirus 16 E5 to the 16 kDa subunit c (proteolipid) of the vacuolar H+-ATPase can be dissociated from the E5-mediated epidermal growth factor receptor overactivation. Oncogene 2000, 19, 3727–3732. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, E.I.; Tugizov, S.; Herrera, R.; Da Costa, M.; Palefsky, J.M. E5 can be expressed in anal cancer and leads to epidermal growth factor receptor-induced invasion in a human papillomavirus 16-transformed anal epithelial cell line. J. Gen. Virol. 2018, 99, 631–644. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Garnett, T.O.; Duerksen-Hughes, P.J. Modulation of apoptosis by human papillomavirus (HPV) oncoproteins. Arch. Virol. 2006, 151, 2321–2335. [Google Scholar] [CrossRef]
- Yuan, C.H.; Filippova, M.; Tungteakkhun, S.S.; Duerksen-Hughes, P.J.; Krstenansky, J.L. Small molecule inhibitors of the HPV16-E6 interaction with caspase 8. Bioorg. Med. Chem. Lett. 2012, 22, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, O.; Veeraraghavalu, K.; Tergaonkar, V.; Liu, Y.; Androphy, E.J.; Stanley, M.A.; Krishna, S. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J. Virol. 2004, 78, 5934–5945. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Münger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol 2010, 84, 9398–9407. [Google Scholar] [CrossRef]
- Niebler, M.; Qian, X.; Höfler, D.; Kogosov, V.; Kaewprag, J.; Kaufmann, A.M.; Ly, R.; Böhmer, G.; Zawatzky, R.; Rösl, F.; et al. Post-translational control of IL-1β via the human papillomavirus type 16 E6 oncoprotein: A novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog. 2013, 9, e1003536. [Google Scholar] [CrossRef] [PubMed]
- Cordano, P.; Gillan, V.; Bratlie, S.; Bouvard, V.; Banks, L.; Tommasino, M.; Campo, M.S. The E6E7 oncoproteins of cutaneous human papillomavirus type 38 interfere with the interferon pathway. Virology 2008, 377, 408–418. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, T.; Ferril, S.; Snider, A.; Barbosa, M. In-vivo analysis of hpv e7 protein association with prb, p107 and p130. Int. J. Oncol. 1995, 6, 167–174. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef]
- Akerman, G.S.; Tolleson, W.H.; Brown, K.L.; Zyzak, L.L.; Mourateva, E.; Engin, T.S.; Basaraba, A.; Coker, A.L.; Creek, K.E.; Pirisi, L. Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Res. 2001, 61, 3837–3843. [Google Scholar] [PubMed]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Chang, H.S.; Lin, C.H.; Yu, W.C. HPV-18 E7 conjugates to c-Myc and mediates its transcriptional activity. Int. J. Biochem. Cell Biol. 2007, 39, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Hellner, K.; Mar, J.; Fang, F.; Quackenbush, J.; Münger, K. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology 2009, 391, 57–63. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.S.; Glenn, W.K.; Salyakina, D.; Clay, R.; Delprado, W.; Cheerala, B.; Tran, D.D.; Ngan, C.C.; Miyauchi, S.; Karim, M.; et al. Human Papilloma Virus Identification in Breast Cancer Patients with Previous Cervical Neoplasia. Front. Oncol. 2015, 5, 298. [Google Scholar] [CrossRef]
- Hennig, E.M.; Suo, Z.; Thoresen, S.; Holm, R.; Kvinnsland, S.; Nesland, J.M. Human papillomavirus 16 in breast cancer of women treated for high grade cervical intraepithelial neoplasia (CIN III). Breast Cancer Res. Treat. 1999, 53, 121–135. [Google Scholar] [CrossRef]
- Widschwendter, A.; Brunhuber, T.; Wiedemair, A.; Mueller-Holzner, E.; Marth, C. Detection of human papillomavirus DNA in breast cancer of patients with cervical cancer history. J. Clin. Virol. 2004, 31, 292–297. [Google Scholar] [CrossRef]
- Islam, M.S.; Chakraborty, B.; Panda, C.K. Human papilloma virus (HPV) profiles in breast cancer: Future management. Ann. Transl. Med. 2020, 8, 650. [Google Scholar] [CrossRef]
- Bodaghi, S.; Wood, L.V.; Roby, G.; Ryder, C.; Steinberg, S.M.; Zheng, Z.M. Could human papillomaviruses be spread through blood? J. Clin. Microbiol. 2005, 43, 5428–5434. [Google Scholar] [CrossRef]
- García-Casas, A.; García-Olmo, D.C.; García-Olmo, D. Further the liquid biopsy: Gathering pieces of the puzzle of genometastasis theory. World J. Clin. Oncol. 2017, 8, 378–388. [Google Scholar] [CrossRef] [PubMed]
- De Carolis, S.; Storci, G.; Ceccarelli, C.; Savini, C.; Gallucci, L.; Sansone, P.; Santini, D.; Seracchioli, R.; Taffurelli, M.; Fabbri, F.; et al. HPV DNA Associates With Breast Cancer Malignancy and It Is Transferred to Breast Cancer Stromal Cells by Extracellular Vesicles. Front. Oncol. 2019, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Carolis, S.; Pellegrini, A.; Santini, D.; Ceccarelli, C.; De Leo, A.; Alessandrini, F.; Arienti, C.; Pignatta, S.; Tesei, A.; Mantovani, V.; et al. Liquid biopsy in the diagnosis of HPV DNA in breast lesions. Future Microbiol. 2018, 13, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, J.; Zhai, K. Detection of Human Papillomavirus DNA in Patients with Breast Tumor in China. PLoS ONE 2015, 10, e0136050. [Google Scholar] [CrossRef]
- De Villiers, E.M.; Sandstrom, R.E.; zur Hausen, H.; Buck, C.E. Presence of papillomavirus sequences in condylomatous lesions of the mamillae and in invasive carcinoma of the breast. Breast Cancer Res. 2005, 7, R1–R11. [Google Scholar] [CrossRef]
- Lindel, K.; Forster, A.; Altermatt, H.J.; Greiner, R.; Gruber, G. Breast cancer and human papillomavirus (HPV) infection: No evidence of a viral etiology in a group of Swiss women. Breast 2007, 16, 172–177. [Google Scholar] [CrossRef]
- De Cremoux, P.; Thioux, M.; Lebigot, I.; Sigal-Zafrani, B.; Salmon, R.; Sastre-Garau, X.; Group, I.C.B. No evidence of human papillomavirus DNA sequences in invasive breast carcinoma. Breast Cancer Res. Treat. 2008, 109, 55–58. [Google Scholar] [CrossRef]
- Chang, P.; Wang, T.; Yao, Q.; Lv, Y.; Zhang, J.; Guo, W.; Wang, L.; Chen, J. Absence of human papillomavirus in patients with breast cancer in north-west China. Med. Oncol. 2012, 29, 521–525. [Google Scholar] [CrossRef]
- Li, N.; Bi, X.; Zhang, Y.; Zhao, P.; Zheng, T.; Dai, M. Human papillomavirus infection and sporadic breast carcinoma risk: A meta-analysis. Breast Cancer Res. Treat. 2011, 126, 515–520. [Google Scholar] [CrossRef]
- Simões, P.W.; Medeiros, L.R.; Simões Pires, P.D.; Edelweiss, M.I.; Rosa, D.D.; Silva, F.R.; Silva, B.R.; Rosa, M.I. Prevalence of human papillomavirus in breast cancer: A systematic review. Int. J. Gynecol. Cancer 2012, 22, 343–347. [Google Scholar] [CrossRef]
- Lawson, J.S.; Glenn, W.K.; Salyakina, D.; Delprado, W.; Clay, R.; Antonsson, A.; Heng, B.; Miyauchi, S.; Tran, D.D.; Ngan, C.C.; et al. Human Papilloma Viruses and Breast Cancer. Front. Oncol. 2015, 5, 277. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M.; Kim, E.H. Human papillomavirus infection and risk of breast cancer: A meta-analysis of case-control studies. Infect. Agent Cancer 2016, 11, 14. [Google Scholar] [CrossRef]
- Choi, J.; Kim, C.; Lee, H.S.; Choi, Y.J.; Kim, H.Y.; Lee, J.; Chang, H.; Kim, A. Detection of Human Papillomavirus in Korean Breast Cancer Patients by Real-Time Polymerase Chain Reaction and Meta-Analysis of Human Papillomavirus and Breast Cancer. J. Pathol. Transl. Med. 2016, 50, 442–450. [Google Scholar] [CrossRef][Green Version]
- Ren, C.; Zeng, K.; Wu, C.; Mu, L.; Huang, J.; Wang, M. Human papillomavirus infection increases the risk of breast carcinoma: A large-scale systemic review and meta-analysis of case-control studies. Gland Surg. 2019, 8, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Al Hamad, M.; Matalka, I.; Al Zoubi, M.S.; Armogida, I.; Khasawneh, R.; Al-Husaini, M.; Sughayer, M.; Jaradat, S.; Al-Nasser, A.D.; Mazzanti, C.M. Human Mammary Tumor Virus, Human Papilloma Virus, and Epstein-Barr Virus Infection Are Associated With Sporadic Breast Cancer Metastasis. Breast Cancer 2020, 14, 1178223420976388. [Google Scholar] [CrossRef]
- Gumus, M.; Yumuk, P.F.; Salepci, T.; Aliustaoglu, M.; Dane, F.; Ekenel, M.; Basaran, G.; Kaya, H.; Barisik, N.; Turhal, N.S. HPV DNA frequency and subset analysis in human breast cancer patients’ normal and tumoral tissue samples. J. Exp. Clin. Cancer Res. 2006, 25, 515–521. [Google Scholar]
- Khodabandehlou, N.; Mostafaei, S.; Etemadi, A.; Ghasemi, A.; Payandeh, M.; Hadifar, S.; Norooznezhad, A.H.; Kazemnejad, A.; Moghoofei, M. Human papilloma virus and breast cancer: The role of inflammation and viral expressed proteins. BMC Cancer 2019, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- El-Sheikh, N.; Mousa, N.O.; Tawfeik, A.M.; Saleh, A.M.; Elshikh, I.; Deyab, M.; Ragheb, F.; Moneer, M.M.; Kawashti, A.; Osman, A.; et al. Assessment of Human Papillomavirus Infection and Risk Factors in Egyptian Women with Breast Cancer. Breast Cancer 2021, 15, 1178223421996279. [Google Scholar] [CrossRef] [PubMed]
- Balci, F.L.; Uras, C.; Feldman, S.M. Is human papillomavirus associated with breast cancer or papilloma presenting with pathologic nipple discharge? Cancer Treat. Res. Commun. 2019, 19, 100122. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Cho, E.Y.; Kim, J.H.; Nam, S.J.; Oh, Y.L.; Song, S.Y.; Yang, J.H.; Kim, D.S. Detection of human papillomavirus DNA by DNA chip in breast carcinomas of Korean women. Tumour Biol. 2007, 28, 327–332. [Google Scholar] [CrossRef]
- Zhang, N.; Ma, Z.P.; Wang, J.; Bai, H.L.; Li, Y.X.; Sun, Q.; Yang, L.; Tao, L.; Zhao, J.; Cao, Y.W.; et al. Human papillomavirus infection correlates with inflammatory Stat3 signaling activity and IL-17 expression in patients with breast cancer. Am. J. Transl Res. 2016, 8, 3214–3226. [Google Scholar]
- Islam, S.; Dasgupta, H.; Roychowdhury, A.; Bhattacharya, R.; Mukherjee, N.; Roy, A.; Mandal, G.K.; Alam, N.; Biswas, J.; Mandal, S.; et al. Study of association and molecular analysis of human papillomavirus in breast cancer of Indian patients: Clinical and prognostic implication. PLoS ONE 2017, 12, e0172760. [Google Scholar] [CrossRef] [PubMed]
- Heng, B.; Glenn, W.K.; Ye, Y.; Tran, B.; Delprado, W.; Lutze-Mann, L.; Whitaker, N.J.; Lawson, J.S. Human papilloma virus is associated with breast cancer. Br. J. Cancer 2009, 101, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Delgado-García, S.; Martínez-Escoriza, J.C.; Alba, A.; Martín-Bayón, T.A.; Ballester-Galiana, H.; Peiró, G.; Caballero, P.; Ponce-Lorenzo, J. Presence of human papillomavirus DNA in breast cancer: A Spanish case-control study. BMC Cancer 2017, 17, 320. [Google Scholar] [CrossRef] [PubMed]
- Habyarimana, T.; Attaleb, M.; Mazarati, J.B.; Bakri, Y.; El Mzibri, M. Detection of human papillomavirus DNA in tumors from Rwandese breast cancer patients. Breast Cancer 2018, 25, 127–133. [Google Scholar] [CrossRef]
- Akil, N.; Yasmeen, A.; Kassab, A.; Ghabreau, L.; Darnel, A.D.; Al Moustafa, A.E. High-risk human papillomavirus infections in breast cancer in Syrian women and their association with Id-1 expression: A tissue microarray study. Br. J. Cancer 2008, 99, 404–407. [Google Scholar] [CrossRef]
- Salman, N.A.; Davies, G.; Majidy, F.; Shakir, F.; Akinrinade, H.; Perumal, D.; Ashrafi, G.H. Association of High Risk Human Papillomavirus and Breast cancer: A UK based Study. Sci. Rep. 2017, 7, 43591. [Google Scholar] [CrossRef]
- Fernandes, A.; Bianchi, G.; Feltri, A.P.; Pérez, M.; Correnti, M. Presence of human papillomavirus in breast cancer and its association with prognostic factors. Ecancermedicalscience 2015, 9, 548. [Google Scholar] [CrossRef]
- Golrokh Mofrad, M.; Sadigh, Z.A.; Ainechi, S.; Faghihloo, E. Detection of human papillomavirus genotypes, herpes simplex, varicella zoster and cytomegalovirus in breast cancer patients. Virol. J. 2021, 18, 25. [Google Scholar] [CrossRef]
- Elagali, A.M.; Suliman, A.A.; Altayeb, M.; Dannoun, A.I.; Parine, N.R.; Sakr, H.I.; Suliman, H.S.; Motawee, M.E. Human papillomavirus, gene mutation and estrogen and progesterone receptors in breast cancer: A cross-sectional study. Pan. Afr. Med. J. 2021, 38, 43. [Google Scholar] [CrossRef]
- Piana, A.F.; Sotgiu, G.; Muroni, M.R.; Cossu-Rocca, P.; Castiglia, P.; De Miglio, M.R. HPV infection and triple-negative breast cancers: An Italian case-control study. Virol. J. 2014, 11, 190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lawson, J.S.; Glenn, W.K.; Heng, B.; Ye, Y.; Tran, B.; Lutze-Mann, L.; Whitaker, N.J. Koilocytes indicate a role for human papilloma virus in breast cancer. Br. J. Cancer 2009, 101, 1351–1356. [Google Scholar] [CrossRef]
- Aguayo, F.; Khan, N.; Koriyama, C.; González, C.; Ampuero, S.; Padilla, O.; Solís, L.; Eizuru, Y.; Corvalán, A.; Akiba, S. Human papillomavirus and Epstein-Barr virus infections in breast cancer from chile. Infect. Agent Cancer 2011, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Castillo, A.; Koriyama, C.; Kijima, Y.; Umekita, Y.; Ohi, Y.; Higashi, M.; Sagara, Y.; Yoshinaka, H.; Tsuji, T.; et al. Human papillomavirus detected in female breast carcinomas in Japan. Br. J. Cancer 2008, 99, 408–414. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef]
- Cheung, J.L.; Cheung, T.H.; Yu, M.Y.; Chan, P.K. Virological characteristics of cervical cancers carrying pure episomal form of HPV16 genome. Gynecol. Oncol. 2013, 131, 374–379. [Google Scholar] [CrossRef]
- de la Cruz-Hernández, E.; Pérez-Cárdenas, E.; Contreras-Paredes, A.; Cantú, D.; Mohar, A.; Lizano, M.; Dueñas-González, A. The effects of DNA methylation and histone deacetylase inhibitors on human papillomavirus early gene expression in cervical cancer, an in vitro and clinical study. Virol. J. 2007, 4, 18. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, Z.; Pan, X.; Uehara, T.; Suzuki, M.; Xie, M. Effects of Methylation Status of CpG Sites within the HPV16 Long Control Region on HPV16-Positive Head and Neck Cancer Cells. PLoS ONE 2015, 10, e0141245. [Google Scholar] [CrossRef]
- Lawson, J.S.; Glenn, W.K.; Whitaker, N.J. Human Papilloma Viruses and Breast Cancer—Assessment of Causality. Front. Oncol. 2016, 6, 207. [Google Scholar] [CrossRef]
- Ngan, C.; Lawson, J.S.; Clay, R.; Delprado, W.; Whitaker, N.J.; Glenn, W.K. Early Human Papilloma Virus (HPV) Oncogenic Influences in Breast Cancer. Breast Cancer 2015, 9, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, N.F.; Platt, R.W.; Duarte-Franco, E.; Costa, M.C.; Sobrinho, J.P.; Prado, J.C.; Ferenczy, A.; Rohan, T.E.; Villa, L.L.; Franco, E.L. Human papillomavirus infection and time to progression and regression of cervical intraepithelial neoplasia. J. Natl. Cancer Inst. 2003, 95, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, N.F.; Trevisan, A.; Duarte-Franco, E.; Rohan, T.E.; Ferenczy, A.; Villa, L.L.; Franco, E.L. Viral load as a predictor of the risk of cervical intraepithelial neoplasia. Int. J. Cancer 2003, 103, 519–524. [Google Scholar] [CrossRef]
- Kroupis, C.; Markou, A.; Vourlidis, N.; Dionyssiou-Asteriou, A.; Lianidou, E.S. Presence of high-risk human papillomavirus sequences in breast cancer tissues and association with histopathological characteristics. Clin. Biochem. 2006, 39, 727–731. [Google Scholar] [CrossRef]
- Wang, Y.W.; Zhang, K.; Zhao, S.; Lv, Y.; Zhu, J.; Liu, H.; Feng, J.; Liang, W.; Ma, R.; Wang, J. HPV Status and Its Correlation with BCL2, p21, p53, Rb, and Survivin Expression in Breast Cancer in a Chinese Population. Biomed. Res. Int. 2017, 2017, 6315392. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, A.; Bismar, T.A.; Kandouz, M.; Foulkes, W.D.; Desprez, P.Y.; Al Moustafa, A.E. E6/E7 of HPV type 16 promotes cell invasion and metastasis of human breast cancer cells. Cell Cycle 2007, 6, 2038–2042. [Google Scholar] [CrossRef]
- Woods Ignatoski, K.M.; Dziubinski, M.L.; Ammerman, C.; Ethier, S.P. Cooperative interactions of HER-2 and HPV-16 oncoproteins in the malignant transformation of human mammary epithelial cells. Neoplasia 2005, 7, 788–798. [Google Scholar] [CrossRef][Green Version]
- Yasmeen, A.; Bismar, T.A.; Dekhil, H.; Ricciardi, R.; Kassab, A.; Gambacorti-Passerini, C.; Al Moustafa, A.E. ErbB-2 receptor cooperates with E6/E7 oncoproteins of HPV type 16 in breast tumorigenesis. Cell Cycle 2007, 6, 2939–2943. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohba, K.; Ichiyama, K.; Yajima, M.; Gemma, N.; Nikaido, M.; Wu, Q.; Chong, P.; Mori, S.; Yamamoto, R.; Wong, J.E.; et al. In vivo and in vitro studies suggest a possible involvement of HPV infection in the early stage of breast carcinogenesis via APOBEC3B induction. PLoS ONE 2014, 9, e97787. [Google Scholar] [CrossRef]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, S.; Meng, Q.; Ma, Y.; Katiyar, P.; Schlegel, R.; Rosen, E.M. BRCA1 interaction with human papillomavirus oncoproteins. J. Biol. Chem. 2005, 280, 33165–33177. [Google Scholar] [CrossRef]
- Wang, Y.X.; Li, Y.Z.; Zhang, Z.Y.; Wang, J.Q.; Cui, J.; Qian, X.L. HPV16 E6 Promotes Breast Cancer Proliferation via Upregulation of COX-2 Expression. Biomed. Res. Int. 2017, 2017, 2948467. [Google Scholar] [CrossRef]
- Frega, A.; Lorenzon, L.; Bononi, M.; De Cesare, A.; Ciardi, A.; Lombardi, D.; Assorgi, C.; Gentile, M.; Moscarini, M.; Torrisi, M.R.; et al. Evaluation of E6 and E7 mRNA expression in HPV DNA positive breast cancer. Eur. J. Gynaecol. Oncol. 2012, 33, 164–167. [Google Scholar]
- Pereira Suarez, A.L.; Lorenzetti, M.A.; Gonzalez Lucano, R.; Cohen, M.; Gass, H.; Martinez Vazquez, P.; Gonzalez, P.; Preciado, M.V.; Chabay, P. Presence of human papilloma virus in a series of breast carcinoma from Argentina. PLoS ONE 2013, 8, e61613. [Google Scholar] [CrossRef] [PubMed]
- Gupta, I.; Jabeen, A.; Vranic, S.; Al Moustafa, A.E.; Al-Thawadi, H. Oncoproteins of High-Risk HPV and EBV Cooperate to Enhance Cell Motility and Invasion of Human Breast Cancer Cells. Front. Oncol. 2021, 11, 630408. [Google Scholar] [CrossRef]
- Deshpande, C.G.; Badve, S.; Kidwai, N.; Longnecker, R. Lack of expression of the Epstein-Barr Virus (EBV) gene products, EBERs, EBNA1, LMP1, and LMP2A, in breast cancer cells. Lab. Investig. 2002, 82, 1193–1199. [Google Scholar] [CrossRef]
- Arbach, H.; Viglasky, V.; Lefeu, F.; Guinebretière, J.M.; Ramirez, V.; Bride, N.; Boualaga, N.; Bauchet, T.; Peyrat, J.P.; Mathieu, M.C.; et al. Epstein-Barr virus (EBV) genome and expression in breast cancer tissue: Effect of EBV infection of breast cancer cells on resistance to paclitaxel (Taxol). J. Virol. 2006, 80, 845–853. [Google Scholar] [CrossRef]
- Hoebe, E.; Wille, C.; Hagemeier, S.; Kenney, S.; Greijer, A.; Middeldorp, J. Epstein-Barr Virus Gene BARF1 Expression is Regulated by the Epithelial Differentiation Factor ΔNp63α in Undifferentiated Nasopharyngeal Carcinoma. Cancers 2018, 10, 76. [Google Scholar] [CrossRef]
- Blanco, R.; Aguayo, F. Role of BamHI-A Rightward Frame 1 in Epstein-Barr Virus-Associated Epithelial Malignancies. Biology 2020, 9, 461. [Google Scholar] [CrossRef]
- James, C.D.; Morgan, I.M.; Bristol, M.L. The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens 2020, 9, 403. [Google Scholar] [CrossRef] [PubMed]
- Jayshree, R.S. The Immune Microenvironment in Human Papilloma Virus-Induced Cervical Lesions-Evidence for Estrogen as an Immunomodulator. Front. Cell Infect. Microbiol. 2021, 11, 649815. [Google Scholar] [CrossRef]
- Ramírez-López, I.G.; Ramírez de Arellano, A.; Jave-Suárez, L.F.; Hernández-Silva, C.D.; García-Chagollan, M.; Hernández-Bello, J.; Lopez-Pulido, E.I.; Macias-Barragan, J.; Montoya-Buelna, M.; Muñoz-Valle, J.F.; et al. Interaction between 17β-estradiol, prolactin and human papillomavirus induce E6/E7 transcript and modulate the expression and localization of hormonal receptors. Cancer Cell Int. 2019, 19, 227. [Google Scholar] [CrossRef] [PubMed]
- Belachew, E.B.; Sewasew, D.T. Molecular Mechanisms of Endocrine Resistance in Estrogen-Positive Breast Cancer. Front. Endocrinol. 2021, 12, 599586. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Giovannelli, P.; Auricchio, F. Cell proliferation regulated by estradiol receptor: Therapeutic implications. Steroids 2010, 75, 524–527. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Chung, S.H.; Wiedmeyer, K.; Shai, A.; Korach, K.S.; Lambert, P.F. Requirement for estrogen receptor alpha in a mouse model for human papillomavirus-associated cervical cancer. Cancer Res. 2008, 68, 9928–9934. [Google Scholar] [CrossRef] [PubMed]
- Mitrani-Rosenbaum, S.; Tsvieli, R.; Tur-Kaspa, R. Oestrogen stimulates differential transcription of human papillomavirus type 16 in SiHa cervical carcinoma cells. J. Gen. Virol. 1989, 70 Pt 8, 2227–2232. [Google Scholar] [CrossRef]
- Park, J.S.; Rhyu, J.W.; Kim, C.J.; Kim, H.S.; Lee, S.Y.; Kwon, Y.I.; Namkoong, S.E.; Sin, H.S.; Um, S.J. Neoplastic change of squamo-columnar junction in uterine cervix and vaginal epithelium by exogenous estrogen in hpv-18 URR E6/E7 transgenic mice. Gynecol. Oncol. 2003, 89, 360–368. [Google Scholar] [CrossRef]
- Ramachandran, B. Functional association of oestrogen receptors with HPV infection in cervical carcinogenesis. Endocr. Relat. Cancer 2017, 24, R99–R108. [Google Scholar] [CrossRef]
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERalpha: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Honda, H.; Kitamura, S. Effects of environmental estrogenic chemicals on AP1 mediated transcription with estrogen receptors alpha and beta. J. Steroid Biochem. Mol. Biol. 2004, 88, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Chan, J.Y.; Liu, P.Y.; Liu, S.T.; Huang, S.M. Human papillomavirus E2 protein associates with nuclear receptors to stimulate nuclear receptor- and E2-dependent transcriptional activations in human cervical carcinoma cells. Int. J. Biochem. Cell Biol. 2007, 39, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.M.; Chung, M.H.; Huang, S.M. Regulation of nuclear receptor activities by two human papillomavirus type 18 oncoproteins, E6 and E7. Biochem. Biophys. Res. Commun. 2003, 303, 932–939. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV Protein | Function | References |
|---|---|---|
| E1 | Initiates viral genome replication. | [51] |
| E2 | Induces viral DNA replication and transcription.Modulates viral gene expression. | [52] |
| E3 | Not known. | [53] |
| E4 | Facilitates the encapsidation and maturation of viral particles. | [53] |
| E5 | Disrupts the v-ATPase-dependent endosomal acidification process, reducing EGFR degradation. | [56,57] |
| Increases both EGFR and pEGFR expression on cell surface. | [54,55,58] | |
| Downregulates the MHC-I, disrupting the host immune response. | [59] | |
| E6 | Protects cells from apoptosis inducing p53, Bak, FADD, and caspase-8 degradation. | [60,61,62] |
| Activates MAPK and mTORC1 signaling. | [63,64] | |
| Disrupts the activation of Th1 CD4+ T cells by means of pro-IL-1ꞵ degradation. | [65] | |
| Downregulates MHC-I expression, targeting STAT1. | [66] | |
| E7 | Induces cell cycle progression by means of Rb degradation. | [67,68] |
| Decreases INFs production targeting STING. | [69] | |
| Activates AKT activity and EGFR expression. | [70] | |
| E6 and E7 | Interact with c-myc, inducing hTERT activation and cell immortalization. | [72,73] |
| Induce EMT, upregulating N-cadherin, Fibronectin, and Vimentin. | [74] | |
| Inhibit INFs and IL-1β production, contributing to immune evasion. | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, R.; Carrillo-Beltrán, D.; Muñoz, J.P.; Corvalán, A.H.; Calaf, G.M.; Aguayo, F. Human Papillomavirus in Breast Carcinogenesis: A Passenger, a Cofactor, or a Causal Agent? Biology 2021, 10, 804. https://doi.org/10.3390/biology10080804
Blanco R, Carrillo-Beltrán D, Muñoz JP, Corvalán AH, Calaf GM, Aguayo F. Human Papillomavirus in Breast Carcinogenesis: A Passenger, a Cofactor, or a Causal Agent? Biology. 2021; 10(8):804. https://doi.org/10.3390/biology10080804
Chicago/Turabian StyleBlanco, Rancés, Diego Carrillo-Beltrán, Juan P. Muñoz, Alejandro H. Corvalán, Gloria M. Calaf, and Francisco Aguayo. 2021. "Human Papillomavirus in Breast Carcinogenesis: A Passenger, a Cofactor, or a Causal Agent?" Biology 10, no. 8: 804. https://doi.org/10.3390/biology10080804
APA StyleBlanco, R., Carrillo-Beltrán, D., Muñoz, J. P., Corvalán, A. H., Calaf, G. M., & Aguayo, F. (2021). Human Papillomavirus in Breast Carcinogenesis: A Passenger, a Cofactor, or a Causal Agent? Biology, 10(8), 804. https://doi.org/10.3390/biology10080804