
Phytochemicals from Leucas zeylanica Targeting Main Protease of SARS-CoV-2: Chemical Profiles, Molecular Docking, and Molecular Dynamics Simulations

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection and Identification of Plant Material
2.2. Preparation of Plant Extract and Decoction Preparation
2.3. Gas Chromatography–Mass Spectrometry (GC-MS) Analysis
2.4. Molecular Docking Study
2.4.1. Ligand Preparation
2.4.2. Protein Preparation
2.4.3. Receptor Grid Generation and Glide Molecular Docking
2.5. ADME/T Properties Analysis
2.6. Molecular Dynamics Simulation
3. Results
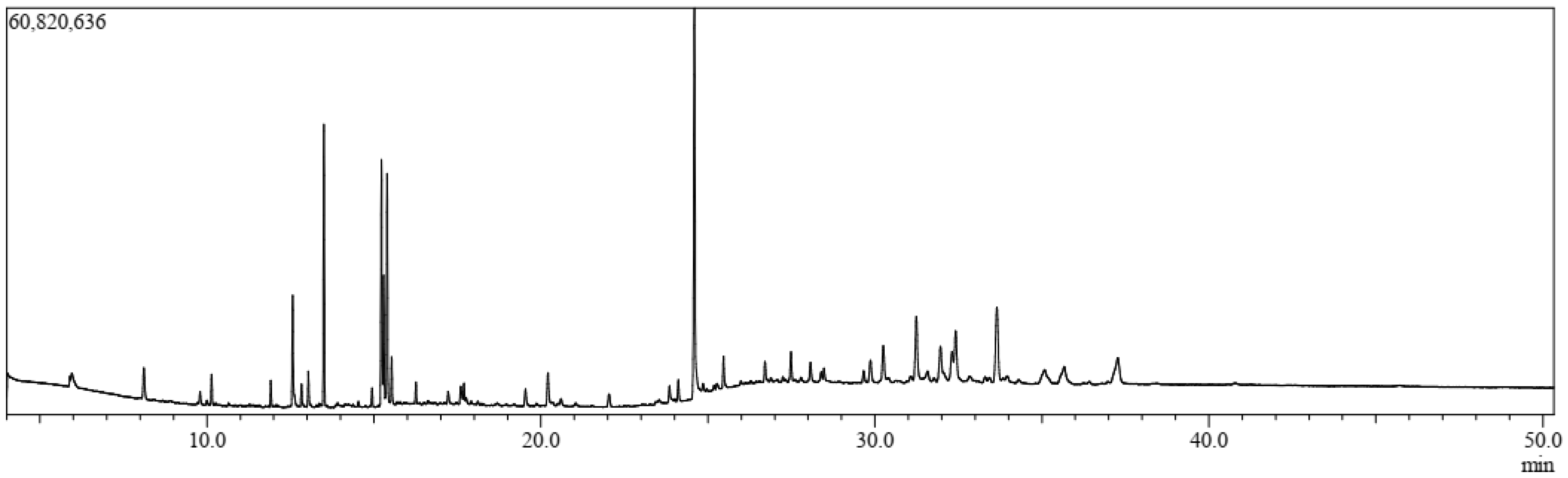
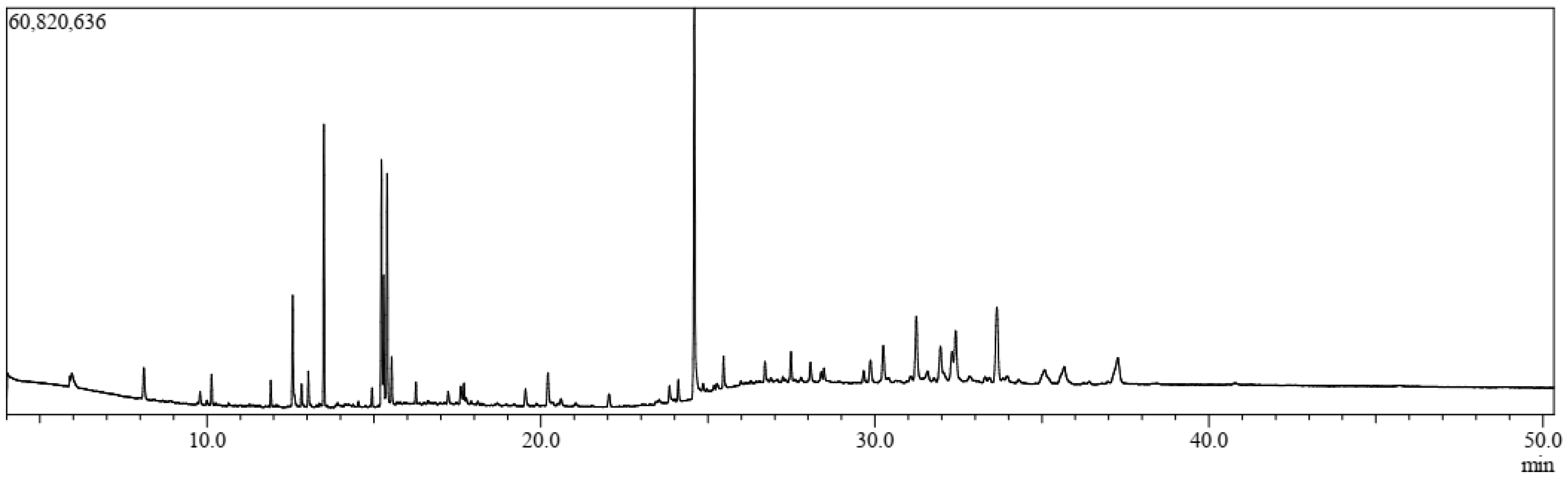
3.1. GC-MS Analysis
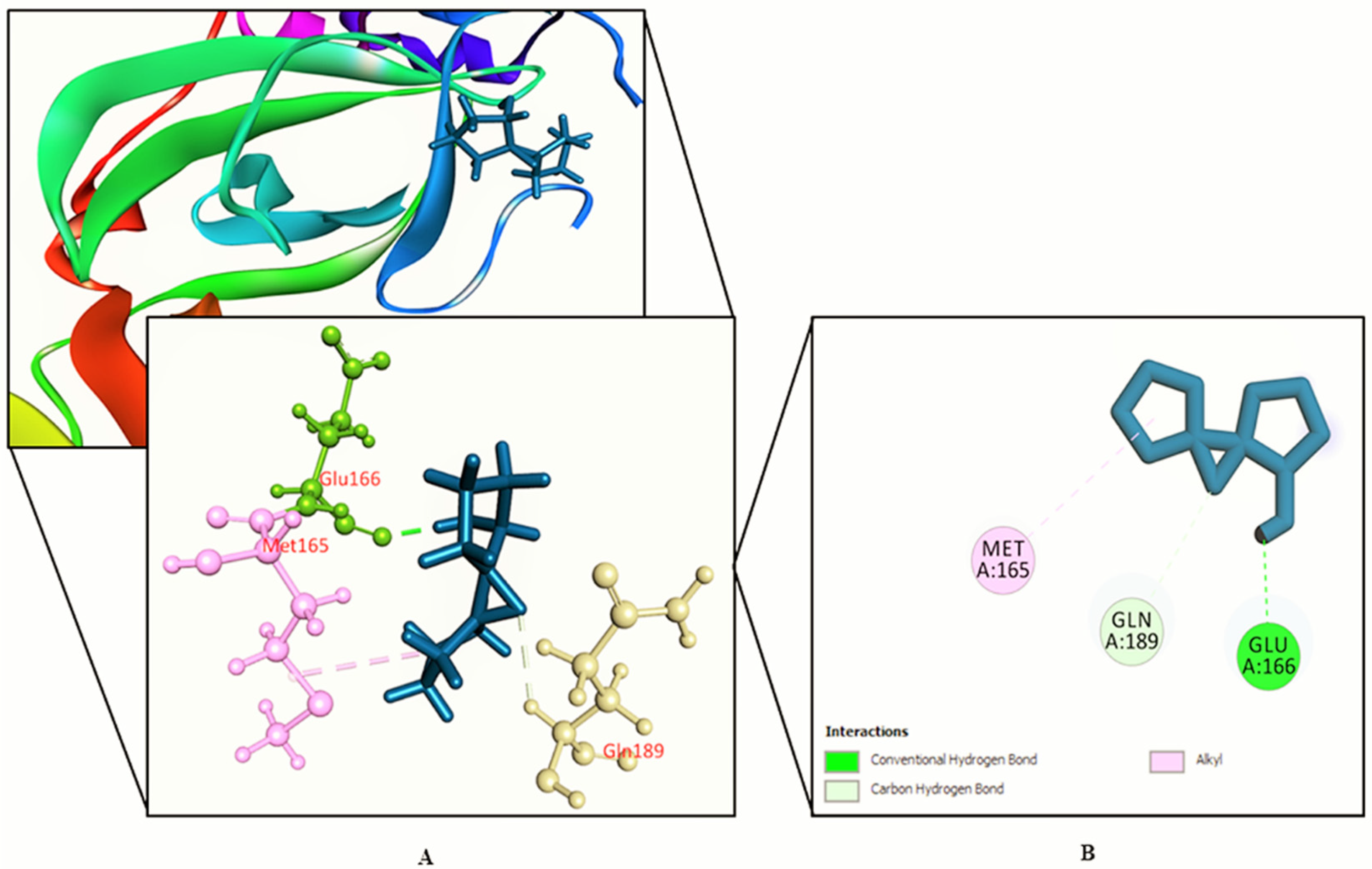
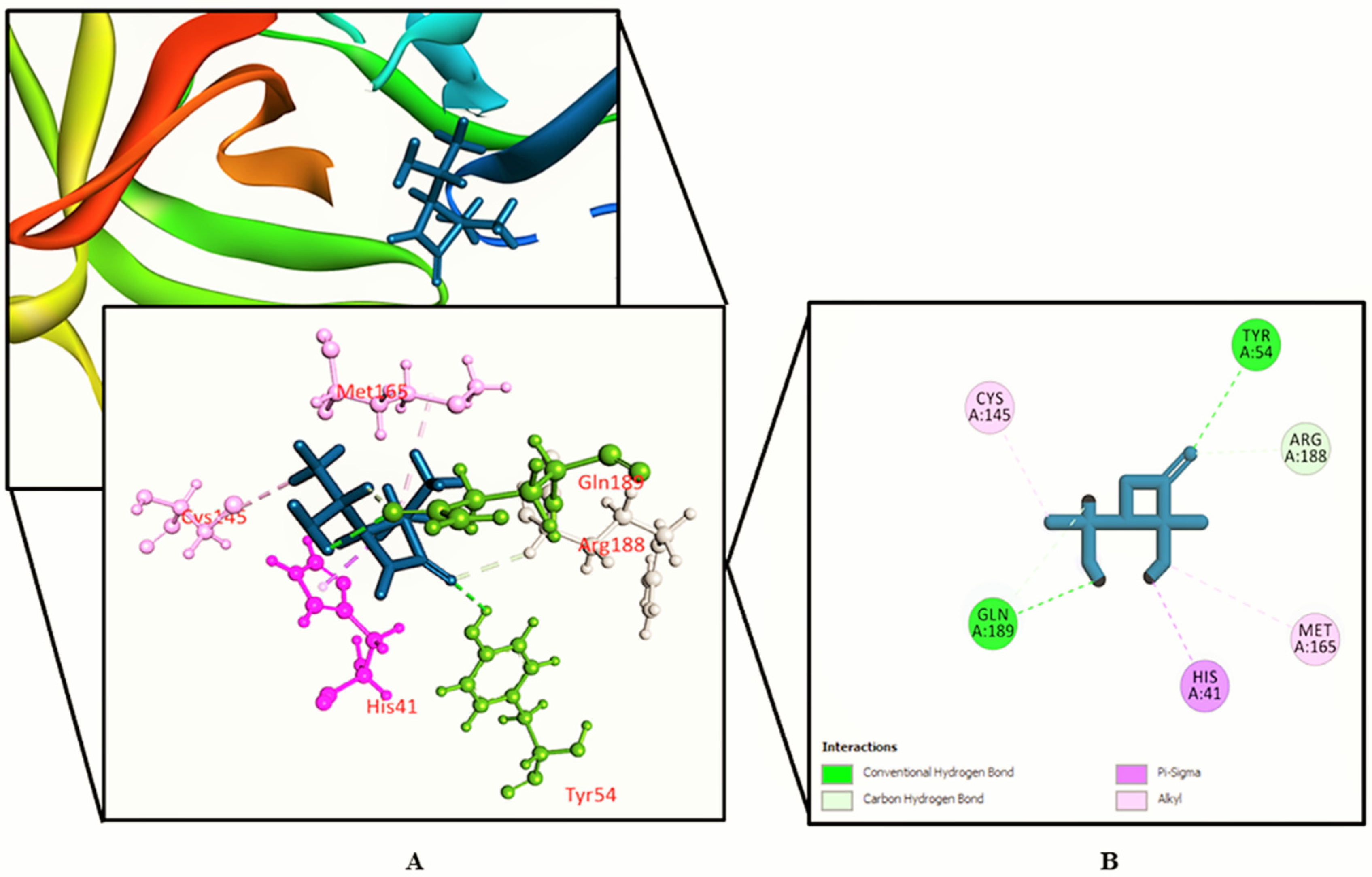
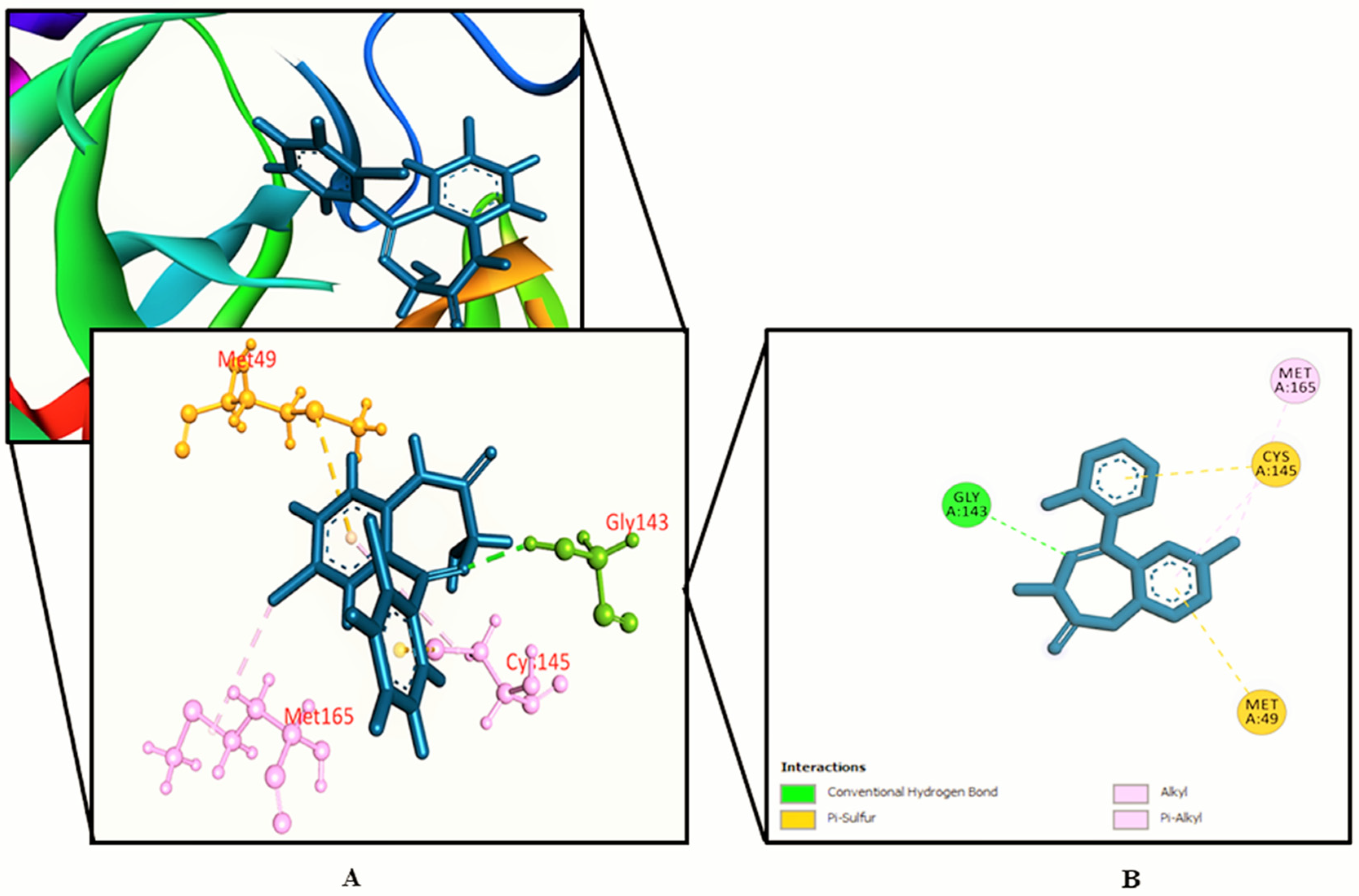
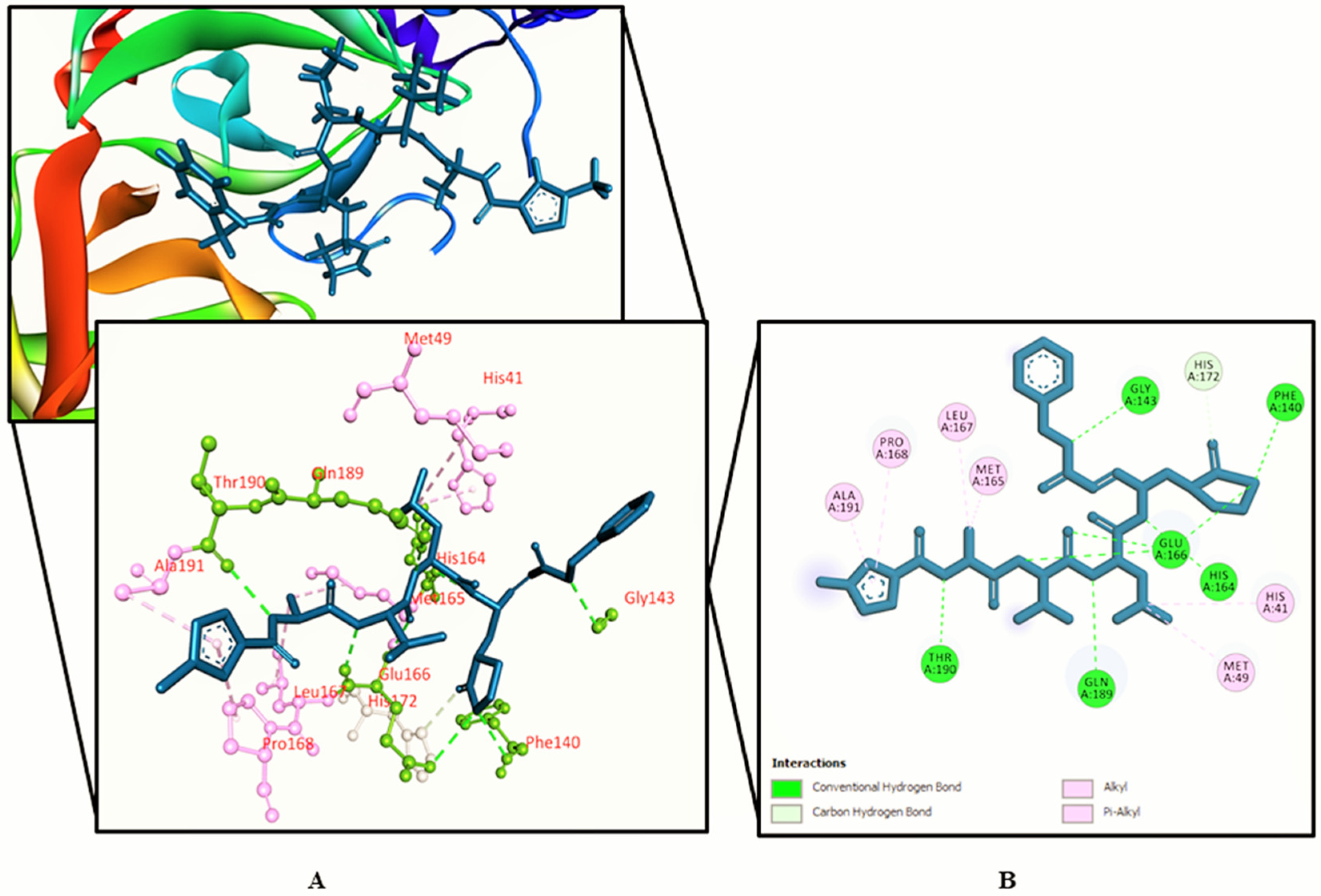
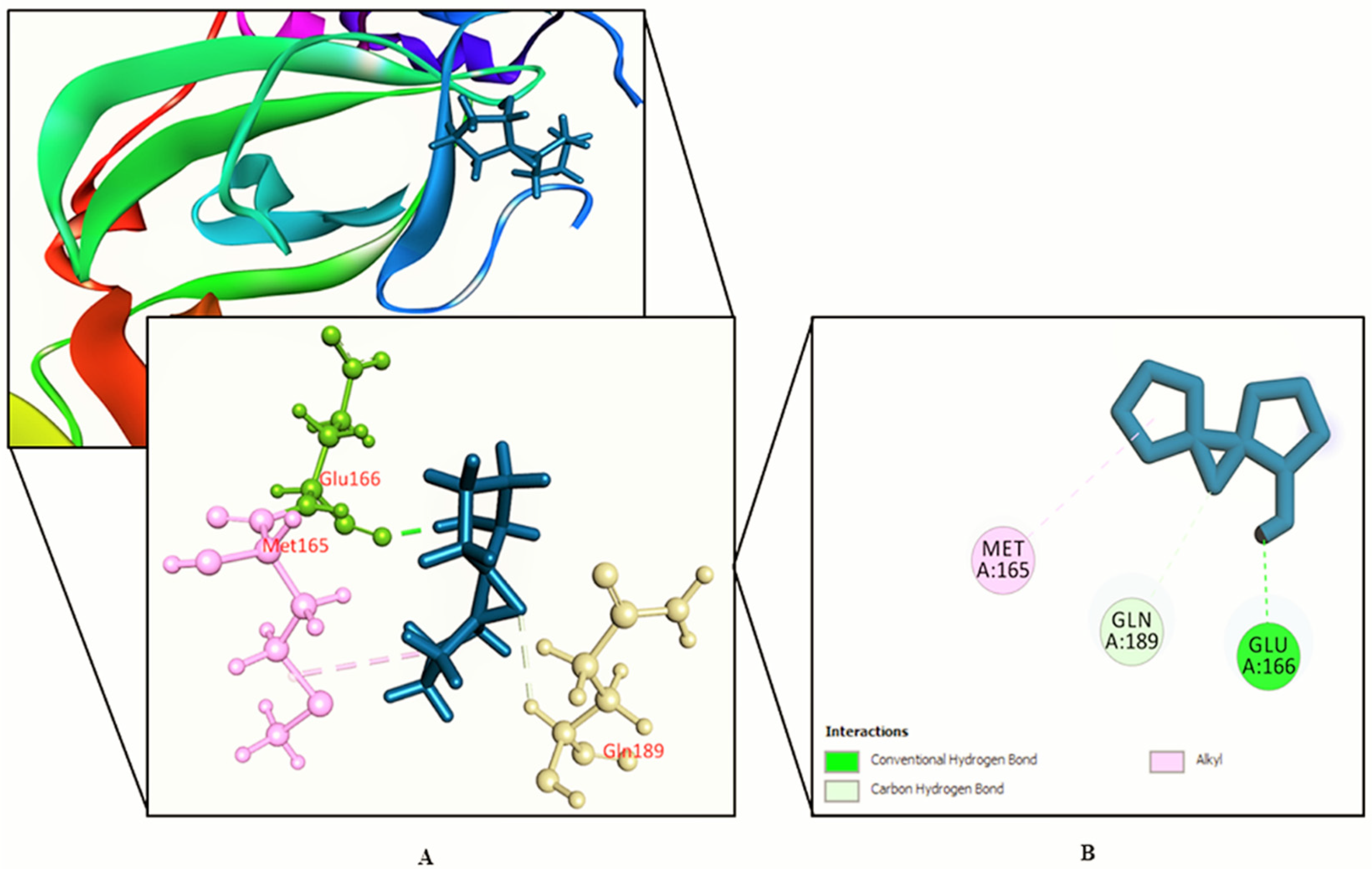
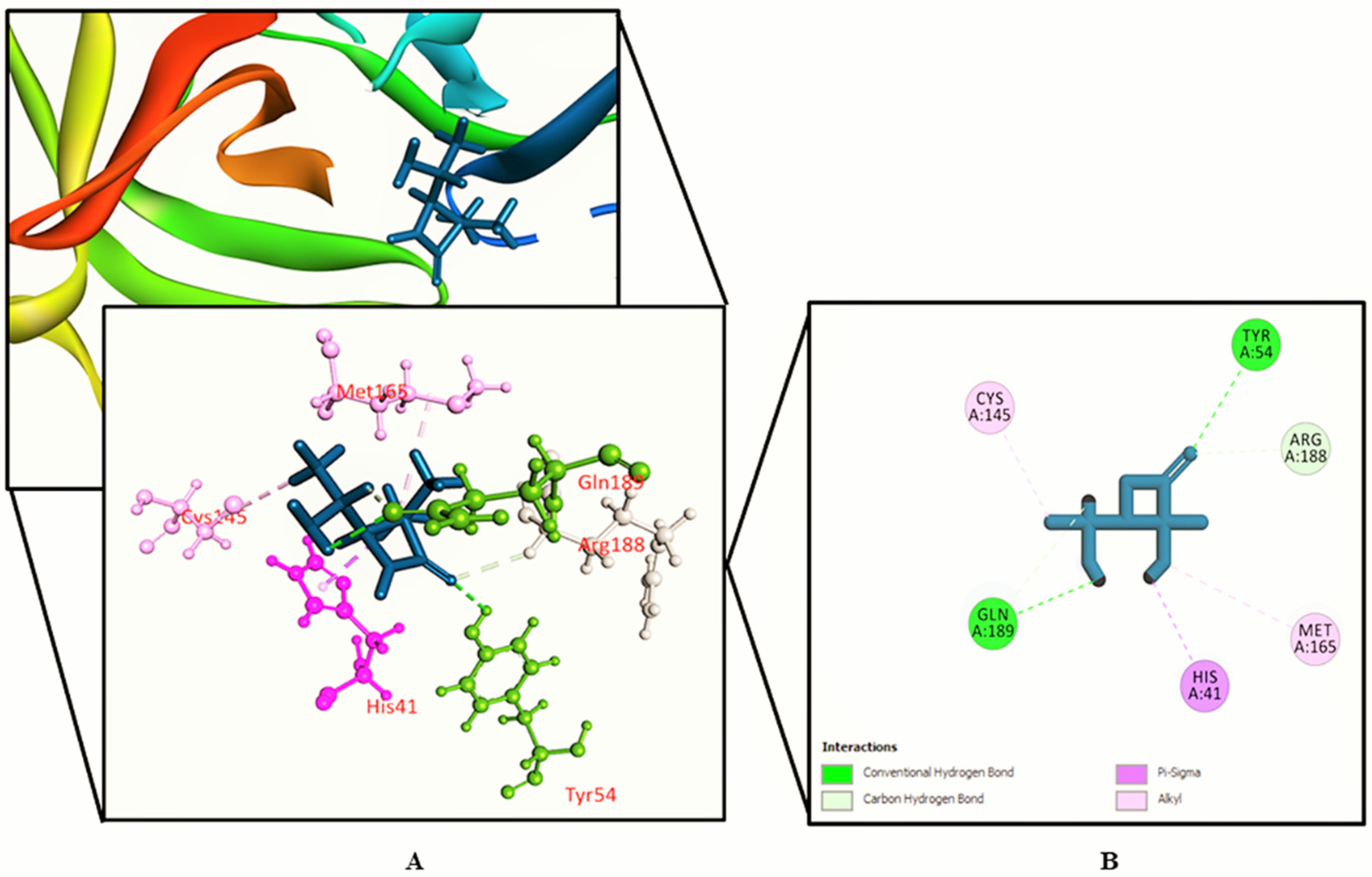
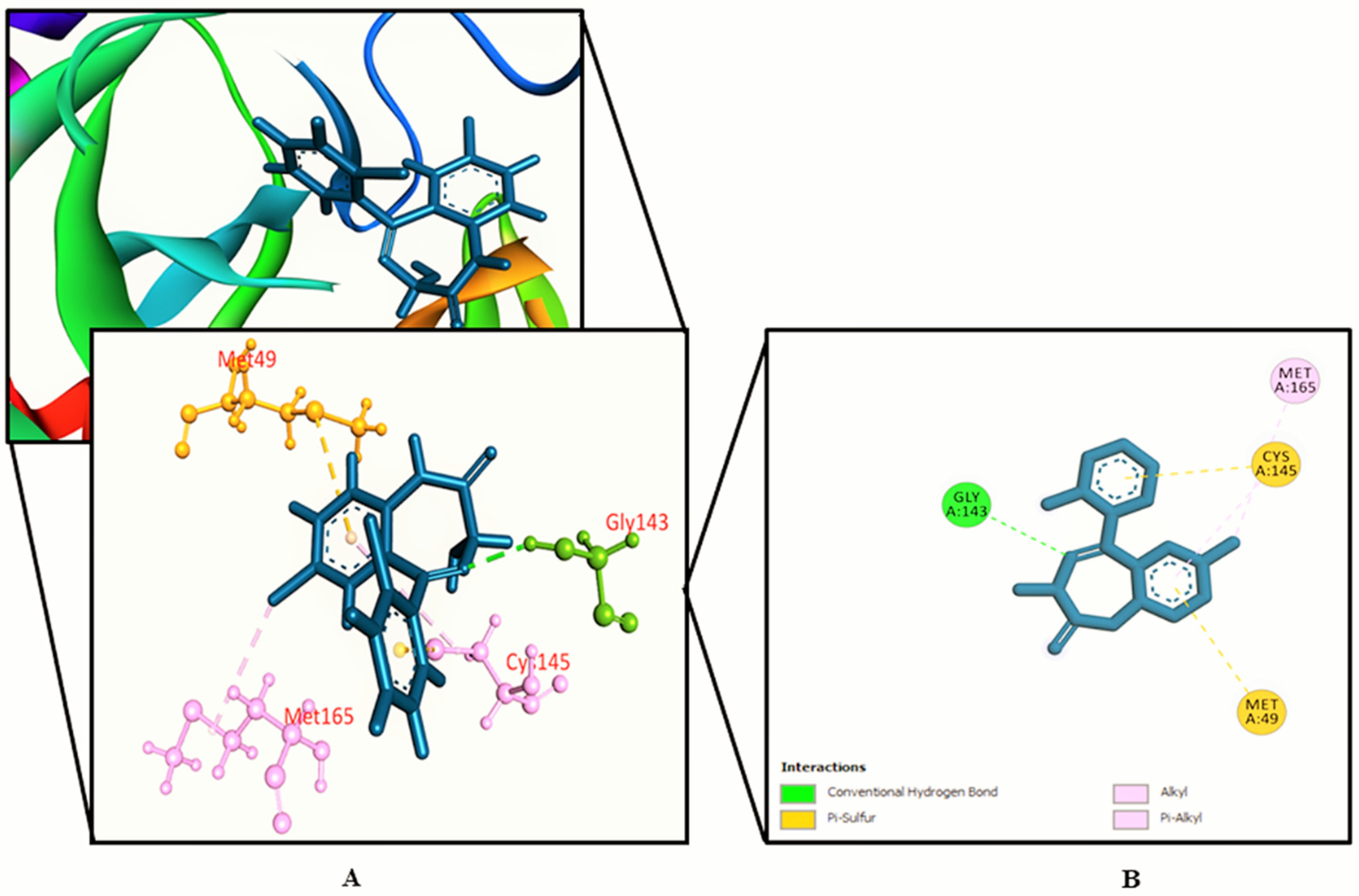
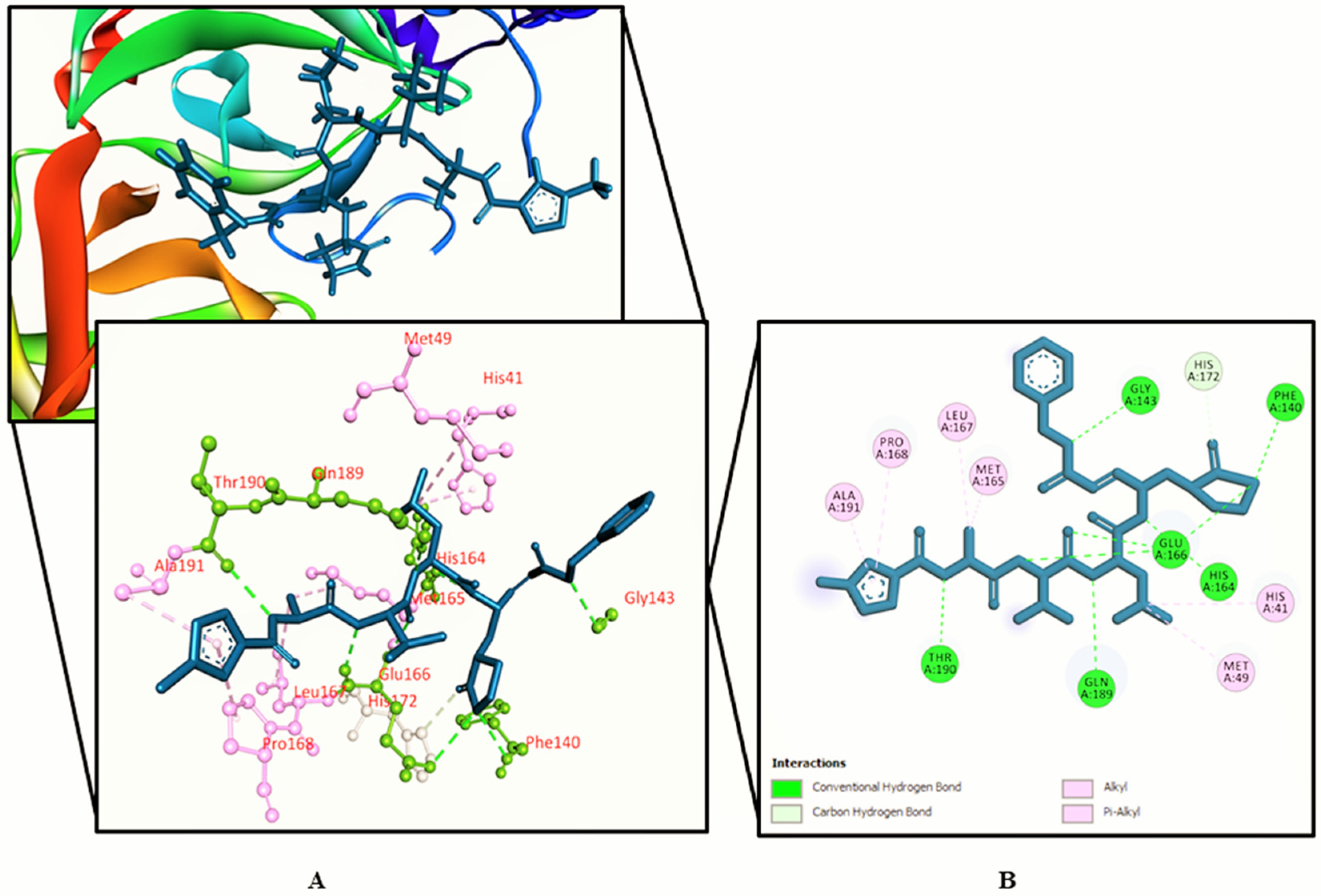
3.2. Molecular Docking Study
3.3. ADME/T Properties Analysis
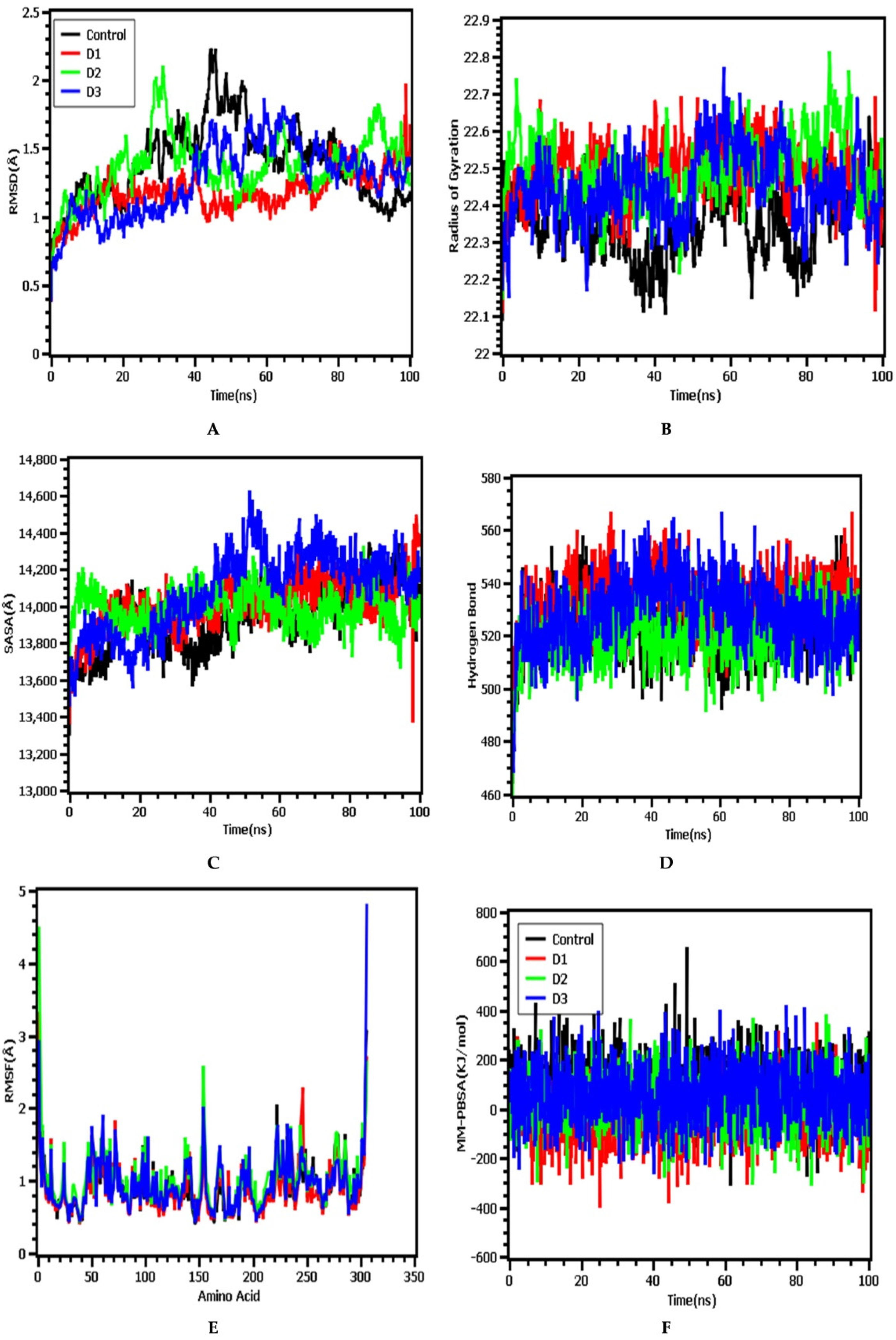
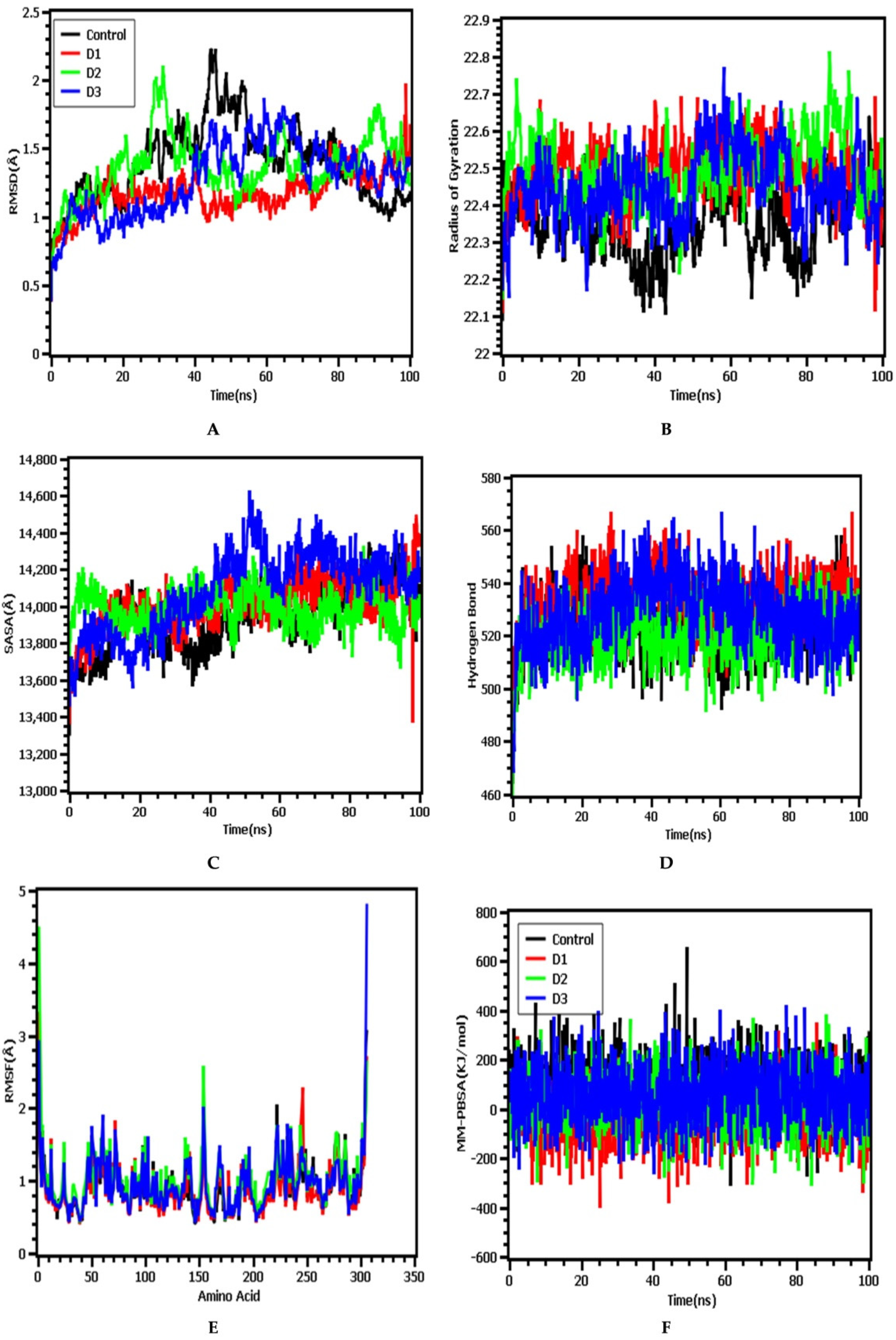
3.4. Molecular Dynamics Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sami, S.A.; Marma, K.K.S.; Chakraborty, A.; Singha, T.; Rakib, A.; Uddin, G.; Hossain, M.K.; Uddin, S.M.N. A comprehensive review on global contributions and recognition of pharmacy professionals amidst COVID-19 pandemic: Moving from present to future. Futur. J. Pharm. Sci. 2021, 7, 1–16. [Google Scholar] [CrossRef]
- Rakib, A.; Sami, S.; Islam, A.; Ahmed, S.; Faiz, F.; Khanam, B.; Marma, K.; Rahman, M.; Uddin, M.; Nainu, F.; et al. Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2. Molecules 2020, 25, 5088. [Google Scholar] [CrossRef]
- Lu, H.; Stratton, C.W.; Tang, Y. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, Z.; Wang, G.; Lau, J.Y.-N.; Zhang, K.; Li, W. COVID-19 in early 2021: Current status and looking forward. Signal Transduct. Target. Ther. 2021, 6, 1–14. [Google Scholar] [CrossRef]
- Rakib, A.; Sami, S.A.; Mimi, N.J.; Chowdhury, M.; Eva, T.A.; Nainu, F.; Paul, A.; Shahriar, A.; Tareq, A.M.; Emon, N.U.; et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput. Biol. Med. 2020, 124, 103967. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Wu, Y.-S.; Lin, W.-H.; Hsu, J.T.-A.; Hsieh, H.-P. Antiviral Drug Discovery Against SARS-CoV. Curr. Med. Chem. 2006, 13, 2003–2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Rabaan, A.A.; Al-Ahmed, S.H.; Garout, M.A.; Al-Qaaneh, A.M.; Sule, A.A.; Tirupathi, R.; Mutair, A.A.; Alhumaid, S.; Al-Omari, A.; Hasan, A.; et al. Diverse Immunological Factors Influencing Pathogenesis in Patients with COVID-19: A Review on Viral Dissemination, Immunotherapeutic Options to Counter Cytokine Storm and Inflammatory Responses. Pathogens 2021, 10, 565. [Google Scholar] [CrossRef]
- Anand, K. Ziebuhr J Fau-Wadhwani P.; Wadhwani P Fau-Mesters JR; Mesters Jr Fau-Hilgenfeld R.; Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Ni, L.; Ye, F.; Chen, M.-L.; Feng, Y.; Deng, Y.-Q.; Zhao, H.; Wei, P.; Ge, J.; Li, X. Characterization of anti-viral immunity in recovered individuals infected by SARS-CoV-2. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4 -derived ROS. Cell. Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahos, R.; Stambas, J.; Bozinovski, S.; Broughton, B.R.S.; Drummond, G.; Selemidis, S. Inhibition of Nox2 Oxidase Activity Ameliorates Influenza A Virus-Induced Lung Inflammation. PLoS Pathog. 2011, 7, e1001271. [Google Scholar] [CrossRef] [Green Version]
- Sebastiano, M.; Chastel, O.; de Thoisy, B.; Eens, M.; Costantini, D. Oxidative stress favours herpes virus infection in vertebrates: A meta-analysis. Curr. Zoöl. 2016, 62, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med Hypotheses 2020, 143, 110102. [Google Scholar] [CrossRef]
- Samir, D. Oxidative Stress Associated with SARS-Cov-2 (COVID-19) Increases the Severity of the Lung Disease–A Systematic Review. J. Infect. Dis. Epidemiology 2020, 6, 121. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Sindhu, J.; Thakur, S.; Rana, A.; Sharma, G.; Mayank; Poduri, R. Recent efforts for drug identification from phytochemicals against SARS-CoV-2: Exploration of the chemical space to identify druggable leads. Food Chem. Toxicol. 2021, 152, 112160. [Google Scholar] [CrossRef]
- Chouhan, H.S.; Singh, S.K. A review of plants of genus Leucas. J. Pharmacogn. Phytother. 2011, 3, 13–26. [Google Scholar] [CrossRef]
- Hossain, S.; Rahman, M.; Fatima, N.; Haque, M.; Islam, J. Leucas zeylanica (L.) R. Br. protects ethanol and hydrogen peroxide-induced oxidative stress on hepatic tissue of rats. Int. Curr. Pharm. J. 2013, 2, 148–151. [Google Scholar] [CrossRef] [Green Version]
- Hung, N.H.; Chuong, N.T.H.; Satyal, P.; Hieu, H.V.; Dai, D.N.; Huong, L.T.; Sinh, L.H.; Ngoc, N.T.B.; Hien, V.T.; Setzer, W.N. Mosquito Larvicidal Activities and Chemical Compositions of the Essential Oils of Leucas zeylanica Growing Wild in Vietnam. Nat. Prod. Commun. 2019, 14, 1–7. [Google Scholar] [CrossRef]
- Mahmud, S.; Biswas, S.; Paul, G.K.; Mita, M.A.; Promi, M.M.; Afrose, S.; Hasan, R.; Zaman, S.; Uddin, M.S.; Dhama, K.; et al. Plant-based phytochemical screening by targeting main protease of SARS-CoV-2 to design effective potent inhibitors. Biology 2021, 10, 589. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; bin Imran, T.; Islam, S. Antioxidative, antimicrobial and cytotoxic effects of the phenolics of Leea indica leaf extract. Saudi J. Biol. Sci. 2013, 20, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-Y.; Zhang, B.; Zhao, T.; Nidhal, N.; Jia-Li, W.; Zhou, X.-M.; Chun-Yan, D. A new triterpenoid glucoside from Leucas zeylanica. Nat. Prod. Res. 2020, 34, 1874–1878. [Google Scholar] [CrossRef]
- Rakib, A.; Paul, A.; Chy, N.U.; Sami, S.A.; Baral, S.K.; Majumder, M.; Tareq, A.M.; Amin, M.N.; Shahriar, A.; Uddin, Z.; et al. Biochemical and Computational Approach of Selected Phytocompounds from Tinospora crispa in the Management of COVID-19. Molecules 2020, 25, 3936. [Google Scholar] [CrossRef]
- Abdullah, F.; Nasir, S.N.A.M.; Han, D.K.; Appalasamy, S.; Nor, M.M.; Rak, A.E. Potential of Leucas zeylanica extract to eliminate E. coli and S. aureus in Corbicula fluminea (“Etak”) tissue. Malays. J. Fundam. Appl. Sci. 2019, 15, 597–599. [Google Scholar] [CrossRef]
- Uddin, Z.; Paul, A.; Rakib, A.; Sami, S.; Mahmud, S.; Rana, S.; Hossain, S.; Tareq, A.; Dutta, M.; Emran, T.; et al. Chemical Profiles and Pharmacological Properties with In Silico Studies on Elatostema papillosum Wedd. Molecules 2021, 26, 809. [Google Scholar] [CrossRef]
- Mahmud, S.; Uddin, M.A.R.; Paul, G.K.; Shimu, M.S.S.; Islam, S.; Rahman, E.; Islam, A.; Islam, M.S.; Promi, M.M.; Emran, T.B.; et al. Virtual screening and molecular dynamics simulation study of plant derived compounds to identify potential inhibitor of main protease from SARS-CoV-2. Brief. Bioinform. 2021, 22, 1402–1414. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Studio, D. Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Accelrys Softw. Inc.: San Diego, CA, USA, 2015. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Kabir, M.S.H.; Hossain, M.M.; Kabir, M.I.; Rahman, M.M.; Hasanat, A.; Emran, T.B.; Rahman, M.A. Phytochemical screening, antioxidant, thrombolytic, α-amylase inhibition and cytotoxic activities of ethanol extract of Steudnera colocasiifolia K. Koch leaves. J. Young Pharmacists 2016, 8, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins: Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G.; Spronk, C. YASARA–yet another scientific artificial reality application. YASARA Org. 2013, 993, 51–78. [Google Scholar]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Mahmud, S.; Parves, R.; Riza, Y.; Sujon, K.M.; Ray, S.; Alam Tithi, F.; Zaoti, Z.F.; Alam, S.; Absar, N. Exploring the potent inhibitors and binding modes of phospholipase A2 through in silico investigation. J. Biomol. Struct. Dyn. 2020, 38, 4221–4231. [Google Scholar] [CrossRef]
- Khan, A.; Mahmud, S.; Alam, A.S.M.R.U.; Rahman, E.; Ahmed, F.; Rahmatullah, M. Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: A molecular docking study. J. Biomol. Struct. Dyn. 2020, 1–7. [Google Scholar] [CrossRef]
- Islam, J.; Parves, R.; Mahmud, S.; Alam Tithi, F.; Reza, A. Assessment of structurally and functionally high-risk nsSNPs impacts on human bone morphogenetic protein receptor type IA (BMPR1A) by computational approach. Comput. Biol. Chem. 2019, 80, 31–45. [Google Scholar] [CrossRef]
- Uddin, M.Z.; Rana, M.S.; Hossain, S.; Dutta, E.; Ferdous, S.; Dutta, M.; Emran, T.B. In vivo neuroprotective, antinociceptive, anti-inflammatory potential in Swiss albino mice and in vitro antioxidant and clot lysis activities of fractionated Holigarna longifolia Roxb. bark extract. J. Complement. Integr. Med. 2019, 17, 1–10. [Google Scholar] [CrossRef]
- Razzaghi-Asl, N.; Mirzayi, S.; Mahnam, K.; Sepehri, S. Identification of COX-2 inhibitors via structure-based virtual screening and molecular dynamics simulation. J. Mol. Graph. Model. 2018, 83, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zheng, Q.; Wang, Z. Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics 2020, 36, 3295–3298. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680. [Google Scholar] [CrossRef]
- Dutta, M.; Nezam, M.; Chowdhury, S.; Rakib, A.; Paul, A.; Sami, S.A.; Uddin, M.Z.; Rana, M.S.; Hossain, S.; Effendi, Y.; et al. Appraisals of the Bangladeshi Medicinal Plant Calotropis gigantea Used by Folk Medicine Practitioners in the Management of COVID-19: A Biochemical and Computational Approach. Front. Mol. Biosci. 2021, 8, 625391. [Google Scholar] [CrossRef]
- Jahan, I.; Tona, M.R.; Sharmin, S.; Sayeed, M.A.; Tania, F.Z.; Paul, A.; Chy, N.U.; Rakib, A.; Bin Emran, T.; Simal-Gandara, J. GC-MS Phytochemical Profiling, Pharmacological Properties, and In Silico Studies of Chukrasia velutina Leaves: A Novel Source for Bioactive Agents. Molecules 2020, 25, 3536. [Google Scholar] [CrossRef]
- Bin Emran, T.; Mowla, T.-E.; Ahmed, S.; Zahan, S.; Rakib, A.; Hasan, M.S.; Amin, M.N.; Mow, T.R.; Uddin, M.M.N. Sedative, Anxiolytic, Antinociceptive, Anti-inflammatory and Antipyretic Effects of a Chloroform Extract from the Leaves of Urena sinuata in Rodents. J. Appl. Life Sci. Int. 2018, 16, 1–19. [Google Scholar] [CrossRef]
- Ahmed, S.; Rakib, A.; Islam, A.; Khanam, B.H.; Faiz, F.B.; Paul, A.; Chy, N.U.; Alam Bhuiya, N.M.M.; Uddin, M.M.N.; Ullah, S.M.A.; et al. In vivo and in vitro pharmacological activities of Tacca integrifolia rhizome and investigation of possible lead compounds against breast cancer through in silico approaches. Clin. Phytosci. 2019, 5, 1–13. [Google Scholar] [CrossRef]
- Obaidullah, A.J.; Alanazi, M.A.; Alsaif, N.A.; Albassam, H.; Almehizia, A.A.; Alqahtani, A.A.; Mahmud, S.; Sami, S.A.; Emran, T.B. Immunoinformatics-guided design of multi-epitope vaccine from structural proteins of severe acute respiratory syndrome-coronavirus-2. RSC Adv. 2021, 11, 18103–18121. [Google Scholar] [CrossRef]
- Rakib, A.; Ahmed, S.; Islam, A.; Uddin, M.M.N.; Paul, A.; Chy, N.U.; Bin Emran, T.; Seidel, V. Pharmacological studies on the antinociceptive, anxiolytic and antidepressant activity of Tinospora crispa. Phytother. Res. 2020, 34, 2978–2984. [Google Scholar] [CrossRef]
- Rakib, A.; Ahmed, S.; Islam, A.; Haye, A.; Uddin, S.M.N.; Uddin, M.M.N.; Hossain, M.K.; Paul, A.; Bin Emran, T. Antipyretic and hepatoprotective potential of Tinospora crispa and investigation of possible lead compounds through in silico approaches. Food Sci. Nutr. 2020, 8, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Islam, S.; Wang, J.; Li, Y.; Chen, X. Traditional Chinese Medicine in the Treatment of Patients Infected with 2019-New Coronavirus (SARS-CoV-2): A Review and Perspective. Int. J. Biol. Sci. 2020, 16, 1708–1717. [Google Scholar] [CrossRef]
- Rakib, A.; Nain, Z.; Islam, M.A.; Sami, S.A.; Mahmud, S.; Islam, A.; Ahmed, S.; Siddiqui, A.B.F.; Babu, S.M.O.F.; Hossain, P.; et al. A molecular modelling approach for identifying antiviral selenium-containing heterocyclic compounds that inhibit the main protease of SARS-CoV-2: An in silico investigation. Brief. Bioinform. 2021, 22, 1476–1498. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Younes, A. Coronavirus COV-19/SARS-CoV-2 affects women less than men: Clinical response to viral infection. J. Biol. Regul. Homeost. Agents 2020, 34, 339–343. [Google Scholar]
- Berg, R.V.D.; Haenen, G.; Bast, A. Transcription factor NF-κB as a potential biomarker for oxidative stress. Br. J. Nutr. 2001, 86, S121–S127. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Biswas, N.N. Phytochemical and Pharmacological Activities of Leucas zeylanica (L.) R. Br. (Family: Lamiaceae). Hamdard Medicus 2010, 53, 19–29. [Google Scholar]
- Chowdhury, K.H.; Chowdhury, M.R.; Mahmud, S.; Tareq, A.M.; Hanif, N.B.; Banu, N.; Reza, A.S.M.A.; Emran, T.B.; Simal-Gandara, J. Drug Repurposing Approach against Novel Coronavirus Disease (COVID-19) through Virtual Screening Targeting SARS-CoV-2 Main Protease. Biology 2020, 10, 2. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Muramatsu, T.; Takemoto, C.; Kim, Y.-T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, M.-F.; Kuo, C.-J.; Chang, K.-T.; Chang, H.-C.; Chou, C.-C.; Ko, T.-P.; Shr, H.-L.; Chang, G.-G.; Wang, A.H.-J.; Liang, P.-H. Mechanism of the Maturation Process of SARS-CoV 3CL Protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallei, T.E.; Tumilaar, S.G.; Niode, N.J.; Fatimawali; Kepel, B.J.; Idroes, R.; Effendi, Y.; Sakib, S.A.; Emran, T.B. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: A molecular docking study. Scientifica 2020, 2020, 6307457. [Google Scholar] [CrossRef] [PubMed]
- Petrera, E.; Níttolo, A.G.; Alché, L.E. Antiviral Action of Synthetic Stigmasterol Derivatives on Herpes Simplex Virus Replication in Nervous CellsIn Vitro. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Marinho, R.D.S.S.; Ramos, C.J.B.; Leite, J.P.G.; Teixeira, V.L.; Paixão, I.C.N.D.P.; Belo, C.A.D.; Pereira, A.B.; Pinto, A.M.V. Antiviral activity of 7-keto-stigmasterol obtained from green Antarctic algae Prasiola crispa against equine herpesvirus 1. Environ. Boil. Fishes 2017, 29, 555–562. [Google Scholar] [CrossRef]
- Tumilaar, S.G.; Siampa, J.P.; Fatimawali; Kepel, B.J.; Niode, N.J.; Idroes, R.; Rakib, A.; Emran, T.B.; Tallei, T.E. Potential of leaf extract of Pangium edule Reinw as HIV-1 protease inhibitor: A computational biology approach. J. Appl. Pharma. Sci. 2021, 11, 101–110. [Google Scholar] [CrossRef]
- Mahmud, S.; Paul, G.K.; Afroze, M.; Islam, S.; Gupt, S.B.R.; Razu, M.H.; Biswas, S.; Zaman, S.; Uddin, M.S.; Khan, M.; et al. Efficacy of Phytochemicals Derived from Avicennia officinalis for the Management of COVID-19: A Combined In Silico and Biochemical Study. Molecules 2021, 28, 2210. [Google Scholar] [CrossRef] [PubMed]
- Gershell, L.J.; Atkins, J.H. A brief history of novel drug discovery technologies. Nat. Rev. Drug Discov. 2003, 2, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.H.; Pichota, A.; Yin, Z. A practical view of ‘druggability’. Curr. Opin. Chem. Biol. 2006, 10, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; Pedretti, A.; Testa, B. Assessing drug-likeness–What are we missing? Drug Discov. Today 2008, 13, 285–294. [Google Scholar] [CrossRef]
- Khan, J.; Sakib, S.A.; Mahmud, S.; Khan, Z.; Islam, M.N.; Sakib, M.A.; Emran, T.B.; Simal-Gandara, J. Identification of potential phytochemicals from Citrus limon against main protease of SARS-CoV-2: Molecular docking, molecular dynamic simulations and quantum computations. J. Biomol. Struct. Dyn. 2021, 2021, 1–12. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. | Name | RT | m/z | Area | Conc. | Peak Area (%) |
|---|---|---|---|---|---|---|
| 1. | 3-Butynoic acid | 5.927 | 40.00 | 36,135 | 0.049 | 0.049299 |
| 2. | Tetradecamethylcycloheptasiloxane | 8.221 | 40.00 | 23,061 | 0.031 | 0.031462 |
| 3. | Trimethylsilyl 2,6-bis[(trimethylsilyl)oxy]benzoate | 10.139 | 73.00 | 1,516,895 | 2.066 | 2.069501 |
| 4. | Bis(heptamethylcyclotetrasiloxy)hexamethyltrisiloxane | 11.914 | 73.00 | 1,205,683 | 1.642 | 1.644915 |
| 5. | Lorazepam, 2TMS derivative | 11.914 | 73.00 | 1,205,683 | 1.642 | 1.644915 |
| 6. | Cyanoacetic acid | 10.669 | 40.00 | 49,231 | 0.067 | 0.067166 |
| 7. | Methyl 11-bromoundecanoate | 11.078 | 40.00 | 45,430 | 0.062 | 0.06198 |
| 8. | Azetidin-2-one 3,3-dimethyl-4-(1-aminoethyl) | 11.078 | 40.00 | 45,430 | 0.062 | 0.06198 |
| 9. | 3-Azabicyclo[3.2.2]nonane | 11.916 | 40.00 | 67,327 | 0.092 | 0.091854 |
| 10. | Phytol acetate | 12.567 | 68.00 | 2,737,101 | 3.728 | 3.73423 |
| 11. | Hexadecanoic acid, methyl ester | 13.502 | 74.00 | 14,101,161 | 19.207 | 19.23823 |
| 12. | 11-Oxa-dispiro[4.0.4.1]undecan-1-ol | 13.503 | 40.00 | 91,678 | 0.125 | 0.125076 |
| 13. | Hexadecamethylcyclooctasiloxane | 14.946 | 73.00 | 789,772 | 1.076 | 1.077487 |
| 14. | 9,12-Octadecadienoic acid (Z,Z)-, methyl ester | 15.225 | 67.00 | 6,028,138 | 8.211 | 8.224195 |
| 15. | 6-Octadecenoic acid, methyl ester, (Z)- | 15.225 | 55.00 | 2,986,886 | 4.068 | 4.075012 |
| 16. | Phytol | 15.393 | 71.00 | 11,551,440 | 15.734 | 15.75964 |
| 17. | Methyl stearate | 15.530 | 74.00 | 2,412,152 | 3.286 | 3.290901 |
| 18. | Octadecamethylcyclononasiloxane | 16.257 | 73.00 | 913,521 | 1.244 | 1.246318 |
| 19. | Pseduosarsasapogenin-5,20-dien | 17.219 | 83.00 | 547,313 | 0.745 | 0.7467 |
| 20. | Cyclodecasiloxane, eicosamethyl- | 19.540 | 73.00 | 929,809 | 1.266 | 1.268539 |
| 21. | Hexadecanoic acid, 2-hydroxy-1-(hydroxymethyl)ethyl ester | 20.211 | 98.00 | 971,087 | 1.323 | 1.324855 |
| 22. | Octadecanoic acid, 2,3-dihydroxypropyl ester | 23.851 | 43.00 | 356,272 | 0.485 | 0.486062 |
| 23. | 13-Docosenamide, (Z)- | 24.596 | 59.00 | 15,302,725 | 20.844 | 20.87752 |
| 24. | N,N′-methylenebis(oleamide), (Z,Z)- | 24.595 | 207.00 | 247,529 | 0.337 | 0.337704 |
| 25. | Squalene | 27.770 | 207.00 | 138,619 | 0.189 | 0.189118 |
| 26. | α-Tocopheryl acetate | 28.073 | 207.00 | 203,341 | 0.277 | 0.277418 |
| 27. | Campesterol | 29.656 | 207.00 | 201,572 | 0.275 | 0.275005 |
| 28. | Stigmasterol | 30.546 | 207.00 | 156,066 | 0.213 | 0.212921 |
| 29. | γ-Sitosterol | 31.581 | 207.00 | 165,310 | 0.225 | 0.225533 |
| 30. | 4-Campestene-3-one | 31.581 | 207.00 | 147,960 | 0.202 | 0.201862 |
| 31. | 9, 19-Cyclolanost-24-en-3-ol, acetate, (3.beta.) | 32.728 | 207.00 | 101,495 | 0.138 | 0.13847 |
| 32. | 4,22-Cholestadien-3-one | 32.728 | 207.00 | 101,495 | 0.138 | 0.13847 |
| 33. | Stigmast-4-en-3-one | 33.655 | 124.00 | 4,766,323 | 6.492 | 6.502699 |
| 34. | 1,2-Bis(trimethylsilyl)benzene | 35.243 | 207.00 | 163,549 | 0.223 | 0.22313 |
| SL. No. | Name | Docking Score | Interactions by H-Bond | Hydrophobic Bonds (Pi–Alkyl/Alkyl Interaction) | Hydrophobic Bonds (Pi–Pi/Pi–Sigma/ Amide–Pi Interaction) | Pi–Sulfur Interaction |
|---|---|---|---|---|---|---|
| 1. | 3-Butynoic acid | −1.08 | Cys 145, Ser 144 (2), Gly 143 | His 172, His 163 | Phe 140 | − |
| 2. | Tetradecamethylcycloheptasiloxane | − | − | − | − | − |
| 3. | Trimethylsilyl 2,6-bis[(trimethylsilyl)oxy]benzoate | − | − | − | − | − |
| 4. | Bis(heptamethylcyclotetrasiloxy)hexamethyltrisiloxane | − | − | − | − | − |
| 5. | Lorazepam, 2TMS derivative | −5.246 | Gly 143 | Cys 145 | − | Cys 145, Met 49 |
| 6. | Cyanoacetic acid | −3.469 | Lys 61 (2), Arg 60, Asp 48 | − | − | − |
| 7. | Methyl 11-bromoundecanoate | −1.792 | Gly 143 | His 163, Cys 145 | − | − |
| 8. | Azetidin-2-one 3,3-dimethyl-4-(1-aminoethyl) | −5.39 | Gln 189, Tyr 54 | Met 165 (2) | His 41 | − |
| 9. | 3-Azabicyclo[3.2.2]nonane | −4.703 | − | His 41 (2), Met 49, Met 165 | − | − |
| 10. | Phytol acetate | −3.357 | Met 165, Glu 166 | Met 165, Cys 145 (2), Leu 27, His 41, Met 49, Met 165 | − | − |
| 11. | Phytol | −1.565 | Asn 142, | His 163, His 172, Met 165 (2), His 41, Met 49 | − | − |
| 12. | Hexadecanoic acid, methyl ester | +0.991 | Asn 142 | Met 165, | His 41 | − |
| 13. | 11-Oxa-dispiro[4.0.4.1]undecan-1-ol | −5.755 | Glu 166 | Met 165 | − | − |
| 14. | Hexadecamethylcyclooctasiloxane | − | − | − | − | − |
| 15. | 9,12-Octadecadienoic acid (Z,Z)-, methyl ester | −1.111 | Glu 166 | Met 165, Leu 167 | − | − |
| 16. | 6-Octadecenoic acid, methyl ester, (Z)- | −0.399 | Ser 144, Gly 143 | Cys 145, His 163 | His 41 | − |
| 17. | Methyl stearate | −0.419 | Gln 189 | His 41, Leu 50, Met 49 | − | − |
| 18. | Octadecamethylcyclononasiloxane | − | − | − | − | − |
| 19. | Pseduosarsasapogenin-5,20-dien | − | − | − | − | − |
| 20. | Cyclodecasiloxane, eicosamethyl- | − | − | − | − | − |
| 21. | Hexadecanoic acid, 2-hydroxy-1-(hydroxymethyl)ethyl ester | −4.152 | Glu 166, Cys 145 | − | − | − |
| 22. | Octadecanoic acid, 2,3-dihydroxypropyl ester | −2.406 | His 163, Gln 143 (2), Ser 144, Cys 145 | − | − | − |
| 23. | 13-Docosenamide, (Z)- | −4.46 | His 163, Cys 145 | His 41, Met 49 | − | − |
| 24. | N,N’′methylenebis(oleamide), (Z, Z)- | −4.057 | Phe 140 | His 41 | − | − |
| 25. | Squalene | −3.609 | − | Met 165 (2), Met 49, Cys 149, His 41 | − | − |
| 26. | α-Tocopheryl acetate | −4.871 | − | Leu 167, Met 165 (2), Met 49, His 41, Leu 27, Cys 145 | − | − |
| 27. | Campesterol | −3.776 | Thr 24 | Met 49 (2), His 41 (2), Met 165 (2), Cys 145 | − | − |
| 28. | Stigmasterol | −4.194 | Cys 145, Ser 144 (2) | Ala 191, Pro 168 | − | − |
| 29. | γ-Sitosterol | −4.854 | Thr 26 | Cys 145 (3), His 41 (2), Met 49, Met 165 (2), Leu 167, Pro 168 | − | − |
| 30. | 4-Campestene-3-one | −3.934 | − | Met 165, Met 49 (2), His 41 (2), Cys 145 | − | − |
| 31. | 9, 19-Cyclolanost-24-en-3-ol, acetate, (3.beta.) | −3.105 | Asn 142, Ser 144 | Cys 145, Pro 168, Ala 191, Leu 50 | − | − |
| 32. | 4,22-Cholestadien-3-one | −3.824 | Ser 144 | Ala 191, | − | − |
| 33. | Stigmast-4-en-3-one | −3.696 | His 41 (2), Cys 145, Leu 27 | − | − | |
| 34. | 1,2-Bis(trimethylsilyl)benzene | − | − | − | − | − |
| 35. | Standard (inhibitor N3) | −7.013 | Phe 140, Gly 143, His 164, Glu 166, Gln 189, Thr 190 | His 41, Met 49, Met 165, Leu 167, Pro 168, Ala 191 | − | − |
| Compounds | Molecular Weight a (g/mol) | Hydrogen Bond Acceptors b | Hydrogen Bond Donors c | MlogP d | Molar Refractivity e | No. of Lipinski Violations f |
|---|---|---|---|---|---|---|
| <500 | ≤10 | ≤5 | <5 | 40–130 | ≤1 | |
| 3-Butynoic acid | 84.07 | 2 | 1 | 0.38 | 21.28 | 1 |
| Tetradecamethylcycloheptasiloxane | 519.08 | 7 | 0 | −1.54 | 129.97 | 1 |
| Trimethylsilyl 2,6-bis[(trimethylsilyl)oxy]benzoate | 370.66 | 4 | 0 | 2.97 | 103.15 | 0 |
| Lorazepam, 2TMS derivative | 465.52 | 4 | 0 | 4.66 | 134.90 | 1 |
| Hexadecanoic acid, 2-hydroxy-1-(hydroxymethyl)ethyl ester | 330.50 | 4 | 2 | 3.18 | 97.06 | 0 |
| Cyanoacetic acid | 85.06 | 3 | 1 | −0.96 | 18.06 | 1 |
| Methyl 11-bromoundecanoate | 279.21 | 2 | 0 | 3.56 | 68.95 | 0 |
| Azetidin-2-one 3,3-dimethyl-4-(1-aminoethyl)- | 142.20 | 2 | 2 | 0.72 | 43.01 | 0 |
| 3-Azabicyclo[3.2.2]nonane | 125.21 | 1 | 1 | 1.83 | 43.06 | 0 |
| Phytol, acetate | 338.57 | 2 | 0 | 5.47 | 108.68 | 1 |
| Hexadecanoic acid, methyl ester | 270.45 | 2 | 0 | 4.44 | 85.12 | 0 |
| 11-Oxa-dispiro[4.0.4.1]undecan-1-ol | 168.23 | 2 | 1 | 1.52 | 46.16 | 0 |
| 9,12-Octadecadienoic acid (Z,Z)-, methyl ester | 294.47 | 2 | 0 | 4.70 | 93.78 | 0 |
| 6-Octadecenoic acid, methyl ester, (Z)- | 296.49 | 2 | 0 | 4.80 | 94.26 | 0 |
| Phytol | 296.53 | 1 | 1 | 5.25 | 98.94 | 1 |
| Methyl stearate | 298.50 | 2 | 0 | 4.91 | 94.73 | 0 |
| Pseduosarsasapogenin-5,20-dien | 414.62 | 3 | 2 | 4.42 | 123.27 | 0 |
| Octadecanoic acid, 2,3-dihydroxypropyl ester | 358.56 | 4 | 2 | 3.63 | 106.67 | 0 |
| 13-Docosenamide, (Z)- | 337.58 | 1 | 1 | 5.06 | 110.30 | 1 |
| Campesterol | 400.68 | 1 | 1 | 6.54 | 128.42 | 1 |
| 4-Campestene-3-one | 398.66 | 1 | 0 | 6.43 | 127.46 | 1 |
| 4,22-Cholestadien-3-one | 382.62 | 1 | 0 | 6.13 | 122.18 | 1 |
| 1,2-Bis(trimethylsilyl)benzene | 222.47 | 0 | 0 | 4.13 | 72.40 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, M.; Tareq, A.M.; Rakib, A.; Mahmud, S.; Sami, S.A.; Mallick, J.; Islam, M.N.; Majumder, M.; Uddin, M.Z.; Alsubaie, A.; et al. Phytochemicals from Leucas zeylanica Targeting Main Protease of SARS-CoV-2: Chemical Profiles, Molecular Docking, and Molecular Dynamics Simulations. Biology 2021, 10, 789. https://doi.org/10.3390/biology10080789
Dutta M, Tareq AM, Rakib A, Mahmud S, Sami SA, Mallick J, Islam MN, Majumder M, Uddin MZ, Alsubaie A, et al. Phytochemicals from Leucas zeylanica Targeting Main Protease of SARS-CoV-2: Chemical Profiles, Molecular Docking, and Molecular Dynamics Simulations. Biology. 2021; 10(8):789. https://doi.org/10.3390/biology10080789
Chicago/Turabian StyleDutta, Mycal, Abu Montakim Tareq, Ahmed Rakib, Shafi Mahmud, Saad Ahmed Sami, Jewel Mallick, Mohammad Nazmul Islam, Mohuya Majumder, Md. Zia Uddin, Abdullah Alsubaie, and et al. 2021. "Phytochemicals from Leucas zeylanica Targeting Main Protease of SARS-CoV-2: Chemical Profiles, Molecular Docking, and Molecular Dynamics Simulations" Biology 10, no. 8: 789. https://doi.org/10.3390/biology10080789
APA StyleDutta, M., Tareq, A. M., Rakib, A., Mahmud, S., Sami, S. A., Mallick, J., Islam, M. N., Majumder, M., Uddin, M. Z., Alsubaie, A., Almalki, A. S. A., Khandaker, M. U., Bradley, D. A., Rana, M. S., & Emran, T. B. (2021). Phytochemicals from Leucas zeylanica Targeting Main Protease of SARS-CoV-2: Chemical Profiles, Molecular Docking, and Molecular Dynamics Simulations. Biology, 10(8), 789. https://doi.org/10.3390/biology10080789