
Sequencing and Chromosome-Scale Assembly of Plant Genomes, Brassica rapa as a Use Case

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction for Nanopore Sequencing
2.2. Nanopore Sequencing
2.3. Nanopore Genome Assembly and Polishing
2.4. Optical Mapping and Hybrid Scaffolding
2.5. Omni-C Library Preparation and Illumina Sequencing
2.6. Scaffolding Using the Omni-C Library
2.7. Pore-C Library Preparation and Nanopore Sequencing
2.8. Scaffolding Using the Pore-C Library
2.9. Super-Scaffolding
2.10. Validation of the Assemblies
2.10.1. Comparison to Reference Genomes
2.10.2. Quality Assessment with Merqury
2.10.3. Gene-Completeness Estimation with Busco
2.11. Construction of a B. rapa Genetic Map and Anchoring
2.12. Putative Position of Peri-Centromere and Sub-Telomere Regions
2.13. Gene Prediction
3. Results
3.1. Nanopore Sequencing and Long Reads Assembly
3.2. Long Range Genome Assembly
3.2.1. Hybrid Scaffolding
3.2.2. Hi-C Scaffolding
3.2.3. Pore-C Scaffolding
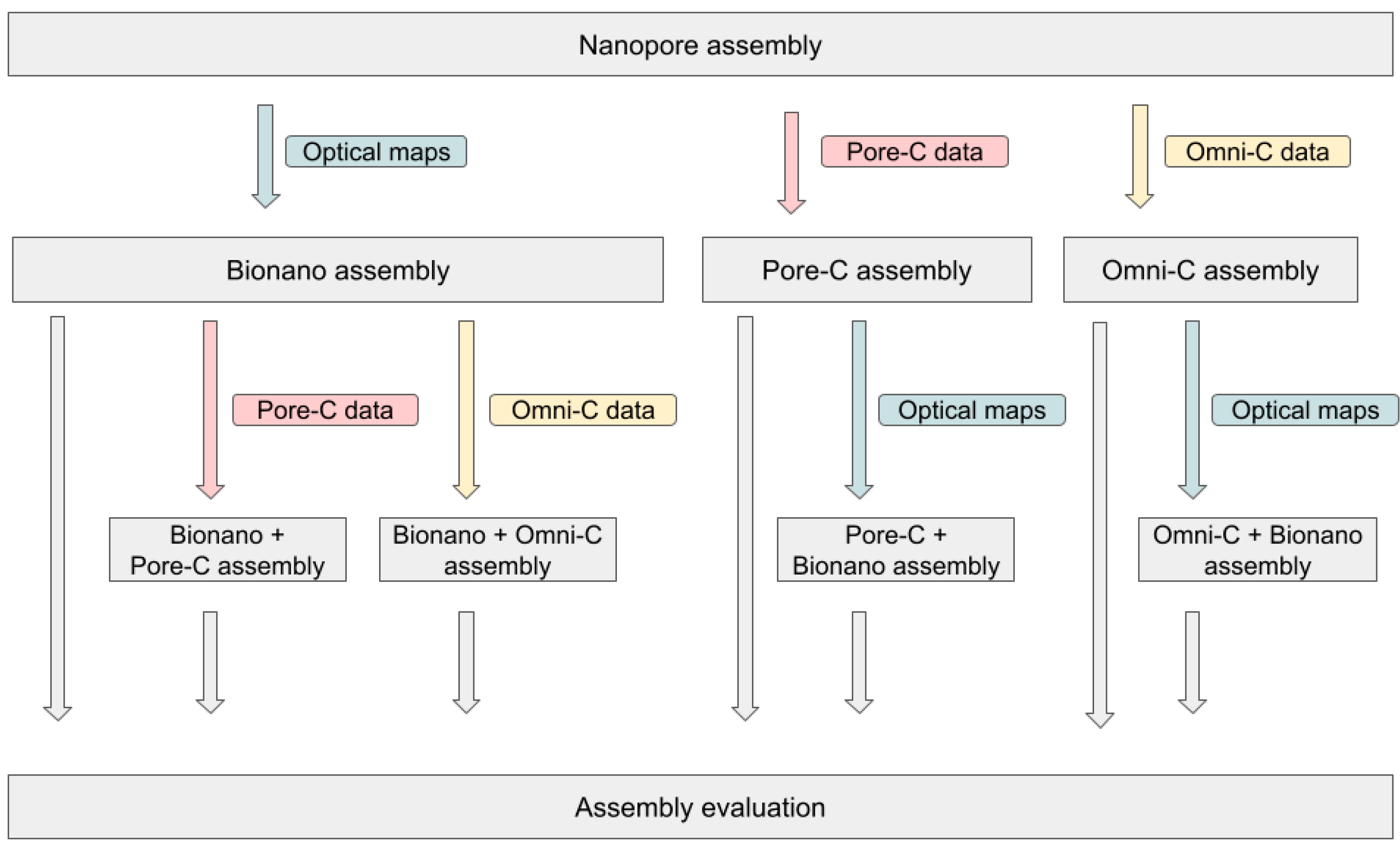
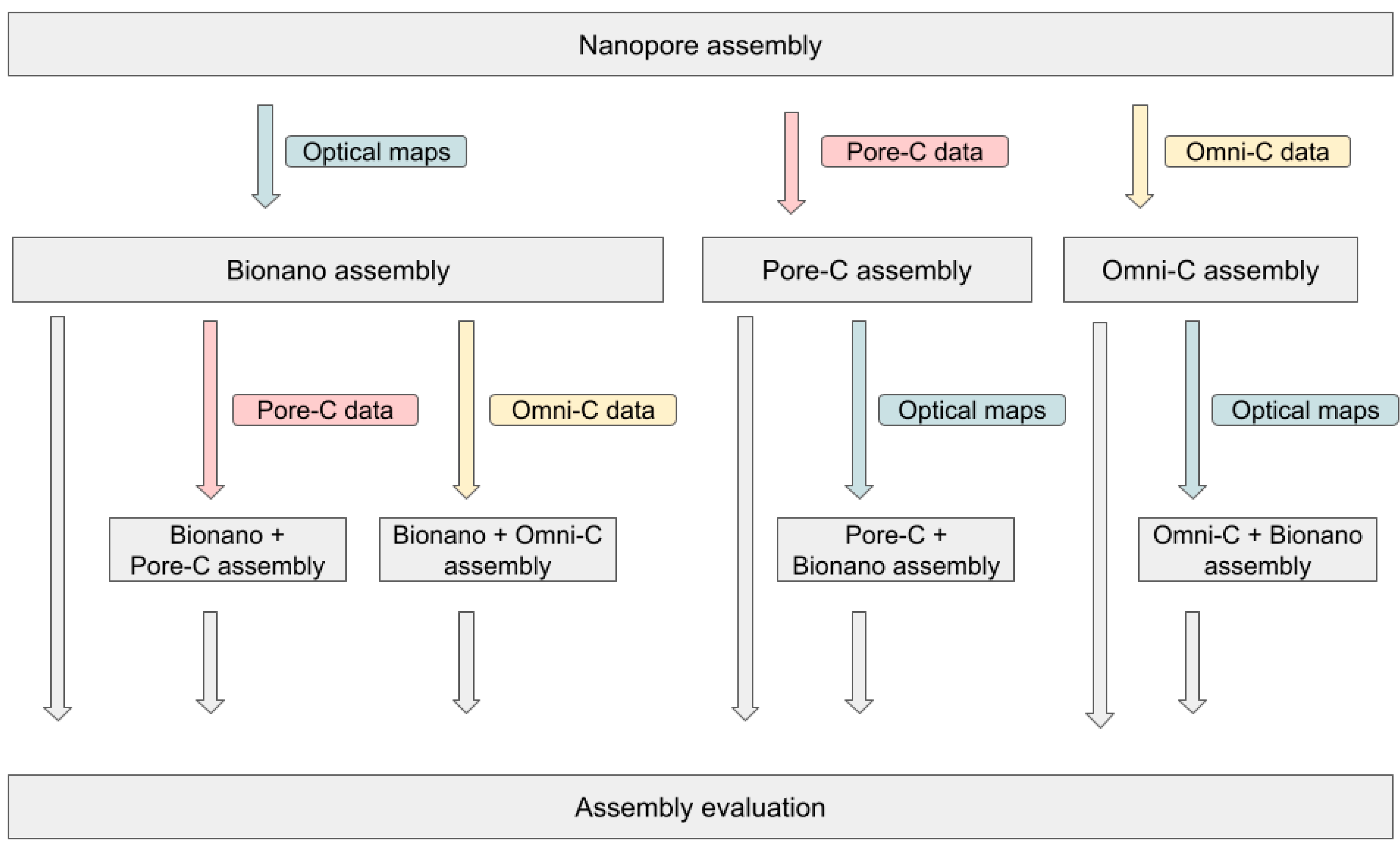
3.3. Combination of Several Long-Range Techniques
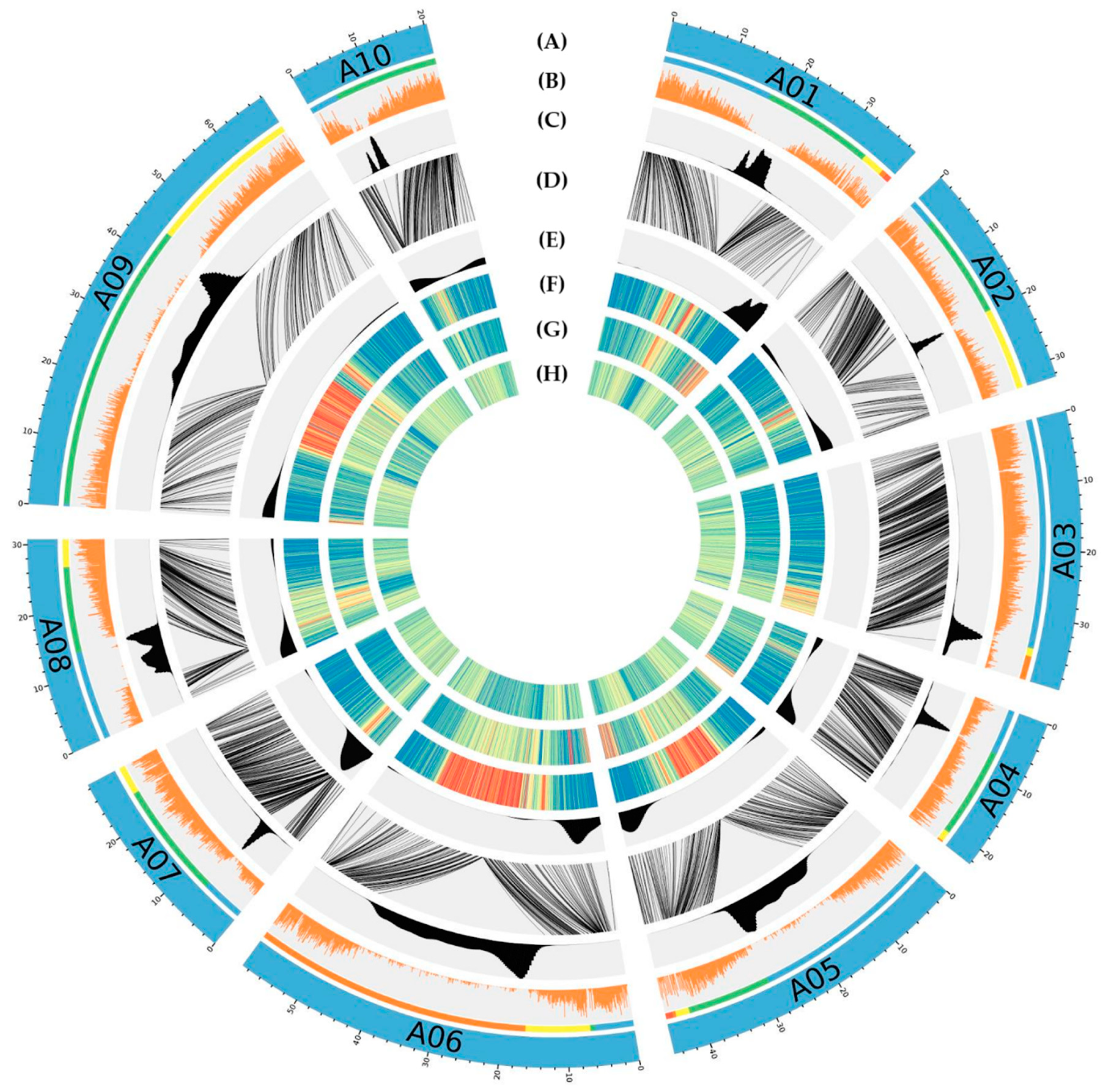
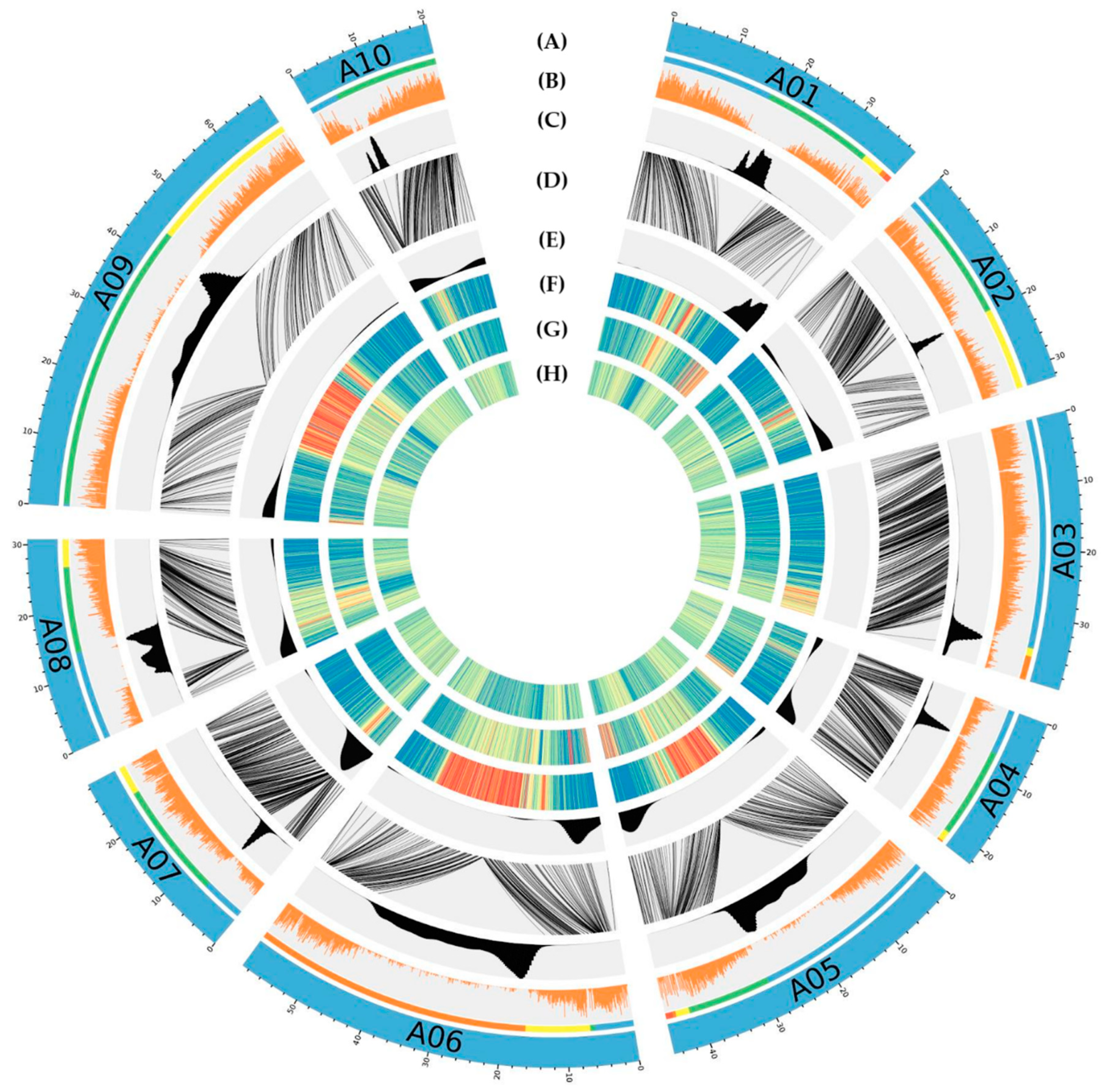
3.4. Anchoring
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimin, A.V.; Stevens, K.A.; Crepeau, M.W.; Puiu, D.; Wegrzyn, J.L.; Yorke, J.A.; Langley, C.H.; Neale, D.B.; Salzberg, S.L. An Improved Assembly of the Loblolly Pine Mega-Genome Using Long-Read Single-Molecule Sequencing. Gigascience 2017, 6, 1–4. [Google Scholar] [PubMed] [Green Version]
- Claros, M.G.; Bautista, R.; Guerrero-Fernández, D.; Benzerki, H.; Seoane, P.; Fernández-Pozo, N. Why Assembling Plant Genome Sequences Is so Challenging. Biology 2012, 1, 439–459. [Google Scholar] [CrossRef] [Green Version]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate Whole Human Genome Sequencing Using Reversible Terminator Chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jain, C.; Aluru, S. A Comprehensive Evaluation of Long Read Error Correction Methods. BMC Genom. 2020, 21, 889. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, K.; Medvedev, P. Error Correction Enables Use of Oxford Nanopore Technology for Reference-Free Transcriptome Analysis. Nat. Commun. 2021, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Warr, A. Errors in Long-Read Assemblies Can Critically Affect Protein Prediction. Nat. Biotechnol. 2019, 37, 124–126. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and Accurate de Novo Genome Assembly from Long Uncorrected Reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Aury, J.-M.; Istace, B. Hapo-G, Haplotype-Aware Polishing of Genome Assemblies with Accurate Reads. NAR Genom. Bioinform. 2021, 3, lqab034. [Google Scholar] [CrossRef] [PubMed]
- Driguez, P.; Bougouffa, S.; Carty, K.; Putra, A.; Jabbari, K.; Reddy, M.; Soppe, R.; Cheung, N.; Fukasawa, Y.; Ermini, L. LeafGo: Leaf to Genome, a Quick Workflow to Produce High-Quality De Novo Genomes with Third Generation Sequencing Technology. bioRxiv 2021. [Google Scholar] [CrossRef]
- Belser, C.; Baurens, F.-C.; Noel, B.; Martin, G.; Cruaud, C.; Istace, B.; Yahiaoui, N.; Labadie, K.; Hřibová, E.; Doležel, J.; et al. Telomere-to-Telomere Gapless Chromosomes of Banana Using Nanopore Sequencing. bioRxiv 2021. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Hon, T.; Mars, K.; Young, G.; Tsai, Y.-C.; Karalius, J.W.; Landolin, J.M.; Maurer, N.; Kudrna, D.; Hardigan, M.A.; Steiner, C.C.; et al. Highly Accurate Long-Read HiFi Sequencing Data for Five Complex Genomes. Sci. Data 2020, 7, 399. [Google Scholar] [CrossRef]
- Sun, H.; Jiao, W.-B.; Krause, K.; Campoy, J.A.; Goel, M.; Folz-Donahue, K.; Kukat, C.; Huettel, B.; Schneeberger, K. Chromosome-Scale and Haplotype-Resolved Genome Assembly of a Tetraploid Potato Cultivar. bioRxiv 2021. [Google Scholar] [CrossRef]
- Campoy, J.A.; Sun, H.; Goel, M.; Jiao, W.-B.; Folz-Donahue, K.; Wang, N.; Rubio, M.; Liu, C.; Kukat, C.; Ruiz, D.; et al. Gamete Binning: Chromosome-Level and Haplotype-Resolved Genome Assembly Enabled by High-Throughput Single-Cell Sequencing of Gamete Genomes. Genome Biol. 2020, 21, 306. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chung, C.Y.-L.; Chan, T.-F. Advances in Optical Mapping for Genomic Research. Comput. Struct. Biotechnol. J. 2020, 18, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Bionano Genomics. Generating Accurate and Contiguous De Novo Genome Assemblies Using Hybrid Scaffolding. 2017. Available online: https://bionanogenomics.com/wp-content/uploads/2017/02/Bionano_HumanPAG_Hybrid-Scaffolding-White-Paper.pdf (accessed on 30 July 2021).
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A Comprehensive Technique to Capture the Conformation of Genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [Green Version]
- McCord, R.P.; Kaplan, N.; Giorgetti, L. Chromosome Conformation Capture and Beyond: Toward an Integrative View of Chromosome Structure and Function. Mol. Cell 2020, 77, 688–708. [Google Scholar] [CrossRef]
- Ulahannan, N.; Pendleton, M.; Deshpande, A.; Schwenk, S.; Behr, J.M.; Dai, X.; Tyer, C.; Rughani, P.; Kudman, S.; Adney, E.; et al. Nanopore Sequencing of DNA Concatemers Reveals Higher-Order Features of Chromatin Structure. bioRxiv 2019. [Google Scholar] [CrossRef]
- Choi, J.Y.; Dai, X.; Peng, J.Z.; Rughani, P.; Hickey, S.; Harrington, E.; Juul, S.; Ayroles, J.; Purugganan, M.; Stacy, E.A. Selection on Old Variants Drives Adaptive Radiation of Metrosideros across the Hawaiian Islands. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fierst, J.L. Using Linkage Maps to Correct and Scaffold de Novo Genome Assemblies: Methods, Challenges, and Computational Tools. Front. Genet. 2015, 6, 220. [Google Scholar] [CrossRef]
- Yu, A.; Li, F.; Xu, W.; Wang, Z.; Sun, C.; Han, B.; Wang, Y.; Wang, B.; Cheng, X.; Liu, A. Application of a High-Resolution Genetic Map for Chromosome-Scale Genome Assembly and Fine QTLs Mapping of Seed Size and Weight Traits in Castor Bean. Sci. Rep. 2019, 9, 11950. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, G.; Yang, S.; Just, J.; Yan, H.; Zhou, N.; Jian, H.; Wang, Q.; Chen, M.; Qiu, X.; et al. The Development of a High-Density Genetic Map Significantly Improves the Quality of Reference Genome Assemblies for Rose. Sci. Rep. 2019, 9, 5985. [Google Scholar] [CrossRef]
- Zhang, W.; Cao, Y.; Wang, K.; Zhao, T.; Chen, J.; Pan, M.; Wang, Q.; Feng, S.; Guo, W.; Zhou, B.; et al. Identification of Centromeric Regions on the Linkage Map of Cotton Using Centromere-Related Repeats. Genomics 2014, 104, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Round, E.K.; Flowers, S.K.; Richards, E.J. Arabidopsis Thaliana Centromere Regions: Genetic Map Positions and Repetitive DNA Structure. Genome Res. 1997, 7, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Belser, C.; Istace, B.; Denis, E.; Dubarry, M.; Baurens, F.-C.; Falentin, C.; Genete, M.; Berrabah, W.; Chèvre, A.-M.; Delourme, R.; et al. Chromosome-Scale Assemblies of Plant Genomes Using Nanopore Long Reads and Optical Maps. Nat. Plants 2018, 4, 879–887. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.-H.; Bancroft, I.; Cheng, F.; et al. The Genome of the Mesopolyploid Crop Species Brassica Rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Nagaharu, U. Genome Analysis in Brassica with Special Reference to the Experimental Formation of B. Napus and Peculiar Mode of Fertilization. J. Jpn. Bot. 1935, 7, 389–452. [Google Scholar]
- Rousseau-Gueutin, M.; Belser, C.; Da Silva, C.; Richard, G.; Istace, B.; Cruaud, C.; Falentin, C.; Boideau, F.; Boutte, J.; Delourme, R.; et al. Long-Read Assembly of the Brassica Napus Reference Genome Darmor-Bzh. Gigascience 2020, 9. [Google Scholar] [CrossRef]
- Wick, R.; Github. Filtlong. Available online: https://github.com/rrwick/Filtlong (accessed on 30 July 2021).
- Liu, H.; Wu, S.; Li, A.; Ruan, J. SMARTdenovo: A de Novo Assembler Using Long Noisy Reads. Gigabyte 2021, 2021, 1–9. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and Accurate Long-Read Assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Chen, Y.; Nie, F.; Xie, S.-Q.; Zheng, Y.-F.; Dai, Q.; Bray, T.; Wang, Y.-X.; Xing, J.-F.; Huang, Z.-J.; Wang, D.-P.; et al. Efficient Assembly of Nanopore Reads via Highly Accurate and Intact Error Correction. Nat. Commun. 2021, 12, 60. [Google Scholar] [CrossRef]
- Github. Medaka. Available online: https://github.com/nanoporetech/medaka (accessed on 30 July 2021).
- Istace, B.; Belser, C.; Aury, J.-M. BiSCoT: Improving Large Eukaryotic Genome Assemblies with Optical Maps. PeerJ 2020, 8, e10150. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De Novo Assembly of the Aedes Aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Github. Juicebox. Available online: https://github.com/aidenlab/Juicebox (accessed on 30 July 2021).
- Github. Juicebox_scripts. Available online: https://github.com/phasegenomics/juicebox_scripts (accessed on 30 July 2021).
- Liu, C. In Situ Hi-C Library Preparation for Plants to Study Their Three-Dimensional Chromatin Interactions on a Genome-Wide Scale. Methods Mol. Biol. 2017, 1629, 155–166. [Google Scholar] [PubMed]
- Github. Pore-C-Snakemake. Available online: https://github.com/nanoporetech/Pore-C-Snakemake/issues/11 (accessed on 30 July 2021).
- Github. Pore-C-Snakemake. Available online: https://github.com/nanoporetech/Pore-C-Snakemake (accessed on 30 July 2021).
- Ghurye, J.; Pop, M.; Koren, S.; Bickhart, D.; Chin, C.-S. Scaffolding of Long Read Assemblies Using Long Range Contact Information. BMC Genom. 2017, 18, 527. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cai, X.; Wu, J.; Liu, M.; Grob, S.; Cheng, F.; Liang, J.; Cai, C.; Liu, Z.; Liu, B.; et al. Improved Brassica Rapa Reference Genome by Single-Molecule Sequencing and Chromosome Conformation Capture Technologies. Hortic. Res. 2018, 5, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabanettes, F.; Klopp, C. D-GENIES: Dot Plot Large Genomes in an Interactive, Efficient and Simple Way. PeerJ 2018, 6, e4958. [Google Scholar] [CrossRef]
- De Givry, S.; Bouchez, M.; Chabrier, P.; Milan, D.; Schiez, T. Cathagene: Multipopulation Integrated Genetic and Radiated Hybrid Mapping. Bioinformatics 2004, 14, 2. [Google Scholar]
- Lim, K.-B.; de Jong, H.; Yang, T.-J.; Park, J.-Y.; Kwon, S.-J.; Kim, J.S.; Lim, M.-H.; Kim, J.A.; Jin, M.; Jin, Y.-M.; et al. Characterization of rDNAs and Tandem Repeats in the Heterochromatin of Brassica Rapa. Mol. Cells 2005, 19, 436–444. [Google Scholar]
- Lim, K.-B.; Yang, T.-J.; Hwang, Y.-J.; Kim, J.S.; Park, J.-Y.; Kwon, S.-J.; Kim, J.; Choi, B.-S.; Lim, M.-H.; Jin, M.; et al. Characterization of the Centromere and Peri-Centromere Retrotransposons in Brassica Rapa and Their Distribution in Related Brassica Species. Plant. J. 2007, 49, 173–183. [Google Scholar] [CrossRef]
- Koo, D.-H.; Hong, C.P.; Batley, J.; Chung, Y.S.; Edwards, D.; Bang, J.-W.; Hur, Y.; Lim, Y.P. Rapid Divergence of Repetitive DNAs in Brassica Relatives. Genomics 2011, 97, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight High-Quality Genomes Reveal Pan-Genome Architecture and Ecotype Differentiation of Brassica Napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Morgulis, A.; Gertz, E.M.; Schäffer, A.A.; Agarwala, R. A Fast and Symmetric DUST Implementation to Mask Low-Complexity DNA Sequences. J. Comput. Biol. 2006, 13, 1028–1040. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [Green Version]
- Dubarry, M.; Noel, B.; Rukwavu, T.; Aury, J.M. Gmove a Tool for Eukaryotic Gene Predictions Using Various Evidences. F1000research Publ. Online 2016. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the Performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Input Contigs (Necat Assembly) | Bionano | Omni-C | Pore-C | |
|---|---|---|---|---|
| Cumulative size | 443,649,441 | 443,951,349 | 439,638,897 | 443,677,941 |
| Number of sequences | 299 | 236 | 590 | 253 |
| N50 (L50) | 10,461,875 (12) | 17,017,634 (8) | 25,523,596 (7) | 20,151,380 (9) |
| N90 (L90) | 857,267 (58) | 3,409,175 (30) | 221,999 (98) | 1,472,408 (41) |
| auN | 14,202,687 | 20,478,883 | 22,995,704 | 16,823,684 |
| Max. size | 45,115,632 | 44,069,534 | 42,018,994 | 32,180,808 |
| Number of Ns (%) | 0 (0%) | 2,914,945 (0.66%) | 20,900 (0.00%) | 28,500 (0.01%) |
| Complete busco genes (%) | 1604 (99.4%) | 1604 (99.4%) | 1604 (99.4%) | 1604 (99.4%) |
| Merqury score | 36.4423 | 37.1176 | 36.4875 | 36.4872 |
| Input Contigs (Necat Assembly) | Bionano + Omni-C | Bionano + Pore-C | Omni-C + Bionano | Pore-C + Bionano | |
|---|---|---|---|---|---|
| Cumulative size | 443,649,441 | 440,038,627 | 443,961,849 | 445,844,245 | 450,760,401 |
| Number of sequences | 299 | 511 | 222 | 401 | 215 |
| N50 (L50) | 10,461,875 (12) | 33,316,896 (5) | 17,017,634 (8) | 20,321,816 (7) | 25,915,290 (7) |
| N90 (L90) | 857,267 (58) | 275,999 (61) | 3,409,175 (29) | 1,763,661 (32) | 3,720,451 (26) |
| auN | 14,202,687 | 34,864,156 | 20,976,778 | 22,582,099 | 22,825,029 |
| Max. size | 45,115,632 | 64,589,792 | 44,069,534 | 51,305,606 | 43,928,997 |
| Number of Ns (%) | 0 (0%) | 2,930,045 (0.67%) | 2,925,445 (0.66%) | 10,143,940 (2.28%) | 3,522,960 (0.78%) |
| Complete buscos genes (%) | 1604 (99.4%) | 1604 (99.4%) | 1604 (99.4%) | 1604 (99.4%) | 1604 (99.4%) |
| Merqury score | 36.4423 | 37.1179 | 37.1176 | 37.0247 | 37.0566 |
| Input Contigs (Necat Assembly) | Bionano | Brassica rapa cv Z1 V2 | Brassica rapa cv Z1 V1 [27] | |
|---|---|---|---|---|
| Cumulative size | 443,649,441 | 443,951,349 | 443,953,949 | 401,164,957 |
| Number of sequences | 299 | 236 | 210 | 237 |
| N50 (L50) | 10,461,875 (12) | 17,017,634 (8) | 39,217,720 (5) | 34,481,996 (5) |
| N90 (L90) | 857,267 (58) | 3,409,175 (30) | 4,034,065 (13) | 2,865,407 (12) |
| auN | 14,202,687 | 20,478,883 | 38,695,451 | 36,201,043 |
| Max. size | 45,115,632 | 44,069,534 | 68,194,707 | 57,670,803 |
| Number of Ns (%) | 0 (0%) | 2,914,945 (0.66%) | 2,917,545 (0.66%) | 32,966,574 (8.22%) |
| Number of contigs | 299 | 301 | 295 | 297 |
| Contigs N50 (L50) | 10,461,875 (12) | 10,256,333 (14) | 10,256,333 (14) | 6,651,009 (14) |
| Complete busco genes (%) | 1604 (99.4%) | 1604 (99.4%) | 1604 (99.4%) | 1594 (98.7%) |
| Merqury score | 36.4423 | 37.1176 | 37.119 | 28.4862 |
| Number of genes | - | - | 56,073 | 46,721 |
| Number of exons/gene | - | - | 4.39 | 4.72 |
| Complete busco genes (%) | - | - | 1573 (97.5%) | 1553 (96.2%) |
| Duplicated busco genes (%) | - | - | 226 (14.0%) | 216 (13.4%) |
| Fragmented busco genes (%) | - | - | 16 (1.0%) | 20 (1.2%) |
| Missing busco genes (%) | - | - | 25 (1.5%) | 41(2.6%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Istace, B.; Belser, C.; Falentin, C.; Labadie, K.; Boideau, F.; Deniot, G.; Maillet, L.; Cruaud, C.; Bertrand, L.; Chèvre, A.-M.; et al. Sequencing and Chromosome-Scale Assembly of Plant Genomes, Brassica rapa as a Use Case. Biology 2021, 10, 732. https://doi.org/10.3390/biology10080732
Istace B, Belser C, Falentin C, Labadie K, Boideau F, Deniot G, Maillet L, Cruaud C, Bertrand L, Chèvre A-M, et al. Sequencing and Chromosome-Scale Assembly of Plant Genomes, Brassica rapa as a Use Case. Biology. 2021; 10(8):732. https://doi.org/10.3390/biology10080732
Chicago/Turabian StyleIstace, Benjamin, Caroline Belser, Cyril Falentin, Karine Labadie, Franz Boideau, Gwenaëlle Deniot, Loeiz Maillet, Corinne Cruaud, Laurie Bertrand, Anne-Marie Chèvre, and et al. 2021. "Sequencing and Chromosome-Scale Assembly of Plant Genomes, Brassica rapa as a Use Case" Biology 10, no. 8: 732. https://doi.org/10.3390/biology10080732
APA StyleIstace, B., Belser, C., Falentin, C., Labadie, K., Boideau, F., Deniot, G., Maillet, L., Cruaud, C., Bertrand, L., Chèvre, A.-M., Wincker, P., Rousseau-Gueutin, M., & Aury, J.-M. (2021). Sequencing and Chromosome-Scale Assembly of Plant Genomes, Brassica rapa as a Use Case. Biology, 10(8), 732. https://doi.org/10.3390/biology10080732