
Aldehyde Dehydrogenase Mutation Exacerbated High-Fat-Diet-Induced Nonalcoholic Fatty Liver Disease with Gut Microbiota Remodeling in Male Mice


 ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
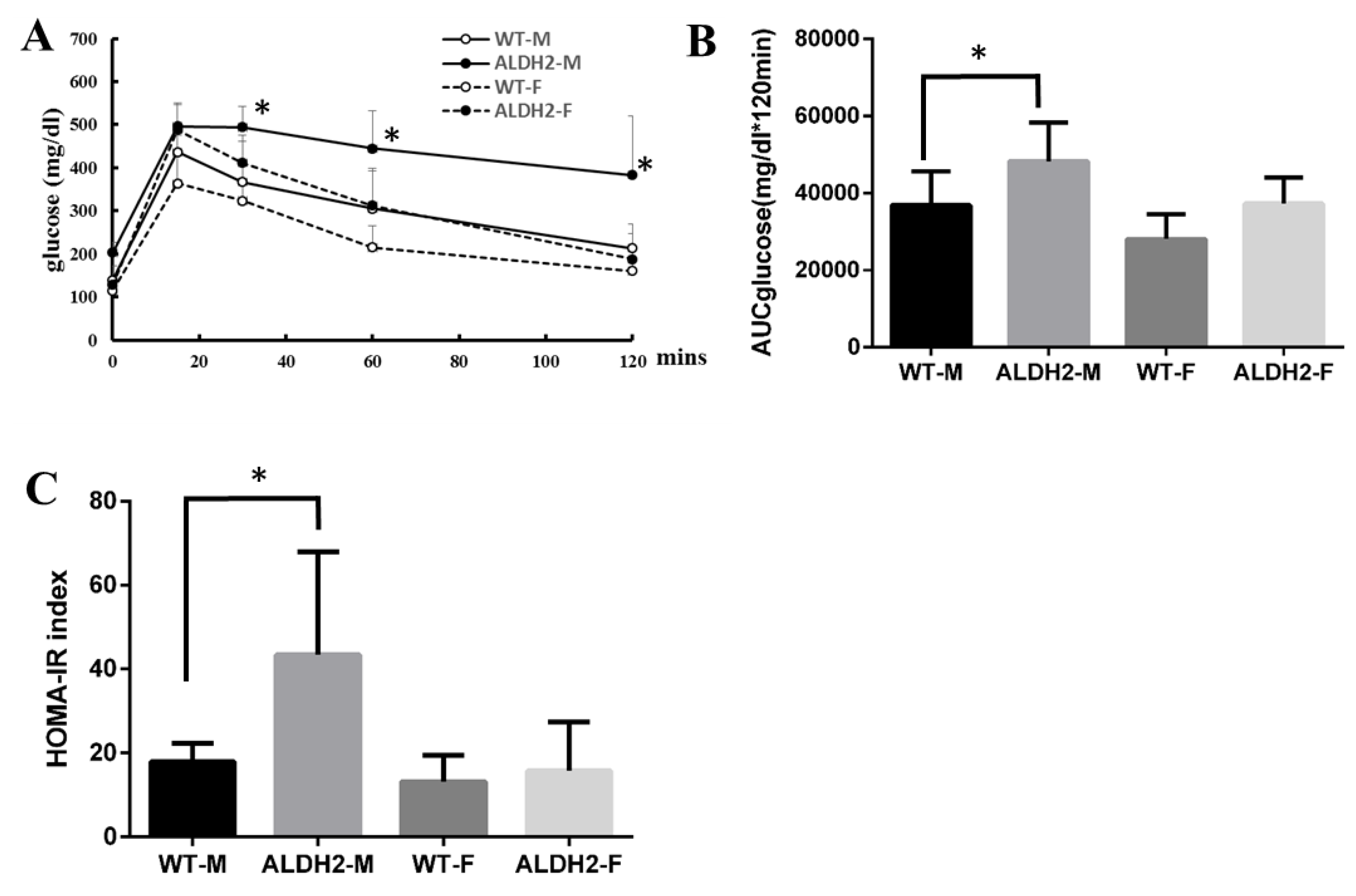
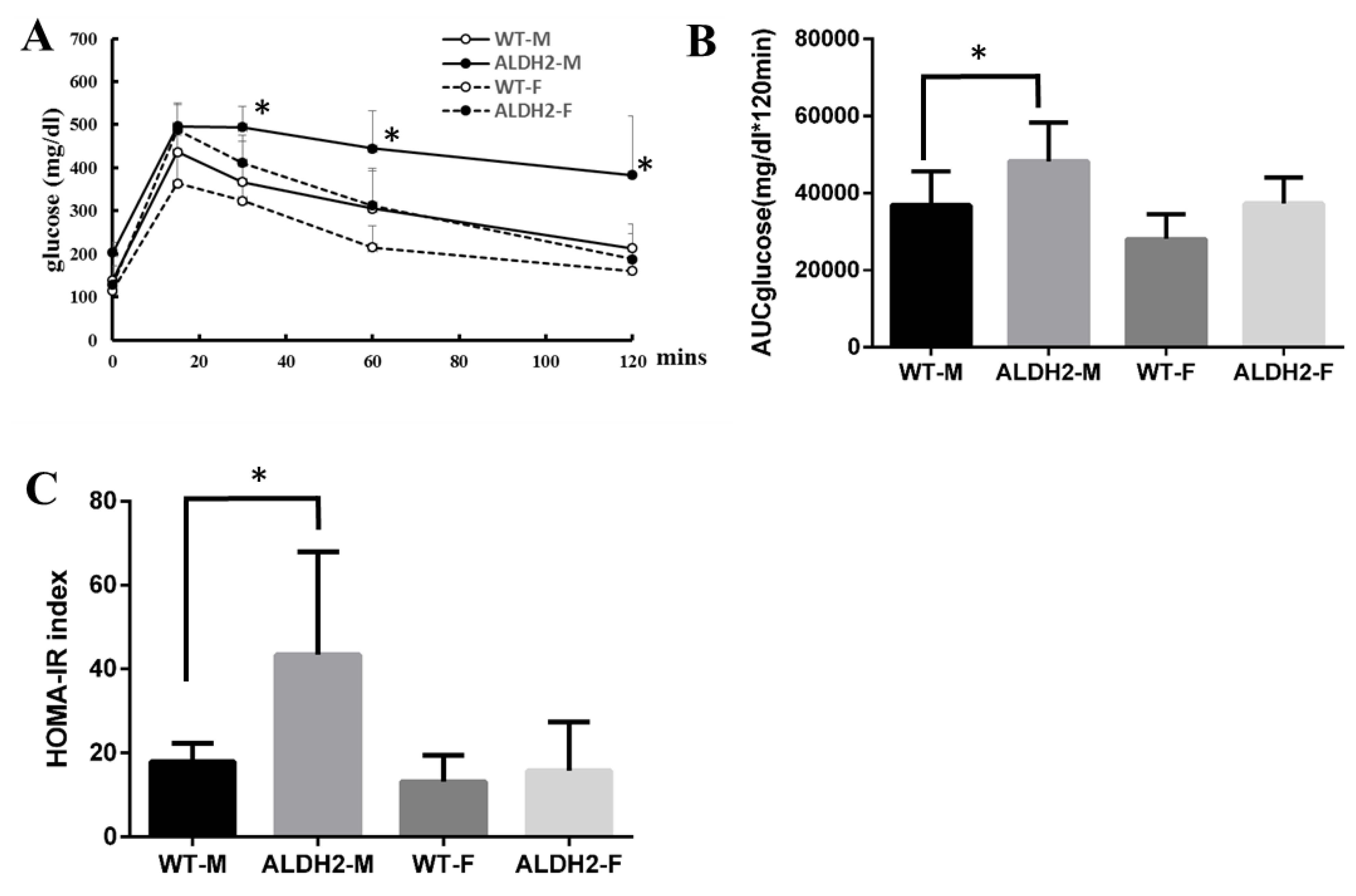
2.3. Oral Glucose-Tolerance Tests and Homeostasis-Model Assessment of Insulin Resistance
2.4. Measurement of Biochemical Parameters
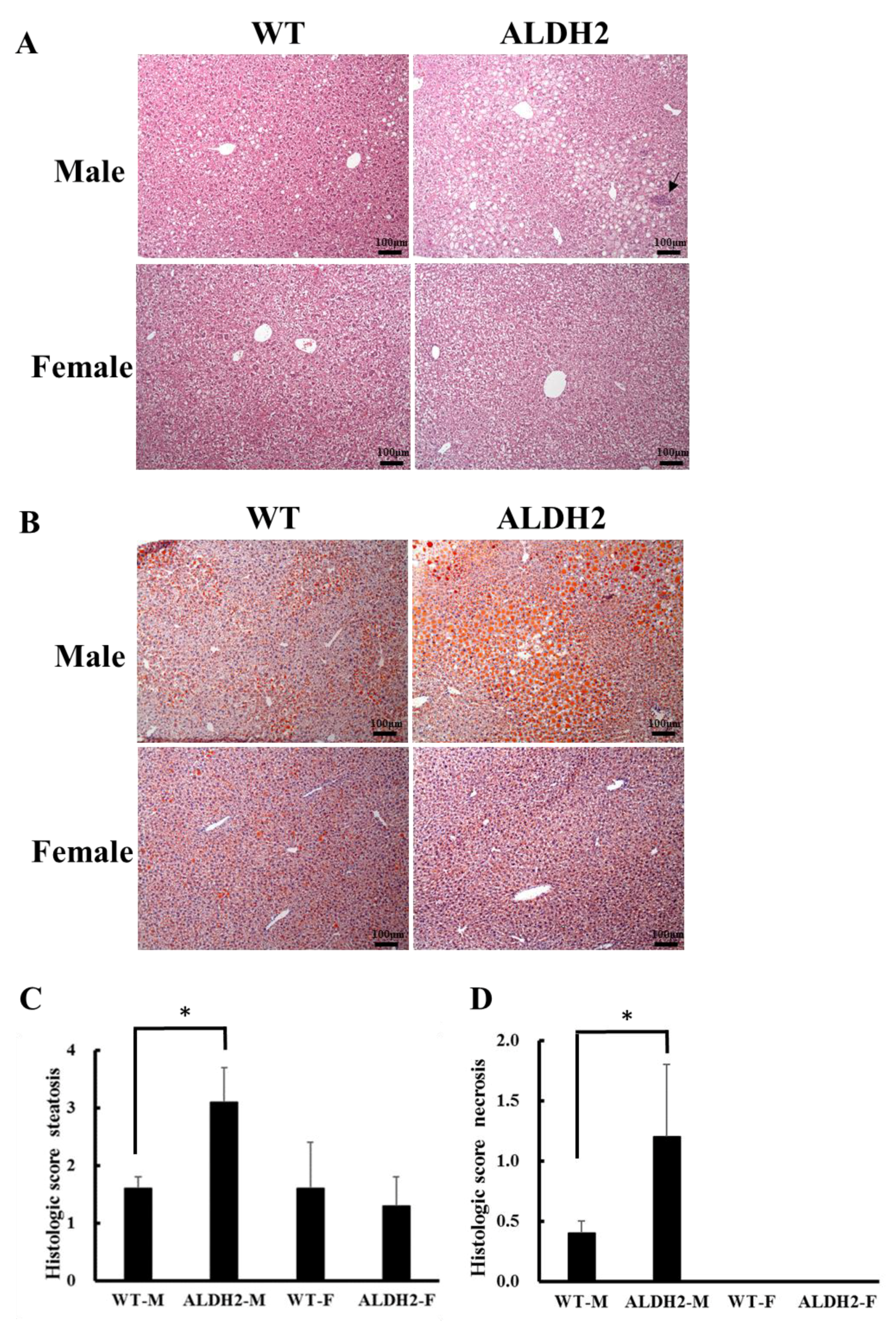
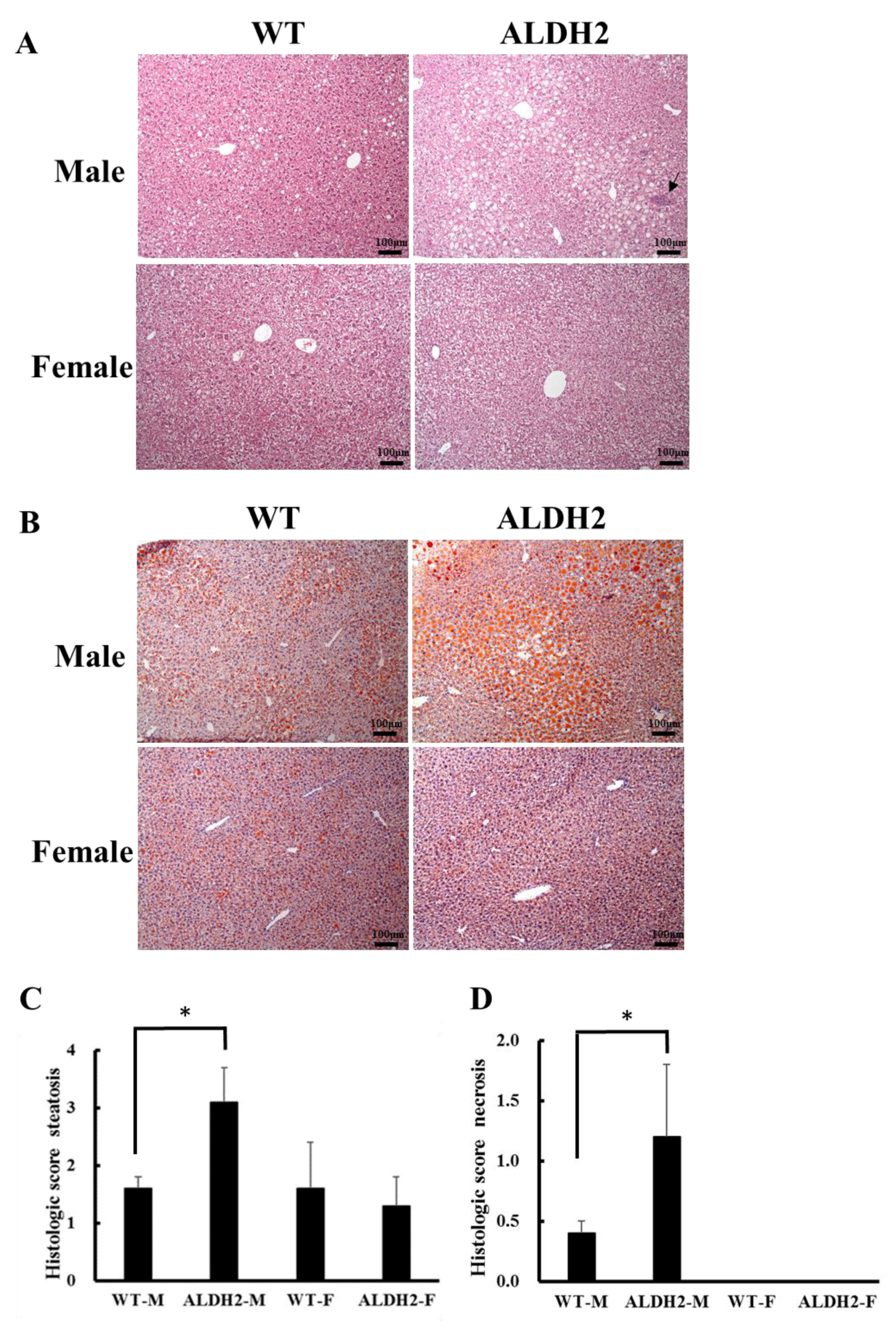
2.5. Histopathological Examination
2.6. Oil Red O Staining
2.7. Measurements of Liver Triglycerides
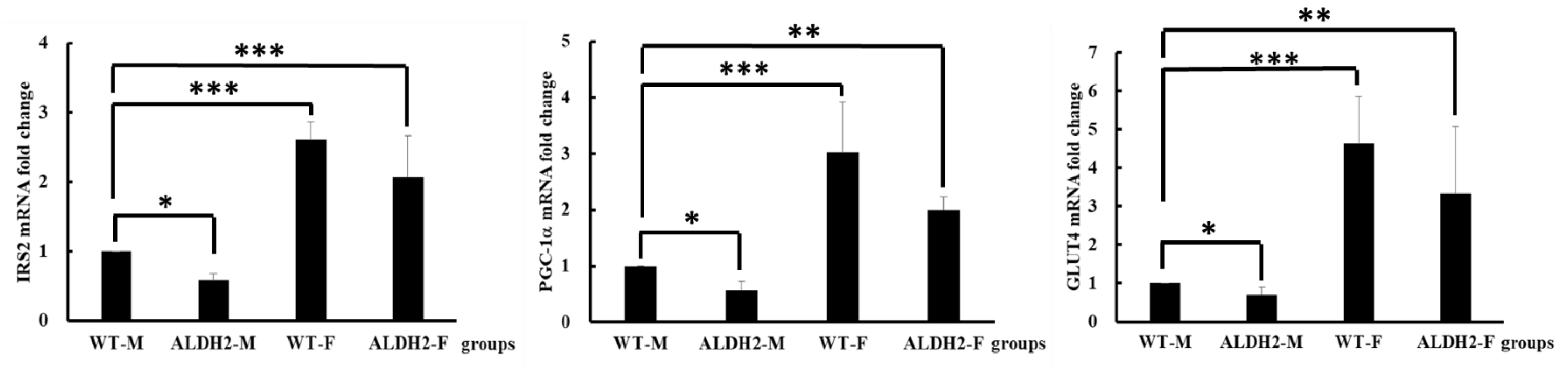
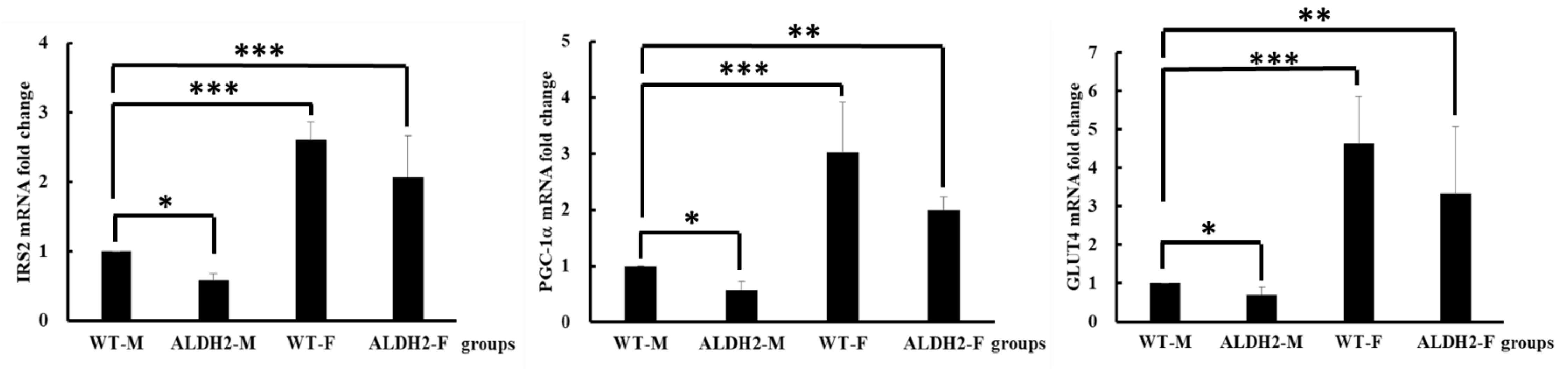
2.8. Insulin-Signaling-Related Gene Expression
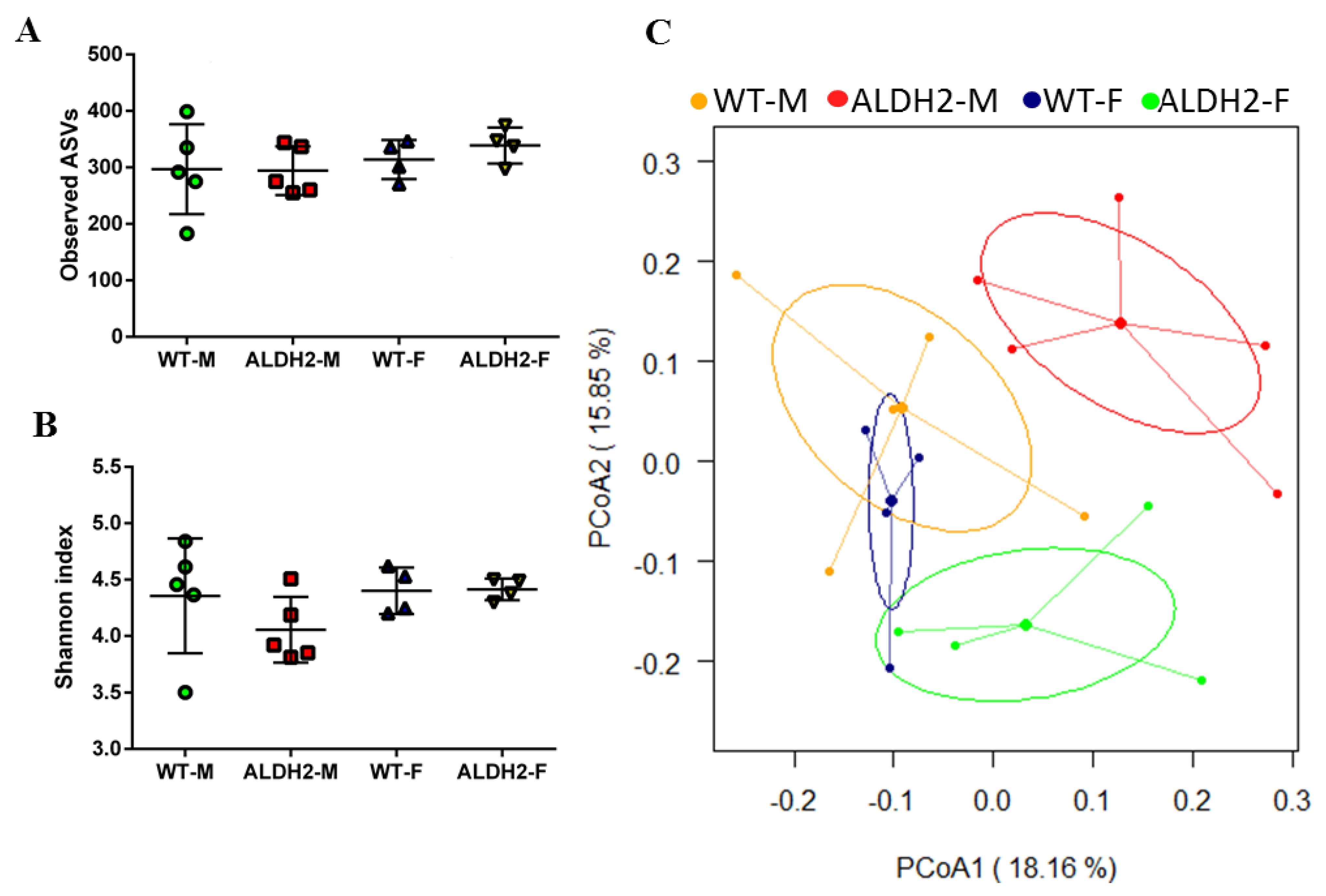
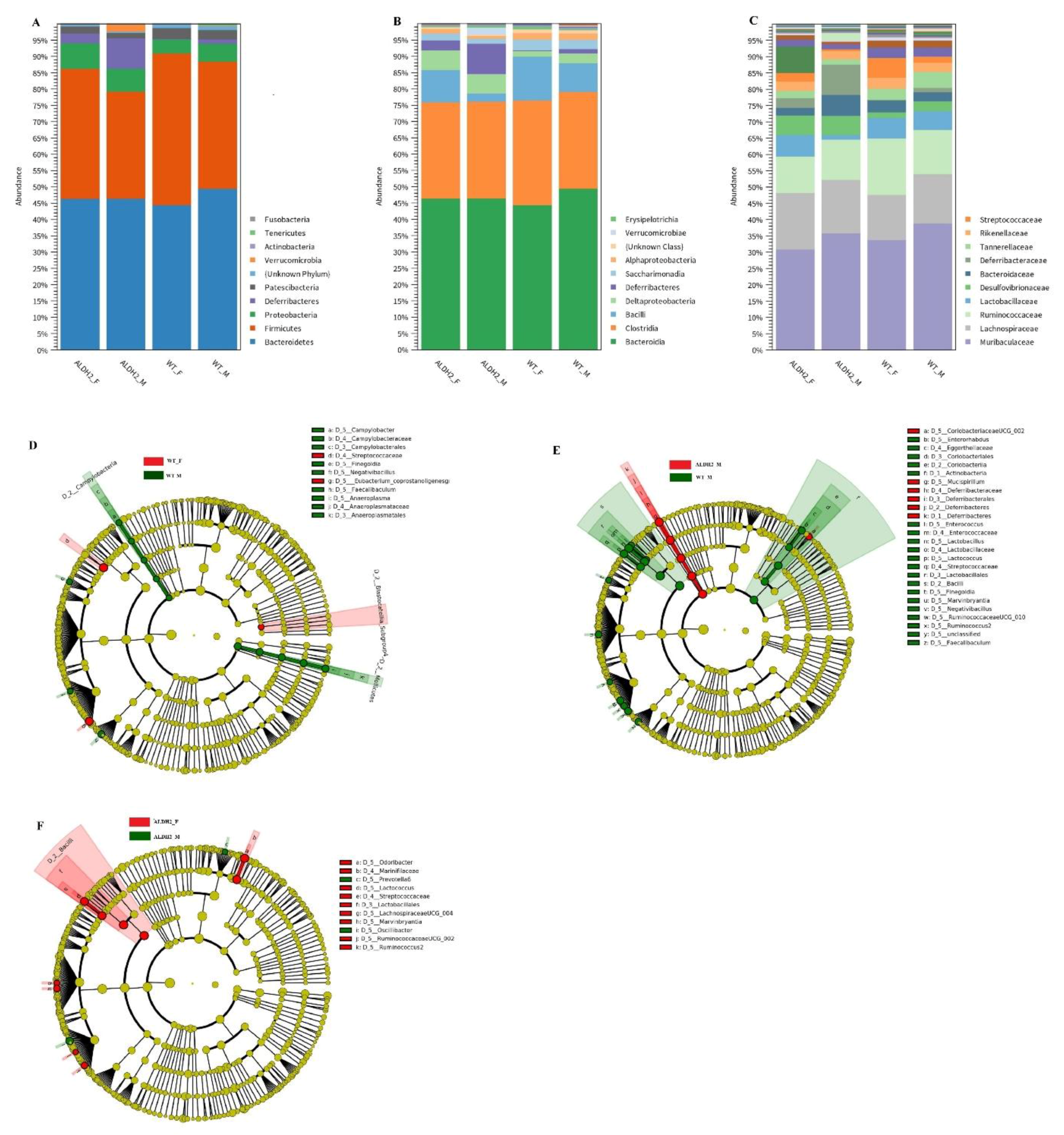
2.9. Fecal Microbiota Analysis
2.10. Statistical Analysis
3. Results
3.1. ALDH2-M Mice Have Higher Terminal Body and White Adipose Tissue Weights
3.2. ALDH2 Male Mice Develop More Severe Hepatic Steatosis
3.3. ALDH2 Gene Mutation Increases Insulin Resistance in ALDH2-M Mice
3.4. Analysis of Gene Expression on Insulin Signaling Pathway
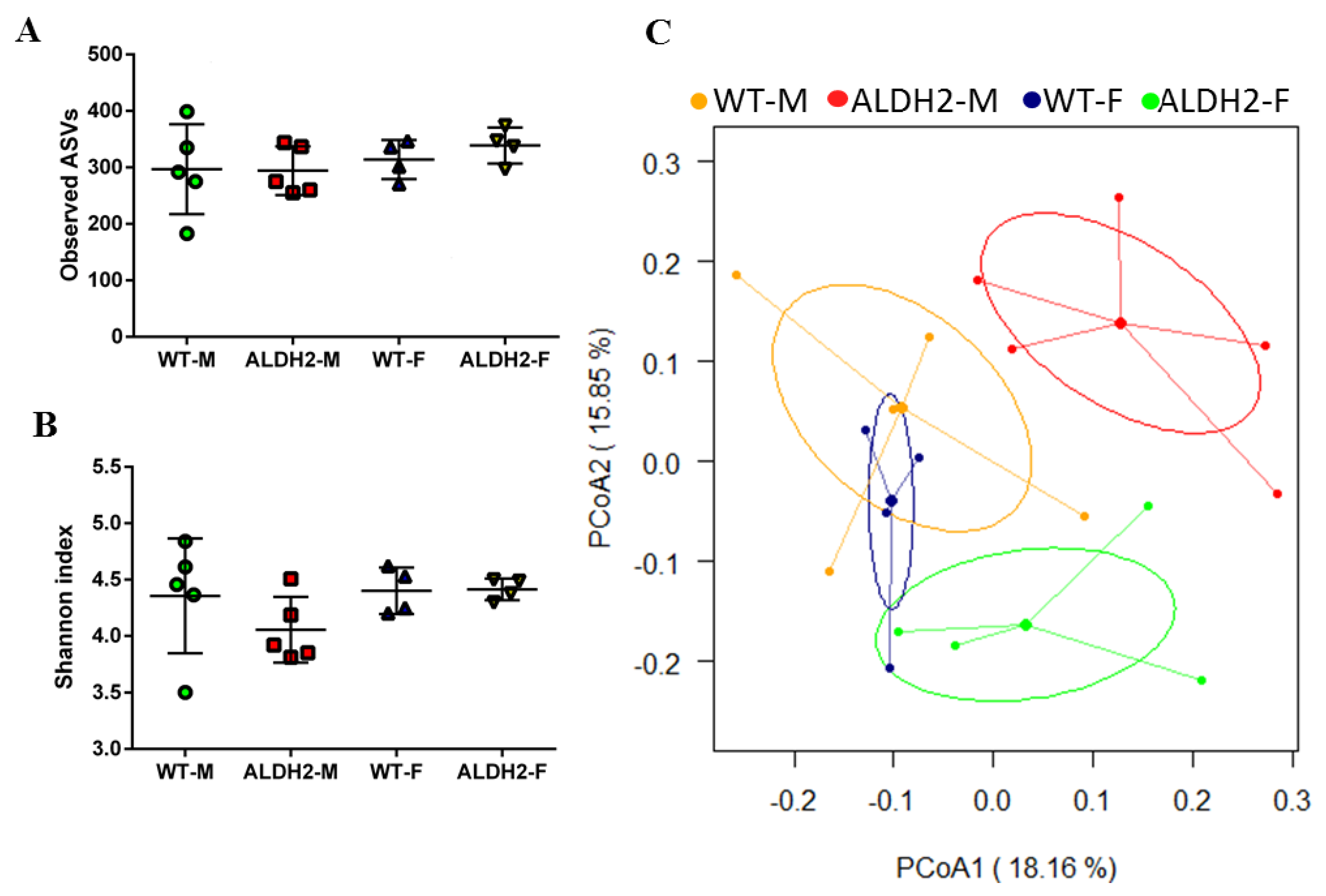
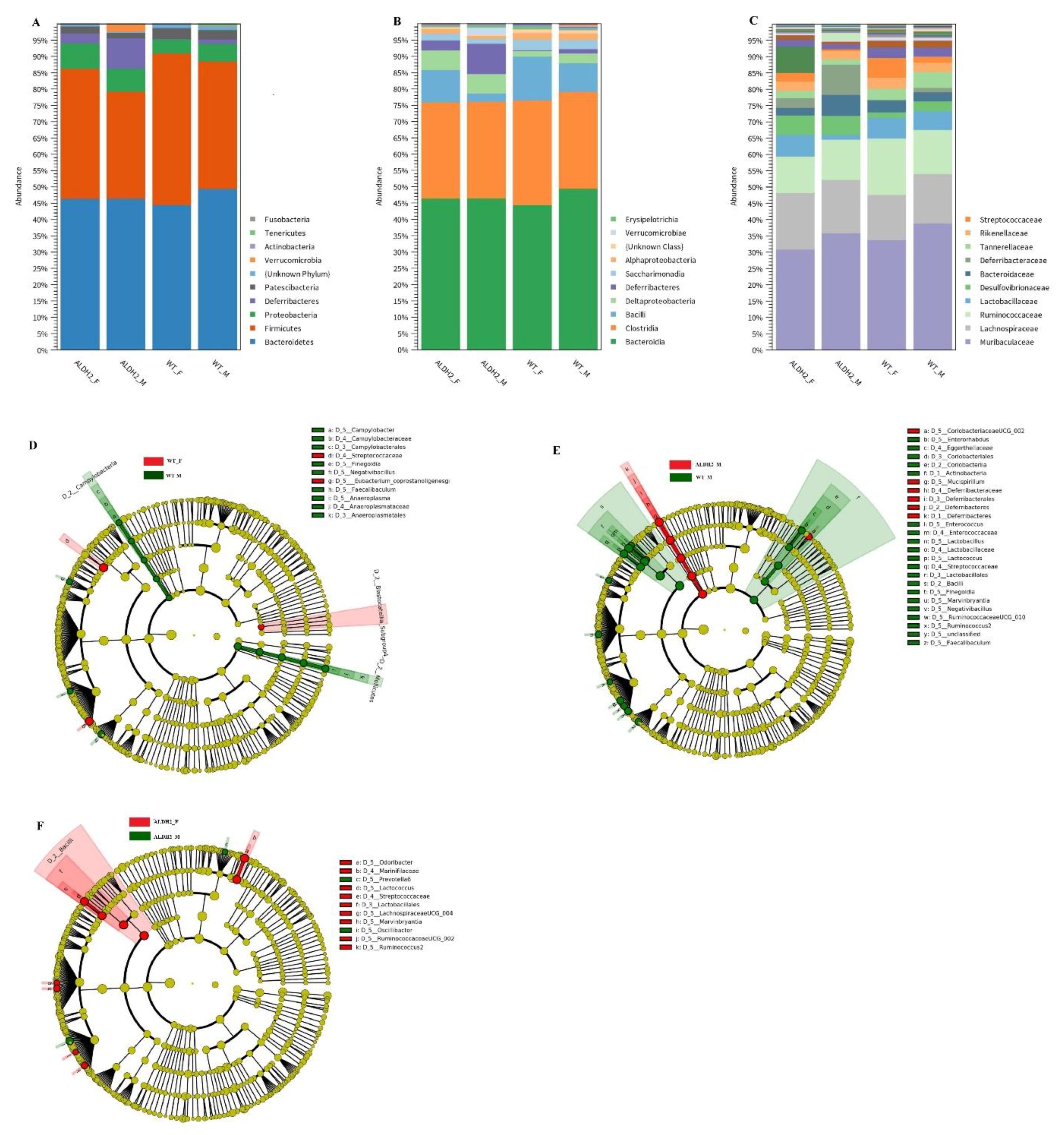
3.5. ALDH2 Gene Mutation Affects Gut-Microbiota Composition and Diversity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klyosov, A.A.; Rashkovetsky, L.G.; Tahir, M.K.; Keung, W.M. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochemistry 1996, 35, 4445–4456. [Google Scholar] [CrossRef]
- Lai, C.L.; Yao, C.T.; Chau, G.Y.; Yang, L.F.; Kuo, T.Y.; Chiang, C.P.; Yin, S.J. Dominance of the inactive Asian variant over activity and protein contents of mitochondrial aldehyde dehydrogenase 2 in human liver. Alcohol. Clin. Exp. Res. 2014, 38, 44–50. [Google Scholar] [CrossRef]
- Chen, Y.C.; Peng, G.S.; Tsao, T.P.; Wang, M.F.; Lu, R.B.; Yin, S.J. Pharmacokinetic and pharmacodynamic basis for overcoming acetaldehyde-induced adverse reaction in Asian alcoholics, heterozygous for the variant ALDH2* 2 gene allele. Pharm. Genom. 2009, 19, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.R.; Wu, G.S.; Pakstis, A.J.; Tong, L.; Oota, H.; Kidd, K.K.; Zhang, Y.P. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2 487Lys. Gene 2009, 435, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Oniki, K.; Morita, K.; Watanabe, T.; Kajiwara, A.; Otake, K.; Nakagawa, K.; Sasaki, Y.; Ogata, Y.; Saruwatari, J. The longitudinal effect of the aldehyde dehydrogenase 2* 2 allele on the risk for nonalcoholic fatty liver disease. Nutr. Diabetes 2016, 6, e210. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Zeng, Q. The Association and Interaction of Aldehyde Dehydrogenase 2 Polymorphisms with Food Group Intake and Probability of Having Non-Alcoholic Fatty Liver Disease. Diabetes Metab. Syndr. Obes. 2020, 13, 5049–5057. [Google Scholar] [CrossRef]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [Green Version]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol. Res. 2017, 38, 147–161. [Google Scholar] [PubMed]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [Green Version]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef]
- Burz, S.; Monnoye, M.; Philippe, C.; Farin, W.; Ratziu, V.; Strozzi, F.; Paillarse, J.M.; Chêne, L.; Blottière, H.; Gérard, P. Fecal Microbiota Transplant from Human to Mice Gives Insights into the Role of the Gut Microbiota in Non-Alcoholic Fatty Liver Disease (NAFLD). Microorganisms 2021, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Fei, N.; Bruneau, A.; Zhang, Z.; Wang, R.; Wang, J.; Rabot, S.; Gérard, P.; Zhao, L. Endotoxin Producers Overgrowing in Human Gut Microbiota as the Causative Agents for Nonalcoholic Fatty Liver Disease. mBio 2020, 11, e03263-19. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.C.; Ching, Y.H.; Li, Y.P.; Liu, J.Y.; Huang, Y.T.; Huang, Y.W.; Yang, S.-S.; Huang, W.C.; Chuang, H.L. Nonalcoholic fatty liver disease is exacerbated in high-fat diet-fed gnotobiotic mice by colonization with the gut microbiota from patients with nonalcoholic steatohepatitis. Nutrients 2017, 9, 1220. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Eur. Gastroenterol. J. 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Johnson, A.M.; Olefsky, J.M. The origins and drivers of insulin resistance. Cell 2013, 152, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.C.; Malik, V.; Jia, W.; Kadowaki, T.; Yajnik, C.S.; Yoon, K.-H.; Hu, F.B. Diabetes in Asia: Epidemiology, risk factors, and pathophysiology. JAMA 2009, 301, 2129–2140. [Google Scholar] [CrossRef]
- Jung, J.G.; Kim, J.S.; Oh, M.K. The role of the flushing response in the relationship between alcohol consumption and insulin resistance. Alcohol. Clin. Exp. Res. 2010, 34, 1699–1704. [Google Scholar] [CrossRef]
- Völzke, H.; Schwarz, S.; Baumeister, S.E.; Wallaschofski, H.; Schwahn, C.; Grabe, H.J.; Kohlmann, T.; John, U.; Dören, M. Menopausal status and hepatic steatosis in a general female population. Gut 2007, 56, 594–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambelli, V.O.; Gross, E.R.; Chen, C.-H.; Gutierrez, V.P.; Cury, Y.; Mochly-Rosen, D. Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci. Transl. Med. 2014, 6, 251ra118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; Hoek, A.M.V.D. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar]
- Armoni, M.; Harel, C.; Karnieli, E. GLUT4 goes abnormal: Regulation of the insulin-responsive GLUT4 transporter in abnormal metabolic states. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 2005, 5, 207–218. [Google Scholar] [CrossRef]
- Wu, H.; Deng, X.; Shi, Y.; Su, Y.; Wei, J.; Duan, H. PGC-1α, glucose metabolism and type 2 diabetes mellitus. J. Endocrinol. 2016, 229, R99–R115. [Google Scholar] [CrossRef]
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A.; Estall, J.L. Estrogen signals through peroxisome proliferator-activated Receptor− γ coactivator 1α to reduce oxidative damage associated with diet-induced fatty liver disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef]
- Kotani, K.; Peroni, O.D.; Minokoshi, Y.; Boss, O.; Kahn, B.B. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization. J. Clin. Investig. 2004, 114, 1666–1675. [Google Scholar] [CrossRef]
- Carvalho, E.; Kotani, K.; Peroni, O.D.; Kahn, B.B. Adipose-specific overexpression of GLUT4 reverses insulin resistance and diabetes in mice lacking GLUT4 selectively in muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E551–E561. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.G.; Kim, S.U.; Wong, V.W. New trends on obesity and NAFLD in Asia. J. Hepatol. 2017, 67, 862–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, Y.; Hyogo, H.; Ono, M.; Mizuta, T.; Ono, N.; Fujimoto, K.; Chayama, K.; Saibara, T. Prevalence and associated metabolic factors of nonalcoholic fatty liver disease in the general population from 2009 to 2010 in Japan: A multicenter large retrospective study. J. Gastroenterol. 2012, 47, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, R.; Ogishi, Y.; Ueda, M.; Matsusaki, M.; Amako, K.; Goda, K.; Park, S.-S. Gender-related differences in mouse hepatic ethanol metabolism. J. Nutr. Sci. Vitaminol. 2002, 48, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Louet, J.F.; LeMay, C.; Mauvais-Jarvis, F. Antidiabetic actions of estrogen: Insight from human and genetic mouse models. Curr. Atheroscler. Rep. 2004, 6, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Safari, Z.; Gérard, P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD). Cell. Mol. Life Sci. 2019, 76, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Nugent, C.A.; Tsompana, M.; Cai, L.; Wang, Y.; Buck, M.J.; Genco, R.J.; Baker, R.D.; Zhu, R.; et al. Gut microbiome may contribute to insulin resistance and systemic inflammation in obese rodents: A meta-analysis. Physiol. Genom. 2018, 50, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, V.V.; Sheng, L.; Jena, P.K. Gender Differences in Bile Acids and Microbiota in Relationship with Gender Dissimilarity in Steatosis Induced by Diet and FXR Inactivation. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Peng, L.; Piekos, S.; Guo, G.L.; Zhong, X.-B. Role of farnesoid X receptor in establishment of ontogeny of phase-I drug metabolizing enzyme genes in mouse liver. Acta Pharm. Sin. B 2016, 6, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Jung, S.R.; Lee, S.Y.; Lee, N.K.; Paik, H.D.; Lim, S.I. Lactobacillus plantarum strain ln4 attenuates diet-induced obesity, insulin resistance, and changes in hepatic mrna levels associated with glucose and lipid metabolism. Nutrients 2018, 10, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagarolli, R.A.; Tobar, N.; Oliveira, A.; Araújo, T.; Carvalho, B.; Rocha, G.; Vecina, J.F.; Calisto, K.; Guadagnini, D.; Prada, P.D.O.; et al. Probiotics modulate gut microbiota and improve insulin sensitivity in DIO mice. J. Nutr. Biochem. 2017, 50, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Okubo, H.; Kobuke, K.; Ohno, H.; Oki, K.; Yoneda, M.; Tanaka, J.; Hattori, N. Alteration of gut microbiota by a Westernized lifestyle and its correlation with insulin resistance in non-diabetic Japanese men. J. Diabetes Investig. 2019, 10, 1463–1470. [Google Scholar] [CrossRef] [Green Version]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press (US): Washington, DC, USA, 2011.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (GenBank ID) | Orientation | Sequence (5′-3′) | UPL |
|---|---|---|---|
| IRS2 (NM_001082121.1) | Forward Reverse | gtccaggcactggagcttt gcgcttcactctttcacga | 53 |
| GLUT4 (NM_009204.2) | Forward Reverse | gacggacactccatctgttg gccacgatggagacatagc | 5 |
| PGC-1α (NM_008904.2) | Forward Reverse | cccatacacaaccgcagtc gaacccttggggtcatttg | 6 |
| β-actin (NM_007393.3) | Forward Reverse | ctaaggccaaccgtgaaaag accagaggcatacagggaca | 64 |
| Characteristic | Male | Female | p Values for Two-Way ANOVA | ||||
|---|---|---|---|---|---|---|---|
| WT | ALDH2 | WT | ALDH2 | Main Effect of Gender | Main Effect of Genotype | Interaction (G × G’) | |
| Diet intake (g/mouse/day) Terminal body weight (g) | 2.74 ± 0.14 38.2 ± 3.6 | 2.79 ± 0.14 44.5 ± 4.8 # | 2.48 ± 0.04 27.2 ± 3.6 | 2.51 ± 0.07 26.6 ± 3.8 | <0.001 <0.0001 | 0.1381 0.0985 | 0.7928 0.471 |
| White adipose tissue weight (g) | 3.16 ± 1.02 | 4.86 ± 0.66 # | 2.05 ± 1.07 | 1.37 ± 0.93 | <0.0001 | 0.203 | 0.007 |
| ALT (U/L) | 61.3 ± 26.2 | 142.9 ± 57.9 | 82.3 ± 37.9 | 92.9 ± 50.3 | 0.4772 | 0.0338 | 0.0939 |
| AST (U/L) | 261.1 ± 104.0 | 346.3 ± 97.6 | 263.6 ± 119.2 | 248.4 ± 90.7 | 0.2697 | 0.4152 | 0.0939 |
| Hepatic TG (mg/dl) | 920.2 ± 151.5 | 1143.1 ± 154.6 | 887.7 ± 162.8 | 871.3 ± 95.9 | 0.0195 | 0.0997 | 0.0594 |
| Taxonomy | WT-M | ALDH-M | WT-F | ALDH-F |
|---|---|---|---|---|
| Actinobacteria (phylum) | 2.896 ± 0.016 | 0.780 ± 0.002 * | 1.167 ± 0.004 | 1.331 ± 0.008 |
| Coriobacteriia (class) | 2.254 ± 0.013 | 0.586 ± 0.002 * | 0.715 ± 0.002 | 1.049 ± 0.008 |
| Coriobacteriales (order) | 2.254 ± 0.013 | 0.586 ± 0.002 * | 0.715 ± 0.002 | 1.049 ± 0.008 |
| Deferribacteres (phylum) | 0.836 ± 0.012 | 5.096 ± 0.028 * | 0.125 ± 0.001 | 1.715 ± 0.017 |
| Deferribacteres (class) | 0.836 ± 0.012 | 5.096 ± 0.028 * | 0.125 ± 0.001 # | 1.715 ± 0.017 |
| Deferribacterales (order) | 0.836 ± 0.012 | 5.096 ± 0.028 * | 0.125 ± 0.001 # | 1.715 ± 0.017 |
| Firmicutes (phylum) | 40.521 ± 0.122 | 37.079 ± 0.114 | 45.770 ± 0.029 | 41.753 ± 0.071 |
| Bacilli (class) | 6.712 ± 0.018 | 2.299 ± 0.018 * | 10.101 ± 0.042 # | 7.380 ± 0.023 # |
| Lactobacillales (order) | 5.497 ± 0.016 | 1.733 ± 0.015 * | 9.055 ± 0.046 # | 6.591 ± 0.022 # |
| Bacillales (order) | 1.215 ± 0.004 | 0.566 ± 0.003 * | 1.046 ± 0.006 | 0.789 ± 0.003 |
| Proteobacteria (phylum) | 5.032 ± 0.025 | 6.786 ± 0.036 | 3.929 ± 0.005 | 7.333 ± 0.033 |
| Deltaproteobacteria (class) | 3.182 ± 0.025 | 5.858 ± 0.038 | 1.861 ± 0.006 | 5.831 ± 0.037 |
| Desulfovibronales (order) | 3.182 ± 0.025 | 5.858 ± 0.038 | 1.856 ± 0.006 # | 5.831 ± 0.037 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.-S.; Chen, Y.-H.; Hu, J.-T.; Chiu, C.-F.; Hung, S.-W.; Chang, Y.-C.; Chiu, C.-C.; Chuang, H.-L. Aldehyde Dehydrogenase Mutation Exacerbated High-Fat-Diet-Induced Nonalcoholic Fatty Liver Disease with Gut Microbiota Remodeling in Male Mice. Biology 2021, 10, 737. https://doi.org/10.3390/biology10080737
Yang S-S, Chen Y-H, Hu J-T, Chiu C-F, Hung S-W, Chang Y-C, Chiu C-C, Chuang H-L. Aldehyde Dehydrogenase Mutation Exacerbated High-Fat-Diet-Induced Nonalcoholic Fatty Liver Disease with Gut Microbiota Remodeling in Male Mice. Biology. 2021; 10(8):737. https://doi.org/10.3390/biology10080737
Chicago/Turabian StyleYang, Sien-Sing, Yi-Hsun Chen, Jui-Ting Hu, Ching-Feng Chiu, Shao-Wen Hung, Yi-Chih Chang, Chien-Chao Chiu, and Hsiao-Li Chuang. 2021. "Aldehyde Dehydrogenase Mutation Exacerbated High-Fat-Diet-Induced Nonalcoholic Fatty Liver Disease with Gut Microbiota Remodeling in Male Mice" Biology 10, no. 8: 737. https://doi.org/10.3390/biology10080737
APA StyleYang, S.-S., Chen, Y.-H., Hu, J.-T., Chiu, C.-F., Hung, S.-W., Chang, Y.-C., Chiu, C.-C., & Chuang, H.-L. (2021). Aldehyde Dehydrogenase Mutation Exacerbated High-Fat-Diet-Induced Nonalcoholic Fatty Liver Disease with Gut Microbiota Remodeling in Male Mice. Biology, 10(8), 737. https://doi.org/10.3390/biology10080737