
Hypoxia Signaling in Parkinson’s Disease: There Is Use in Asking “What HIF?”


Abstract
:Simple Summary
Abstract
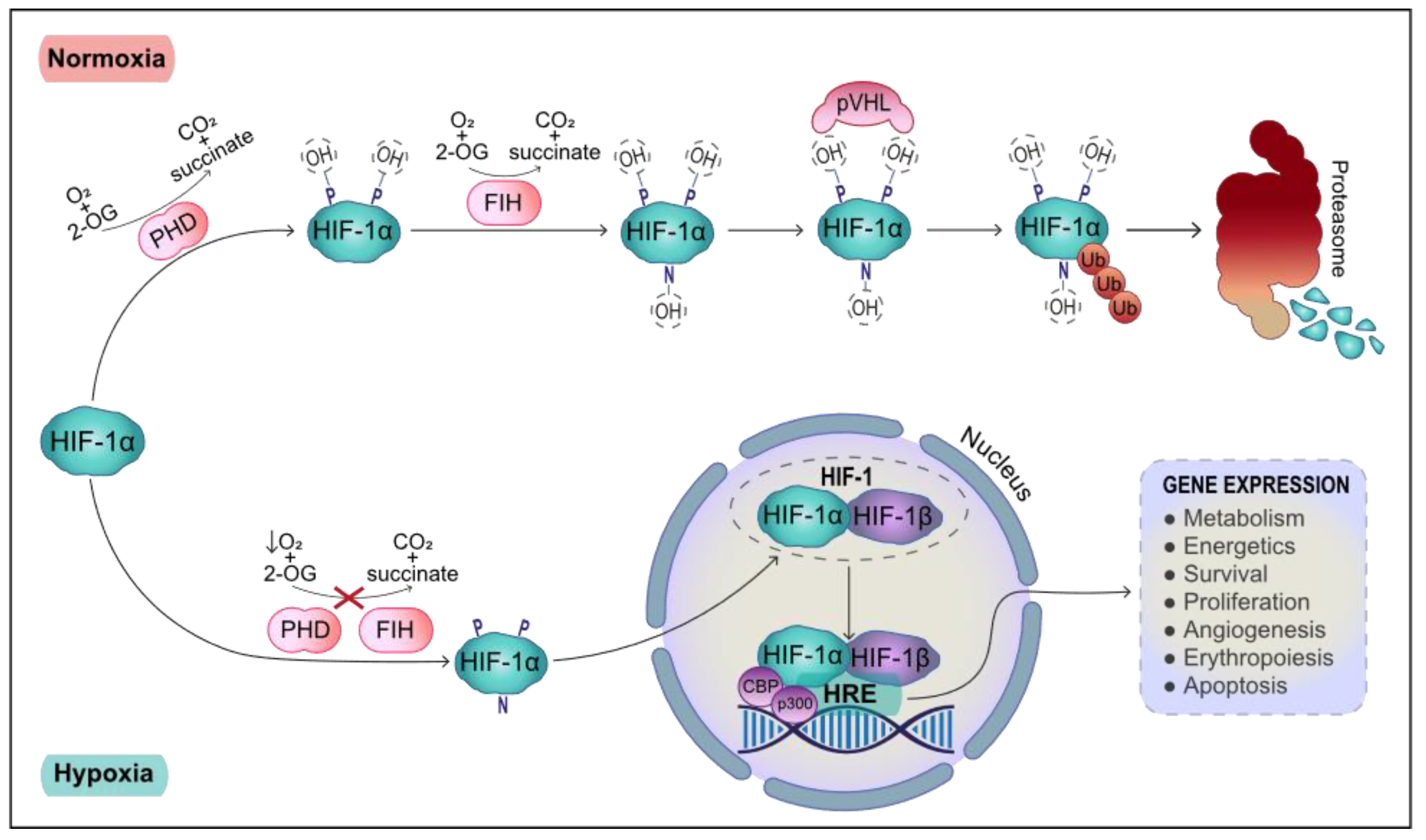
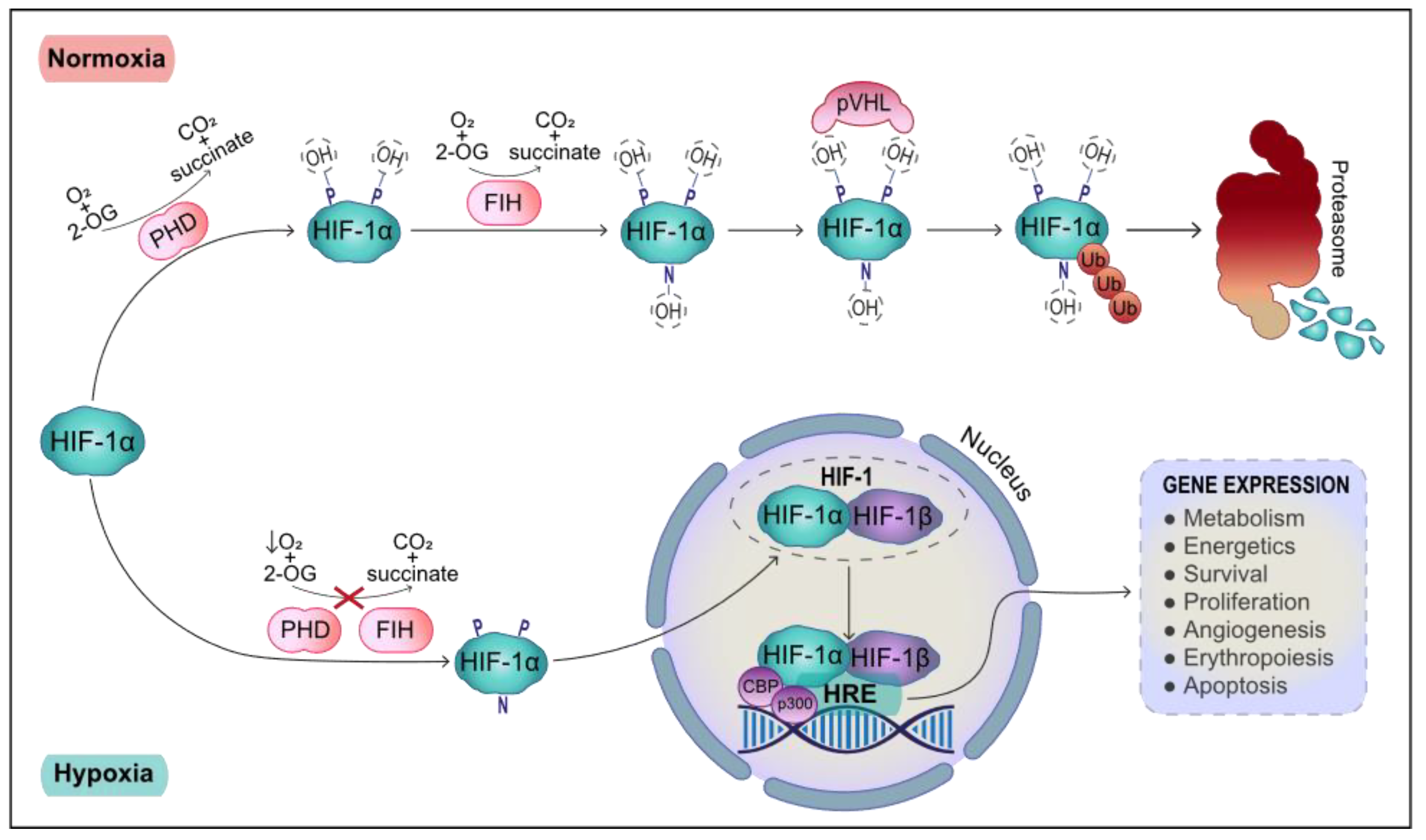
1. Systemic and Cellular Response to Hypoxia
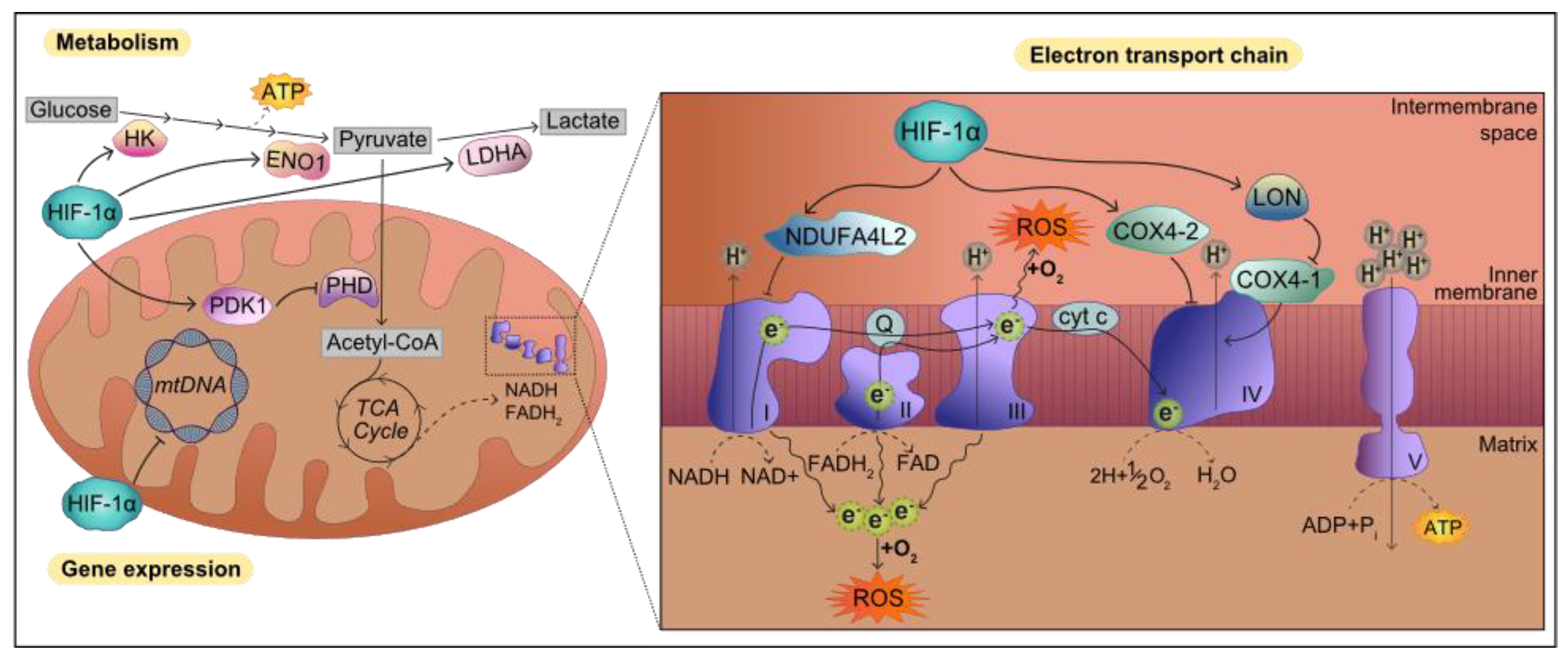
2. Transcriptional Response to Hypoxia: HIF-1 Target Genes
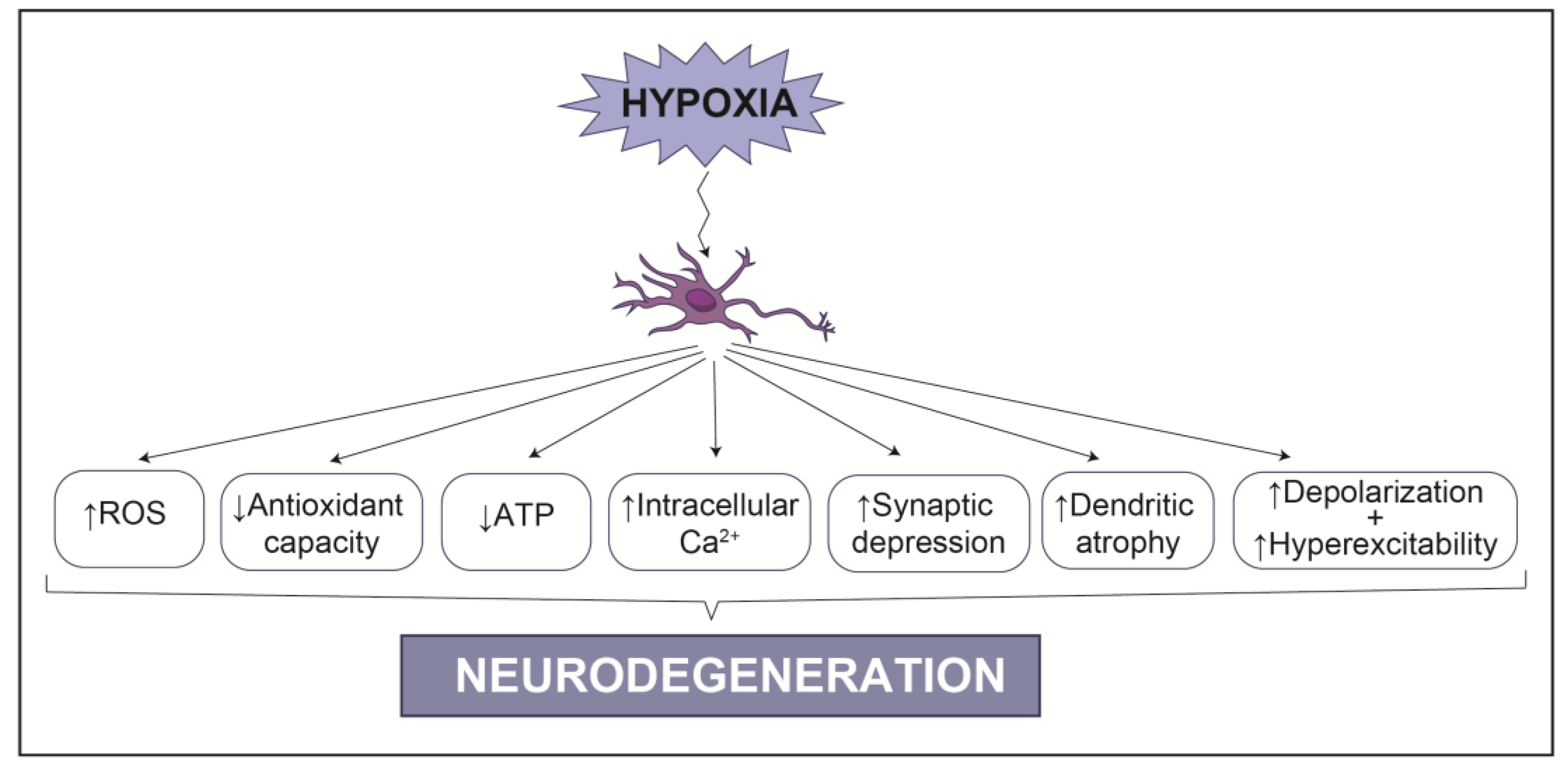
3. Hypoxia in the Nervous System: Links to Neurodegeneration
4. Parkinson’s Disease
5. Crosstalk between Hypoxia, HIF-1α and PD Related Genes
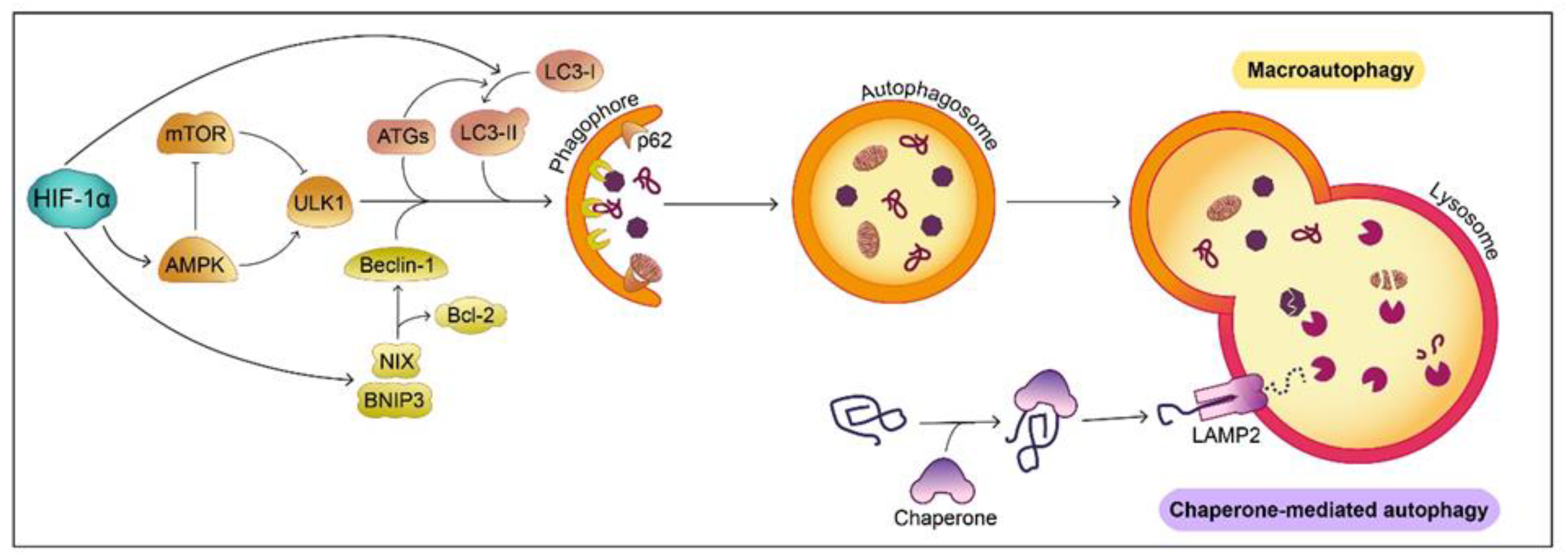
6. Hypoxia and HIF-1α Signaling in Pathways Linked to PD
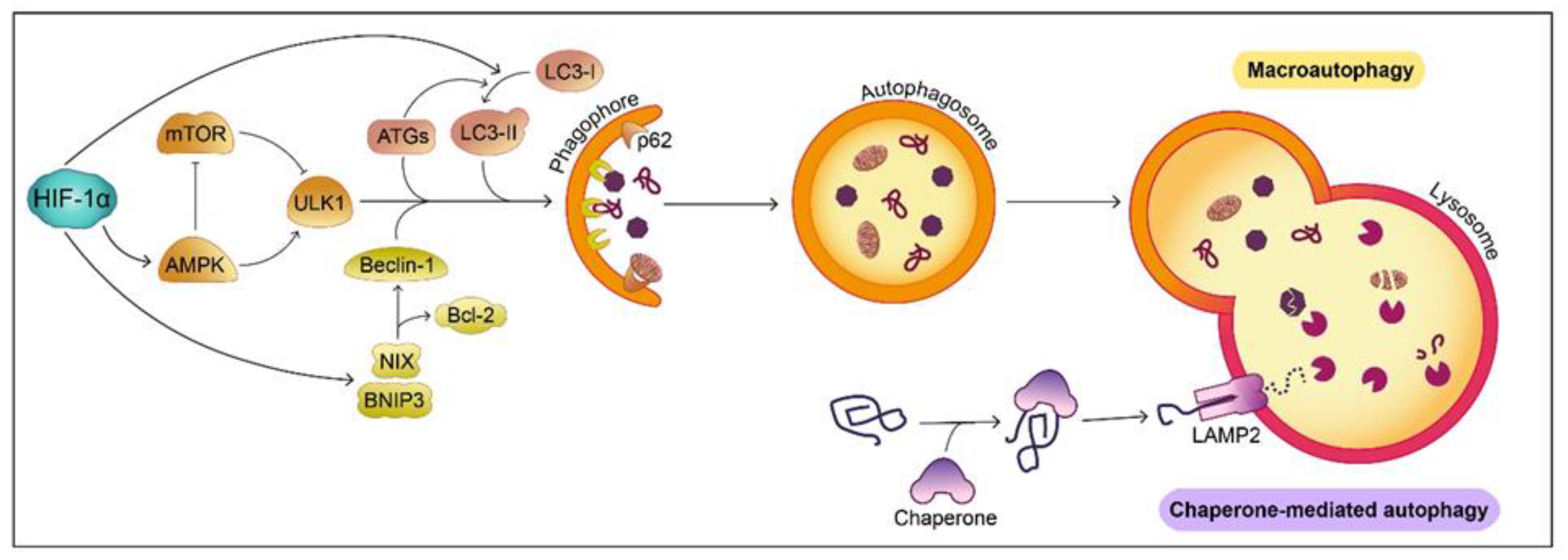
6.1. Protein Degradation
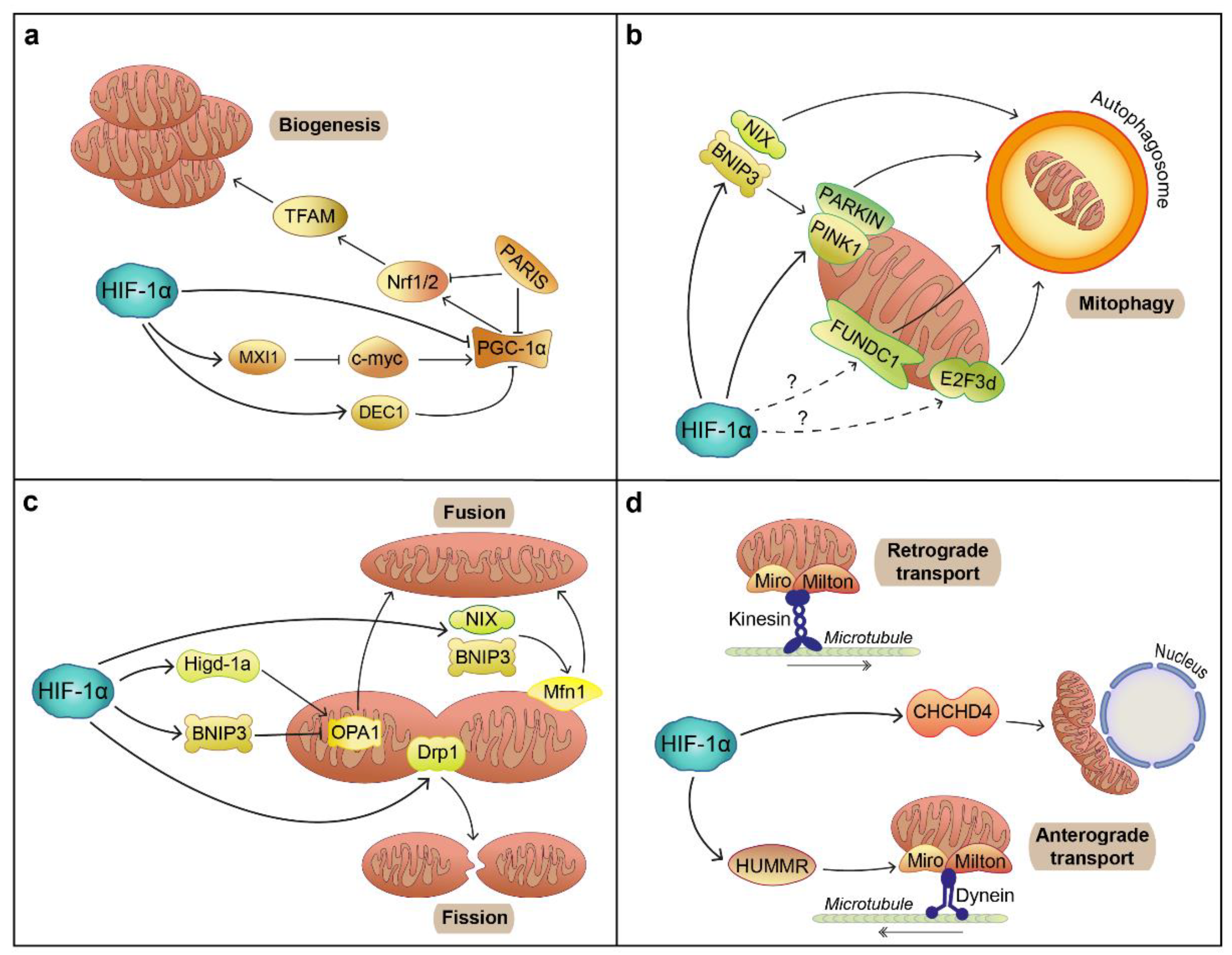
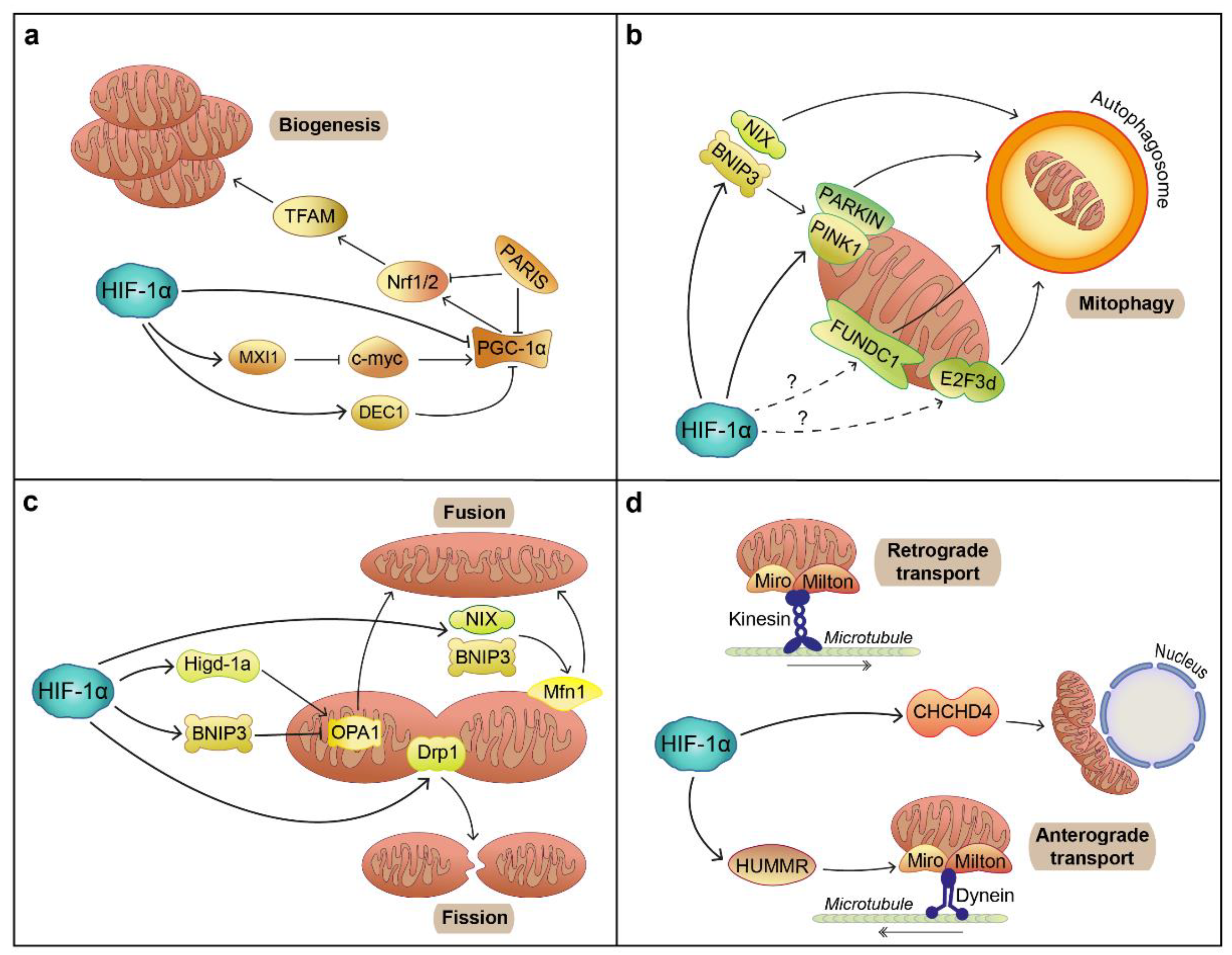
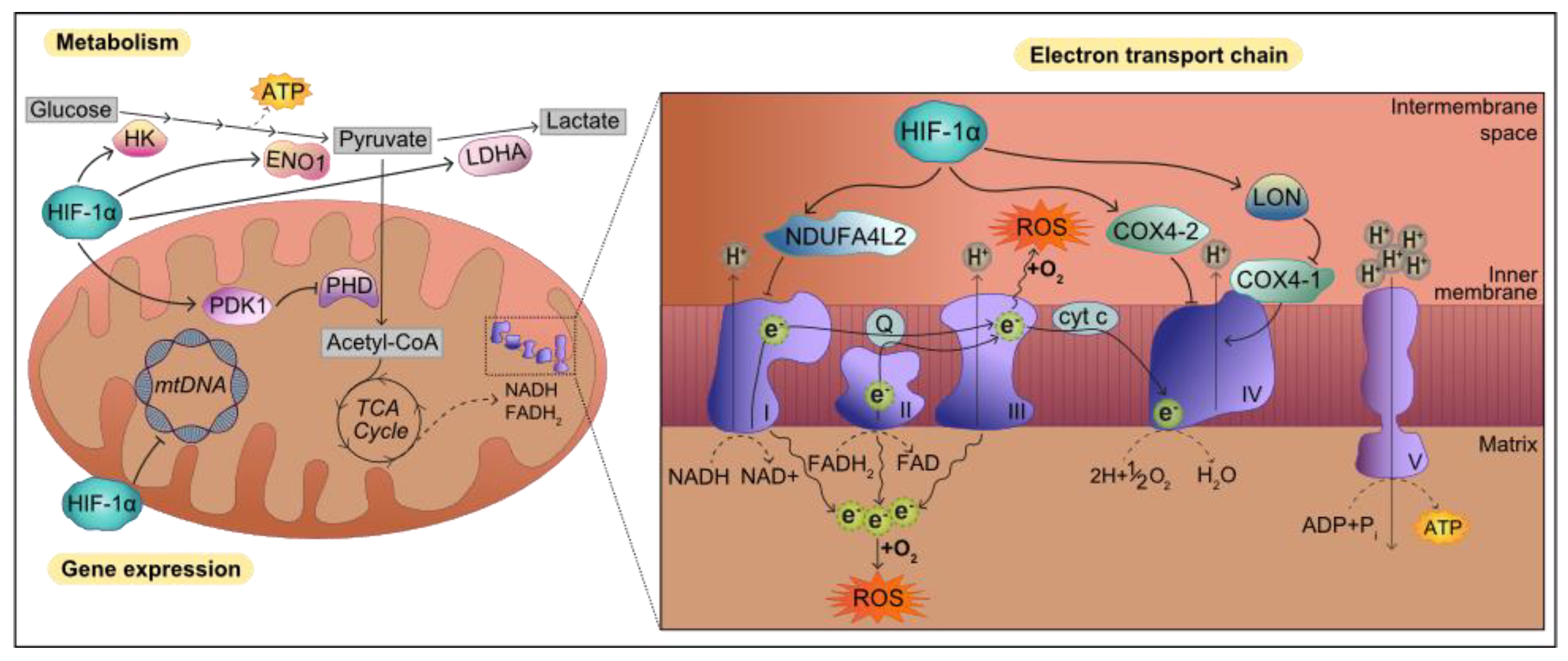
6.2. Mitochondrial Function
6.3. Oxidative Stress
7. PD Risk Factors and Hypoxic Stress
8. Evidence of Hypoxic Injury in the PD Brain
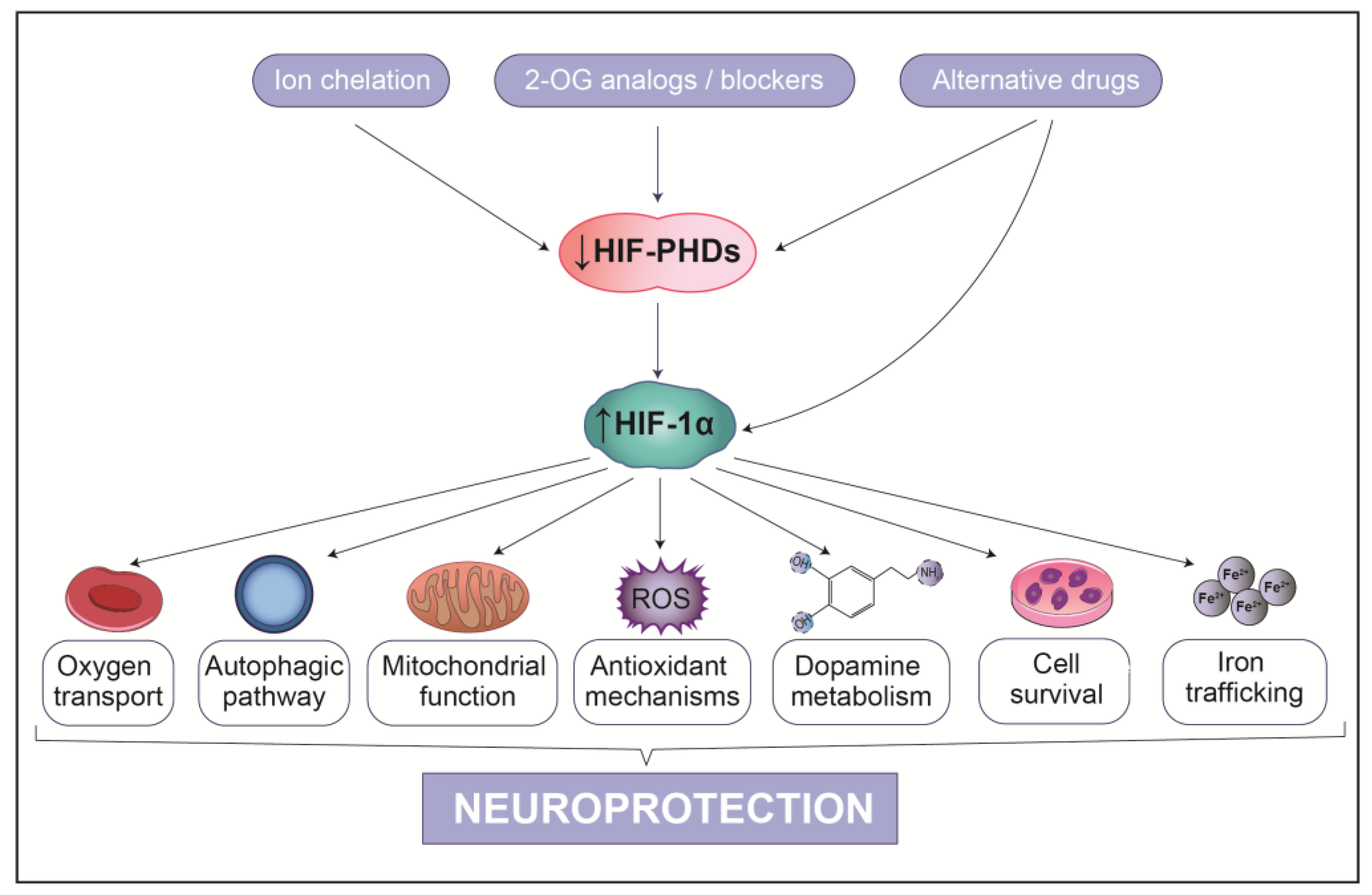
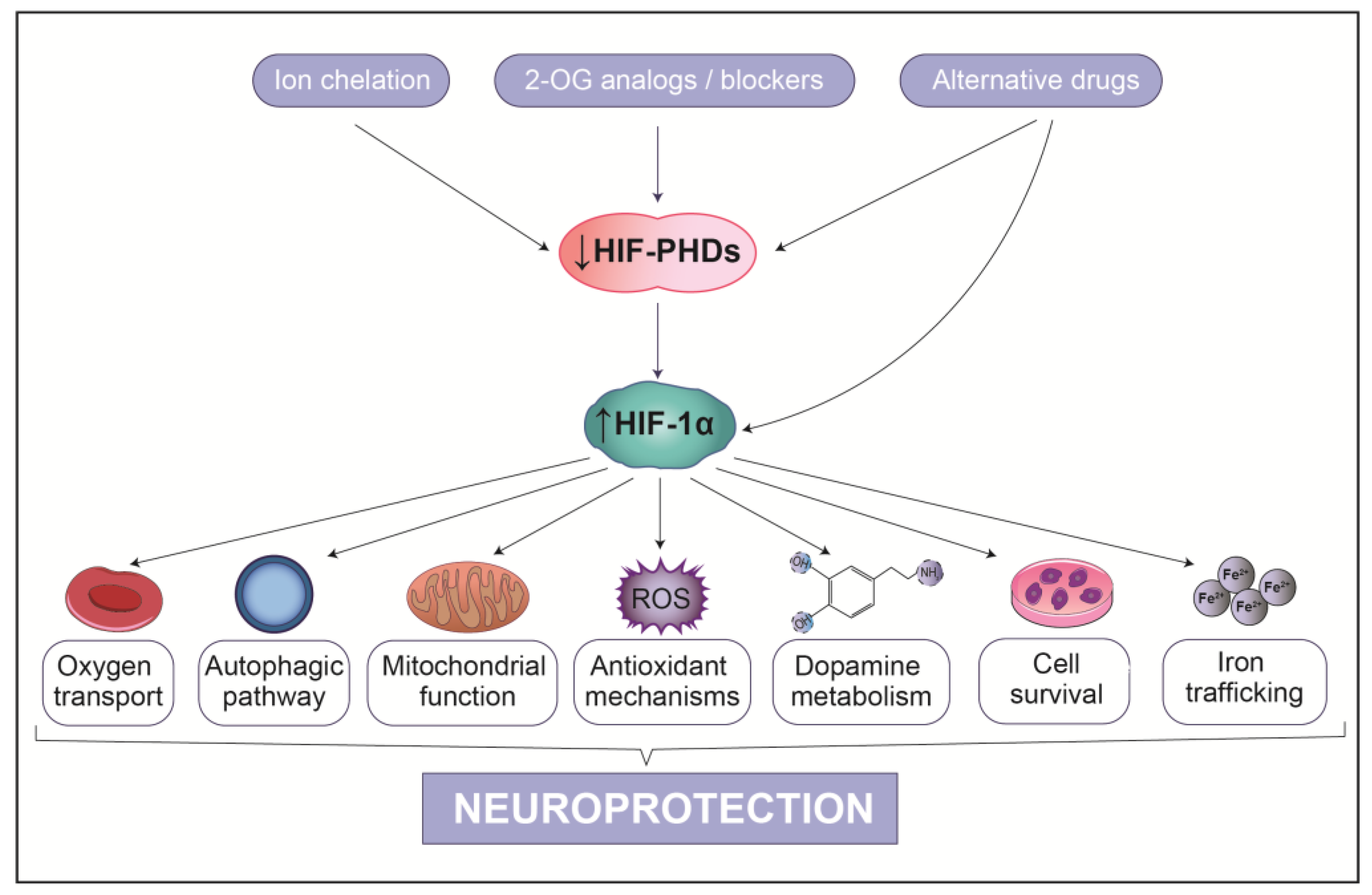
9. HIF-1α-Based Therapeutic Strategies for PD
9.1. Indirect PHD Inhibitors
9.2. Competitive PHD Inhibitors
9.3. Atypical HIF-1α Inducers
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López-Barneo, J.; González-Rodríguez, P.; Gao, L.; Fernández-Agüera, M.C.; Pardal, R.; Ortega-Sáenz, P. Oxygen sensing by the carotid body: Mechanisms and role in adaptation to hypoxia. Am. J. Physiol. Cell Physiol. 2016, 310, C629–C642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutz, E.; Pan, J.; Yeger, H.; Domnik, N.J.; Fisher, J.T. Recent advances and controversies on the role of pulmonary neuroepithelial bodies as airway sensors. Semin. Cell Dev. Biol. 2013, 24, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Robbins, P.A.; Ratcliffe, P.J. The human side of hypoxia-inducible factor. Br. J. Haematol. 2008, 141, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA Binding, and Transactivation Properties of Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of Hypoxia-Inducible Factor 1α is Mediated by an O2-dependent Degradation Domain via the Ubiquitin-Proteasome Pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Ratcliffe, P.J.; Willam, C.; Pugh, C.W.; Masson, N. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef] [Green Version]
- Epstein, A.C.R.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Benita, Y.; Kikuchi, H.; Smith, A.D.; Zhang, M.Q.; Chung, D.C.; Xavier, R.J. An integrative genomics approach identifies Hypoxia Inducible Factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009, 37, 4587–4602. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. How Cells Obtain Energy from Food. In Molecular Biology of the Cell, 4th ed.; Garland Science: Oxford, UK, 2002. [Google Scholar]
- Ebert, B.L.; Gleadle, J.M.; O’Rourke, J.F.; Bartlett, S.M.; Poulton, J.; Ratcliffe, P.J. Isoenzyme-specific regulation of genes involved in energy metabolism by hypoxia: Similarities with the regulation of erythropoietin. Biochem. J. 1996, 313 Pt 3, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mole, D.R.; Schlemminger, I.; McNeill, L.A.; Hewitson, K.S.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. 2-Oxoglutarate analogue inhibitors of hif prolyl hydroxylase. Bioorg. Med. Chem. Lett. 2003, 13, 2677–2680. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.; Shimoda, L.; Dang, C.V.; Semenza, G. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Semenza, G. Molecular basis of hypoxia-induced erythropoietin expression. Curr. Opin. Hematol. 1996, 3, 156–162. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, M.; Liu, L.X.; Date, T.; Belanger, A.J.; Vincent, K.A.; Akita, G.Y.; Kuriyama, T.; Cheng, S.H.; Gregory, R.J.; Jiang, C. Hypoxia-Inducible Factor-1 Mediates Activation of Cultured Vascular Endothelial Cells by Inducing Multiple Angiogenic Factors. Circ. Res. 2003, 93, 664–673. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.J.; Jiang, B.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.K. Hypoxia-inducible Factor-1 Mediates Transcriptional Activation of the Heme Oxygenase-1 Gene in Response to Hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [Green Version]
- Krishnamachary, B.; Berg-Dixon, S.; Kelly, B.; Agani, F.; Feldser, D.; Ferreira, G.; Iyer, N.; LaRusch, J.; Pak, B.; Taghavi, P.; et al. Regulation of Colon Carcinoma Cell Invasion by Hypoxia-Inducible Factor 1. Cancer Res. 2003, 63, 1138. [Google Scholar] [PubMed]
- Rolfs, A.; Kvietikova, I.; Gassmann, M.; Wenger, R.H. Oxygen-regulated Transferrin Expression Is Mediated by Hypoxia-inducible Factor-1. J. Biol. Chem. 1997, 272, 20055–20062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, I.; Huang, L.E.; Koshiji, M.; Pete, E.A.; Kageyama, Y.; Barrett, J.C. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent Regulation of Hypoxic Induction of the Cell Death Factors BNIP3 and NIX in Human Tumors. Cancer Res. 2001, 61, 6669. [Google Scholar] [PubMed]
- Kim, J.; Ahn, H.; Ryu, J.; Suk, K.; Park, J. BH3-only Protein Noxa Is a Mediator of Hypoxic Cell Death Induced by Hypoxia-inducible Factor 1α. J. Exp. Med. 2004, 199, 113–124. [Google Scholar] [CrossRef]
- Erecińska, M.; Silver, I.A. Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 2001, 128, 263–276. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.N.; Klein-Flügge, M.C.; Howarth, C.; Attwell, D. Oxidative Phosphorylation, Not Glycolysis, Powers Presynaptic and Postsynaptic Mechanisms Underlying Brain Information Processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef]
- Nieber, K. Hypoxia and Neuronal Function under in Vitro Conditions. Pharmacol. Ther. 1999, 82, 71–86. [Google Scholar] [CrossRef]
- Lall, R.; Mohammed, R.; Ojha, U. What are the links between hypoxia and Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 2019, 15, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, H.; Lee, J.; Park, K.S.; Jeon, G.S.; Shon, J.; Ahn, S.; Kim, S.H.; Lee, K.M.; Sung, J.; et al. Intermittent hypoxia can aggravate motor neuronal loss and cognitive dysfunction in ALS mice. PLoS ONE 2013, 8, e81808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrowski, R.P.; Zhang, J.H. The insights into molecular pathways of hypoxia-inducible factor in the brain. J. Neurosci. Res. 2020, 98, 57–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yan, J.; Chang, Y.; ShiDu Yan, S.; Shi, H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem. 2011, 18, 4335–4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tysnes, O.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Mehan, S.; Singh, S. Understanding multifactorial architecture of Parkinson’s disease: Pathophysiology to management. Neurol. Sci. 2019, 40, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Karimi-Moghadam, A.; Charsouei, S.; Bell, B.; Jabalameli, M. Parkinson Disease from Mendelian Forms to Genetic Susceptibility: New Molecular Insights into the Neurodegeneration Process. Cell. Mol. Neurobiol. 2018, 38, 1153–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Benskey, M.J.; Perez, R.G.; Manfredsson, F.P. The contribution of alpha synuclein to neuronal survival and function—Implications for Parkinson’s disease. J. Neurochem. 2016, 137, 331–359. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Chen, T.; Wang, Q.; Wang, Q.; Chao, D.; Chao, D.; Xia, T.; Xia, T.; Sheng, S.; Sheng, S.; et al. δ-Opioid Receptor Activation Attenuates the Oligomer Formation Induced by Hypoxia and/or α-Synuclein Overexpression/Mutation Through Dual Signaling Pathways. Mol. Neurobiol. 2019, 56, 3463–3475. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; He, X.; Zhang, Q.; Zhang, N.; Li, L.; Hui, Y.; Li, W.; Wang, D.; Wu, Z. α-synuclein induces microglial cell migration through stimulating HIF-1α accumulation. J. Neurosci. Res. 2017, 95, 1809–1817. [Google Scholar] [CrossRef]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s Disease-Associated Mutations in Leucine-Rich Repeat Kinase 2 Augment Kinase Activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Li, C.; Xing, Z.; Hu, Q.; Liang, K.; Han, L.; Wang, C.; Hawke, D.H.; Wang, S.; Zhang, Y.; et al. The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat. Cell Biol. 2016, 18, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Joo, H.; Bae, J.; Hyeon, S.J.; Her, S.; Ko, E.; Choi, H.G.; Ryu, H.; Hur, E.; Bu, Y.; et al. Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Dis. 2018, 9, 1125. [Google Scholar] [CrossRef]
- van Veen, S.; Sørensen, D.M.; Holemans, T.; Holen, H.W.; Palmgren, M.G.; Vangheluwe, P. Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front. Mol. Neurosci. 2014, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Al-Din, A.; Hillmer, A.M.; Gründemann, J.; Goebel, I.; Woods, C.G.; Stiller, B.; Wriekat, A.; Heimbach, A.; Mubaidin, A.F.; Behrens, M.I.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Xu, Q.; Guo, H.; Zhang, X.; Tang, B.; Cai, F.; Zhou, W.; Song, W. Hypoxia regulation of ATP13A2 (PARK9) gene transcription. J. Neurochem. 2012, 122, 251–259. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Rane, A.; Chinta, S.J.; Andersen, J.K. Regulation of ATP13A2 via PHD2-HIF1 Signaling Is Critical for Cellular Iron Homeostasis: Implications for Parkinson’s Disease. J. Neurosci. 2016, 36, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Sandrelli, F.; Beltramini, M.; Greggio, E.; Bubacco, L.; Bisaglia, M. Recent findings on the physiological function of DJ-1: Beyond Parkinson’s disease. Neurobiol. Dis. 2017, 108, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. DJ-1/PARK7 Is an Important Mediator of Hypoxia-Induced Cellular Responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Sheng, C.; Heng, X.; Zhang, G.; Xiong, R.; Li, H.; Zhang, S.; Chen, S. DJ-1 deficiency perturbs microtubule dynamics and impairs striatal neurite outgrowth. Neurobiol. Aging 2013, 34, 489–498. [Google Scholar] [CrossRef]
- Parsanejad, M.; Zhang, Y.; Qu, D.; Irrcher, I.; Rousseaux, M.W.C.; Aleyasin, H.; Kamkar, F.; Callaghan, S.; Slack, R.S.; Mak, T.W.; et al. Regulation of the VHL/HIF-1 Pathway by DJ-1. J. Neurosci. 2014, 34, 8043–8050. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Zhou, C.; Lu, X.; Liu, Q.; Liu, M.; Chen, G.; Chen, W.; Wang, S.; Qiu, Y. DJ-1 promotes survival of human colon cancer cells under hypoxia by modulating HIF-1α expression through the PI3K-AKT pathway. Cancer Manag. Res. 2018, 10, 4615–4629. [Google Scholar] [CrossRef] [PubMed]
- De Lazzari, F.; Prag, H.A.; Gruszczyk, A.V.; Whitworth, A.J.; Bisaglia, M. DJ-1: A promising therapeutic candidate for ischemia-reperfusion injury. Redox Biol. 2021, 41, 101884. [Google Scholar] [CrossRef]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Jimenez-Blasco, D.; Fernandez, E.; Almeida, A.; Bolaños, J.P. DJ1 represses glycolysis and cell proliferation by transcriptionally upregulating PINK1. Biochem. J. 2015, 467, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deas, E.; Plun-Favreau, H.; Wood, N.W. PINK1 function in health and disease. EMBO Mol. Med. 2009, 1, 152–165. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Fernandez, E.; Martins, L.M.; Almeida, A.; Bolaños, J.P. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun. 2014, 5, 4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Wadlington, N.L.; Chen, L.; Zhuang, X.; Brorson, J.R.; Kang, U.J. Loss of PINK1 attenuates HIF-1α induction by preventing 4E-BP1-dependent switch in protein translation under hypoxia. J. Neurosci. 2014, 34, 3079–3089. [Google Scholar] [CrossRef] [Green Version]
- Kung-Chun Chiu, D.; Pui-Wah Tse, A.; Law, C.; Ming-Jing Xu, I.; Lee, D.; Chen, M.; Kit-Ho Lai, R.; Wai-Hin Yuen, V.; Wing-Sum Cheu, J.; Wai-Hung Ho, D.; et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Mizuno, Y.; Yamamura, Y.; Hattori, N.; Matsumine, H.; Yokochi, M.; Asakawa, S.; Shimizu, N.; Minoshima, S. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Shen, J.; Zhang, S.; Liu, A.; Chen, X.; Wang, Y.; Sun, J.; Dai, S.; Xu, J. Inactivating the ubiquitin ligase Parkin suppresses cell proliferation and induces apoptosis in human keloids. J. Cell. Physiol. 2019, 234, 16601–16608. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Torii, S.; Kakita, A.; Sogawa, K. Inhibitory PAS domain protein is a substrate of PINK1 and Parkin and mediates cell death in a Parkinson’s disease model. Cell Death Dis. 2015, 6, e1886. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, G.; D’Amico, A.; Reitano, R.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. Parkin modulates expression of HIF-1α and HIF-3α during hypoxia in gliobastoma-derived cell lines in vitro. Cell Tissue Res. 2016, 364, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Malkus, K.A.; Tsika, E.; Ischiropoulos, H. Oxidative modifications, mitochondrial dysfunction, and impaired protein degradation in Parkinson’s disease: How neurons are lost in the Bermuda triangle. Mol. Neurodegener. 2009, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakis, N.; Xilouri, M.; Koros, C.; Simitsi, A.; Stamelou, M.; Maniati, M.; Stefanis, L. Autophagy dysfunction in peripheral blood mononuclear cells of Parkinson’s disease patients. Neurosci. Lett. 2019, 704, 112–115. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25. [Google Scholar] [PubMed]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted Autophagy Leads to Dopaminergic Axon and Dendrite Degeneration and Promotes Presynaptic Accumulation of α-Synuclein and LRRK2 in the Brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winslow, A.R.; Chen, C.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Exp. Med. 2010, 207, i29. [Google Scholar] [CrossRef]
- Michiorri, S.; Gelmetti, V.; Giarda, E.; Lombardi, F.; Romano, F.; Marongiu, R.; Nerini-Molteni, S.; Sale, P.; Vago, R.; Arena, G.; et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010, 17, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced Basal Autophagy and Impaired Mitochondrial Dynamics Due to Loss of Parkinson’s Disease-Associated Protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orenstein, S.J.; Kuo, S.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanese, F.; Novello, S.; Morari, M. Autophagy and LRRK2 in the Aging Brain. Front. Neurosci. 2019, 13, 1352. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Ramírez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bézard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [Green Version]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef]
- Fowler, A.J.; Moussa, C.E. Activating Autophagy as a Therapeutic Strategy for Parkinson’s Disease. CNS Drugs 2018, 32, 1–11. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, L.; Liu, B.; Lv, G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp. Ther. Med. 2016, 11, 2233–2239. [Google Scholar] [CrossRef] [Green Version]
- Bohensky, J.; Leshinsky, S.; Srinivas, V.; Shapiro, I. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 2010, 25, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, C.; Jiang, X.; Li, L.; Zhang, D.; Tang, D.; Yan, T.; Zhang, Q.; Yuan, H.; Jia, J.; et al. Involvement of autophagy in hypoxia-BNIP3 signaling to promote epidermal keratinocyte migration. Cell Death Dis. 2019, 10, 234. [Google Scholar] [CrossRef] [Green Version]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef]
- Koike, M.; Tanida, I.; Ueno, T.; Iwata, J.; Chiba, T.; Tanaka, K.; Uchiyama, Y.; Waguri, S.; Komatsu, M.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2009, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia–ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Hao, L.; Guo, Y.; Yang, G.; Mei, H.; Li, X.; Zhai, Q. Chloroquine inhibits autophagy and deteriorates the mitochondrial dysfunction and apoptosis in hypoxic rat neurons. Life Sci. 2018, 202, 70–77. [Google Scholar] [CrossRef]
- Chu, C.T. Eaten Alive: Autophagy and Neuronal Cell Death after Hypoxia-Ischemia. Am. J. Pathol. 2008, 172, 284–287. [Google Scholar] [CrossRef]
- Koike, M.; Shibata, M.; Tadakoshi, M.; Gotoh, K.; Komatsu, M.; Waguri, S.; Kawahara, N.; Kuida, K.; Nagata, S.; Kominami, E.; et al. Inhibition of Autophagy Prevents Hippocampal Pyramidal Neuron Death after Hypoxic-Ischemic Injury. Am. J. Pathol. 2008, 172, 454–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaught, K.S.P.; Olanow, C.W.; Halliwell, B.; Isacson, O.; Jenner, P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 589–594. [Google Scholar] [CrossRef]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shao, M.; Guo, B.; Li, C.; Lu, Y.; Yang, X.; Li, S.; Li, H.; Zhu, Q.; Zhong, H.; et al. Tetramethylpyrazine Analogue T-006 Promotes the Clearance of Alpha-synuclein by Enhancing Proteasome Activity in Parkinson’s Disease Models. Neurotherapeutics 2019, 16, 1225–1236. [Google Scholar] [CrossRef]
- Li, T.; Feng, Y.; Yang, R.; Wu, L.; Li, R.; Huang, L.; Yang, Q.; Chen, J. Salidroside Promotes the Pathological α-Synuclein Clearance through Ubiquitin-Proteasome System in SH-SY5Y Cells. Front. Pharmacol. 2018, 9, 377. [Google Scholar] [CrossRef] [Green Version]
- Um, J.W.; Im, E.; Lee, H.J.; Min, B.; Yoo, L.; Yoo, J.; Lübbert, H.; Stichel-Gunkel, C.; Cho, H.; Yoon, J.B.; et al. Parkin Directly Modulates 26S Proteasome Activity. J. Neurosci. 2010, 30, 11805–11814. [Google Scholar] [CrossRef] [Green Version]
- Abu-El-Rub, E.; Sequiera, G.L.; Sareen, N.; Yan, W.; Moudgil, M.; Sabbir, M.G.; Dhingra, S. Hypoxia-induced 26S proteasome dysfunction increases immunogenicity of mesenchymal stem cells. Cell Death Dis. 2019, 10, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Lee, Y.; Stevens, D.A.; Kang, S.; Jiang, H.; Lee, Y.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Acevedo-Cintrón, J.; Jhaldiyal, A.; Wang, H.; Andrabi, S.A.; Eacker, S.; Karuppagounder, S.S.; Brahmachari, S.; Chen, R.; Kim, H.; et al. Defects in Mitochondrial Biogenesis Drive Mitochondrial Alterations in PARKIN-Deficient Human Dopamine Neurons. Stem Cell Rep. 2020, 15, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, Q.; Zhang, L.; Fang, Z.; Zhao, F.; Lv, Z.; Gu, Z.; Zhang, J.; Wang, J.; Zen, K.; et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010, 20, 676–687. [Google Scholar] [CrossRef]
- Tohme, S.; Yazdani, H.O.; Liu, Y.; Loughran, P.; Windt, D.J.; Huang, H.; Simmons, R.L.; Shiva, S.; Tai, S.; Tsung, A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth via HMGB1 and TLR9 interaction. Hepatology 2017, 66, 182–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutsaeva, D.R.; Carraway, M.S.; Suliman, H.B.; Demchenko, I.T.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Transient Hypoxia Stimulates Mitochondrial Biogenesis in Brain Subcortex by a Neuronal Nitric Oxide Synthase-Dependent Mechanism. J. Neurosci. 2008, 28, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Foo, S.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hagan, K.A.; Cocchiglia, S.; Zhdanov, A.V.; Tambuwala, M.M.; Cummins, E.P.; Monfared, M.; Agbor, T.A.; Garvey, J.F.; Papkovsky, D.B.; Taylor, C.T.; et al. PGC-1 is coupled to HIF-1-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2188–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaGory, E.; Wu, C.; Taniguchi, C.; Ding, C.; Chi, J.; von Eyben, R.; Scott, D.; Richardson, A.; Giaccia, A. Suppression of PGC-1α Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.; Semenza, G.L. HIF-1 Inhibits Mitochondrial Biogenesis and Cellular Respiration in VHL-Deficient Renal Cell Carcinoma by Repression of C-MYC Activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Lin, Q.; Shao, X.; Zhu, X.; Wu, J.; Wu, B.; Zhang, M.; Zhou, W.; Zhou, Y.; Jin, H.; et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, H.; Peng, J.; Zhu, Y.; Zhang, H.; Tang, F.; Jian, Z.; Xiao, Y. Multinucleated polyploid cardiomyocytes undergo an enhanced adaptability to hypoxia via mitophagy. J. Mol. Cell. Cardiol. 2020, 138, 115–135. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Araki, K.; Kawauchi, K.; Sugimoto, W.; Tsuda, D.; Oda, H.; Yoshida, R.; Ohtani, K. Mitochondrial protein E2F3d, a distinctive E2F3 product, mediates hypoxia-induced mitophagy in cancer cells. Commun. Biol. 2019, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct Membrane Association Drives Mitochondrial Fission by the Parkinson Disease-associated Protein α-Synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [Green Version]
- Dagda, R.K.; Cherra, S.J.; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 Function Promotes Mitophagy through Effects on Oxidative Stress and Mitochondrial Fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of Parkin or PINK1 Function Increases Drp1-dependent Mitochondrial Fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Dorn, G.W. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryde, K.R.; Smith, H.L.; Chau, K.; Schapira, A.H.V. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef]
- Wan, Y.; Zhang, J.; Yang, Z.; Jiang, L.; Wei, Y.; Lai, Q.; Wang, J.; Xin, H.; Han, X.-J. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol. Rep. 2014, 32, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Scimia, M.; Wilkinson, D.; Trelles, R.; Wood, M.; Bowtell, D.; Dillin, A.; Mercola, M.; Ronai, Z. Fine-Tuning of Drp1/Fis1 Availability by AKAP121/Siah2 Regulates Mitochondrial Adaptation to Hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef] [Green Version]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Cho, G.; Lee, J.; Paik, S.; Kim, Y.S.; Lee, H. Higd-1a interacts with Opa1 and is required for the morphological and functional integrity of mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 13014–13019. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Rouleau, M.; Gounon, P.; Brahimi-Horn, M.C.; Pouysségur, J.; Mazure, N.M. Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J. Cell. Physiol. 2010, 222, 648–657. [Google Scholar] [CrossRef]
- Song, P.; Li, S.; Wu, H.; Gao, R.; Rao, G.; Wang, D.; Chen, Z.; Ma, B.; Wang, H.; Sui, N.; et al. Parkin promotes proteasomal degradation of p62: Implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson’s disease. Protein Cell 2016, 7, 114–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Zhang, X.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihofen, A.; Thomas, K.J.; Ostaszewski, B.L.; Cookson, M.R.; Selkoe, D.J. Pink1 Forms a Multiprotein Complex with Miro and Milton, Linking Pink1 Function to Mitochondrial Trafficking. Biochemistry 2009, 48, 2045–2052. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.; Schwarz, T. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St. Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Prots, I.; Grosch, J.; Brazdis, R.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [Green Version]
- Al-Mehdi, A.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear Mitochondrial Clustering Creates an Oxidant-Rich Nuclear Domain Required for Hypoxia-Induced Transcription. Sci. Signal 2012, 5, ra47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.W.; Staples, O.; Turmaine, M.; Ashcroft, M. CHCHD4 Regulates Intracellular Oxygenation and Perinuclear Distribution of Mitochondria. Front. Oncol. 2017, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lim, S.; Hoffman, D.; Aspenstrom, P.; Federoff, H.J.; Rempe, D.A. HUMMR, a Hypoxia- and HIF-1α-Inducible Protein, Alters Mitochondrial Distribution and Transport. J. Cell Biol. 2009, 185, 1065–1081. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Cooper, J.M.; Schapira, A.H.V.; Taanman, J. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 2009, 4, e4756. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.; Leung, S.W.; Passantino, R.; Concordet, J.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhou, Y.; Li, L.; Li, S.; Long, D.; Chen, X.; Zhang, J.; Feng, L.; Li, Y. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species—The good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.; Zhang, C.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Parkinson’s Dis. 2018, 2018, 9163040. [Google Scholar] [CrossRef] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Dolgacheva, L.; Berezhnov, A.; Fedotova, E.; Zinchenko, V.; Abramov, A. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Takahashi-Niki, K.; Niki, T.; Taira, T.; Iguchi-Ariga, S.M.M.; Ariga, H. Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson’s disease patients. Biochem. Biophys. Res. Commun. 2004, 320, 389–397. [Google Scholar] [CrossRef]
- Kim, J.H.; Song, S.; Park, S.G.; Song, S.U.; Xia, Y.; Sung, J. Primary Involvement of NADPH Oxidase 4 in Hypoxia-Induced Generation of Reactive Oxygen Species in Adipose-Derived Stem Cells. Stem Cells Dev. 2012, 21, 2212–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naranjo-Suarez, S.; Carlson, B.A.; Tsuji, P.A.; Yoo, M.; Gladyshev, V.N.; Hatfield, D.L. HIF-Independent Regulation of Thioredoxin Reductase 1 Contributes to the High Levels of Reactive Oxygen Species Induced by Hypoxia. PLoS ONE 2012, 7, e30470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán-Pla, A.; Cervera, A.M.; Apostolova, N.; Garcia-Bou, R.; Víctor, V.M.; Murphy, M.P.; McCreath, K.J. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1α. FEBS Lett. 2005, 579, 2669–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parandavar, E.; Yazdanparast, R. Differential impact of various reactive oxygen species (ROS) on HIF-1α/p53 direct interaction in SK-N-MC neuroblastoma cells. Cell Biosci. 2017, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.; Zucchelli, S.; Biagioli, M.; Roncaglia, P.; Vilotti, S.; Calligaris, R.; Krmac, H.; Girardini, J.E.; Del Sal, G.; Gustincich, S. Parkinson Disease-associated DJ-1 Is Required for the Expression of the Glial Cell Line-derived Neurotrophic Factor Receptor RET in Human Neuroblastoma Cells. J. Biol. Chem. 2010, 285, 18565–18574. [Google Scholar] [CrossRef] [Green Version]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Yeo, E. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Frenkel-Denkberg, G.; Gershon, D.; Levy, A.P. The function of hypoxia-inducible factor 1 (HIF-1) is impaired in senescent mice. FEBS Lett. 1999, 462, 341–344. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, D.; Choi, J.; Kim, J.; Park, S.C.; Youn, H. Analysis of the effect of aging on the response to hypoxia by cDNA microarray. Mech. Ageing Dev. 2003, 124, 941–949. [Google Scholar] [CrossRef]
- Hoenig, M.R.; Bianchi, C.; Rosenzweig, A.; Sellke, F.W. Decreased Vascular Repair and Neovascularization with Ageing: Mechanisms and Clinical Relevance with an Emphasis on Hypoxia-Inducible Factor-1. Curr. Mol. Med. 2008, 8, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Sandhir, R.; Hamilton, E.S.; Berman, N.E.J. Impaired Expression of Neuroprotective Molecules in the HIF-1α Pathway following Traumatic Brain Injury in Aged Mice. J. Neurotrauma 2009, 26, 1557–1566. [Google Scholar] [CrossRef]
- Ndubuizu, O.I.; Chavez, J.C.; LaManna, J.C. Increased prolyl 4-hydroxylase expression and differential regulation of hypoxia-inducible factors in the aged rat brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.; Steinkraus, K.A.; Sutphin, G.L.; Ramos, F.J.; Shamieh, L.S.; Huh, A.; Davis, C.; Chandler-Brown, D.; Kaeberlein, M. Proteasomal Regulation of the Hypoxic Response Modulates Aging in C. elegans. Science 2009, 324, 1196–1198. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Nakada, T.; Kyo, S.; Inoue, M.; Shay, J.W.; Isaka, K. Hypoxia-Inducible Factor 1 Mediates Upregulation of Telomerase (hTERT). Mol. Cell. Biol. 2004, 24, 6076–6083. [Google Scholar] [CrossRef] [Green Version]
- Hatcher, J.M.; Pennell, K.D.; Miller, G.W. Parkinson’s disease and pesticides: A toxicological perspective. Trends Pharmacol. Sci. 2008, 29, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Tan, J.; Wan, Z.; Zou, Y.; Afewerky, H.K.; Zhang, Z.; Zhang, T. Effects of Commonly Used Pesticides in China on the Mitochondria and Ubiquitin-Proteasome System in Parkinson’s Disease. Int. J. Mol. Sci. 2017, 18, 2507. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, A.; Liu, N.; Zhang, Y.; Zhao, H.; Liu, X. The relationship between obstructive sleep apnea and Parkinson’s disease: A systematic review and meta-analysis. Neurol. Sci. 2020, 41, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sun, B.; Chen, D.; Chen, Y.; Li, W.; Xu, M.; Shen, Y.; Xu, Z.; Wang, Y.; Bu, X. Plasma α-synuclein levels are increased in patients with obstructive sleep apnea syndrome. Ann. Clin. Transl. Neurol. 2019, 6, 788–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, B.; Shell, B.; Cunningham, J.T.; Cunningham, R.L. Chronic intermittent hypoxia induces oxidative stress and inflammation in brain regions associated with early-stage neurodegeneration. Physiol. Rep. 2017, 5, e13258. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Byers, A.; Barnes, D.; Li, Y.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology 2018, 90, e1771–e1779. [Google Scholar] [CrossRef]
- Uryu, K.; Chen, X.; Martinez, D.; Browne, K.D.; Johnson, V.E.; Graham, D.I.; Lee, V.M.-Y.; Trojanowski, J.Q.; Smith, D.H. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 2007, 208, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondello, S.; Buki, A.; Italiano, D.; Jeromin, A. α-Synuclein in CSF of patients with severe traumatic brain injury. Neurology 2013, 80, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Onodera, H.; Okabe, S.; Kikuchi, Y.; Tsuda, T.; Itoyama, Y. Impaired chemosensitivity and perception of dyspnoea in Parkinson’s disease. Lancet 2000, 356, 739–740. [Google Scholar] [CrossRef]
- Lin, W.; Chen, P.; Huang, C.; Tsai, N.; Chen, H.; Wang, H.; Chou, K.; Chen, M.; Chen, Y.; Lu, C. Autonomic Function Impairment and Brain Perfusion Deficit in Parkinson’s Disease. Front. Neurol. 2017, 8, 246. [Google Scholar] [CrossRef] [Green Version]
- Baille, G.; Perez, T.; Devos, D.; Deken, V.; Defebvre, L.; Moreau, C. Early occurrence of inspiratory muscle weakness in Parkinson’s disease. PLoS ONE 2018, 13, e0190400. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Burke, R.E.; Macaya, A.; DeVivo, D.; Kenyon, N.; Janec, E.M. Neonatal hypoxic-ischemic or excitotoxic striatal injury results in a decreased adult number of substantia nigra neurons. Neuroscience 1992, 50, 559–569. [Google Scholar] [CrossRef]
- Oo, T.F.; Henchcliffe, C.; Burke, R.E. Apoptosis in substantia nigra following developmental hypoxic-ischemic injury. Neuroscience 1995, 69, 893–901. [Google Scholar] [CrossRef]
- Milosevic, J.; Maisel, M.; Wegner, F.; Leuchtenberger, J.; Wenger, R.H.; Gerlach, M.; Storch, A.; Schwarz, J. Lack of hypoxia-inducible factor-1 alpha impairs midbrain neural precursor cells involving vascular endothelial growth factor signaling. J. Neurosci. 2007, 27, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Kim, H.; Bang, Y.; Seol, W.; Choi, H.; Choi, H. Hypoxia-inducible factor-1α upregulates tyrosine hydroxylase and dopamine transporter by nuclear receptor ERRγ in SH-SY5Y cells. NeuroReport 2015, 26, 380–386. [Google Scholar] [CrossRef]
- Grünblatt, E.; Mandel, S.; Jacob-Hirsch, J.; Zeligson, S.; Amariglo, N.; Rechavi, G.; Li, J.; Ravid, R.; Roggendorf, W.; Riederer, P.; et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural Transm. 2004, 111, 1543–1573. [Google Scholar] [CrossRef]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.M.; Lockner, R.J.; Palaniappan, C.; Federoff, H.J. Robust dysregulation of gene expression in substantia nigra and striatum in Parkinson’s disease. Neurobiol. Dis. 2006, 21, 305–313. [Google Scholar] [CrossRef]
- Elstner, M.; Morris, C.M.; Heim, K.; Bender, A.; Mehta, D.; Jaros, E.; Klopstock, T.; Meitinger, T.; Turnbull, D.M.; Prokisch, H. Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 2011, 122, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, J.; Rogers, J.; Xie, J. Brain Iron Metabolism Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 3078–3101. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Nizet, V.; Johnson, R.S. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008, 7, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Bergström, A.; Fog, K.; Sager, T.N.; Bruun, A.T.; Thirstrup, K. Competitive HIF Prolyl Hydroxylase Inhibitors Show Protection against Oxidative Stress by a Mechanism Partially Dependent on Glycolysis. ISRN Neurosci. 2012, 2013, 598587. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, J.; Priyam, A.; Gowrishetty, K.K.; Mishra, S.; Bandyopadhyay, J. Iron chelator Deferoxamine protects human neuroblastoma cell line SH-SY5Y from 6-Hydroxydopamine-induced apoptosis and autophagy dysfunction. J. Trace Elem. Med. Biol. 2020, 57, 126406. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Xie, W.; Jankovic, J.; Le, W.; Pan, T. Neuroprotection of deferoxamine on rotenone-induced injury via accumulation of HIF-1α and induction of autophagy in SH-SY5Y cells. Neurochem. Int. 2010, 57, 198–205. [Google Scholar] [CrossRef]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural Transm. 2011, 118, 223–231. [Google Scholar] [CrossRef]
- Fine, J.M.; Forsberg, A.C.; Renner, D.B.; Faltesek, K.A.; Mohan, K.G.; Wong, J.C.; Arneson, L.C.; Crow, J.M.; Frey, W.H.; Hanson, L.R. Intranasally-administered deferoxamine mitigates toxicity of 6-OHDA in a rat model of Parkinson’s disease. Brain Res. 2014, 1574, 96–104. [Google Scholar] [CrossRef]
- Febbraro, F.; Andersen, K.J.; Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. Chronic intranasal deferoxamine ameliorates motor defects and pathology in the α-synuclein rAAV Parkinson’s model. Exp. Neurol. 2013, 247, 45–58. [Google Scholar] [CrossRef]
- Guo, C.; Hao, L.; Yang, Z.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.; Zhong, M.; Wang, T.; Li, J.; et al. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 2016, 280, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Gal, S.; Gal, S.; Zheng, H.; Zheng, H.; Fridkin, M.; Fridkin, M.; Youdim, M.; Youdim, M. Restoration of Nigrostriatal Dopamine Neurons in Post-MPTP Treatment by the Novel Multifunctional Brain-Permeable Iron Chelator-Monoamine Oxidase Inhibitor Drug, M30. Neurotox Res. 2010, 17, 15–27. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Weinreb, O.; Amit, T.; Mandel, S.; Bar-Am, O.; Youdim, M.B.H. Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 2011, 189, 345–358. [Google Scholar] [CrossRef]
- Lee, D.W.; Rajagopalan, S.; Siddiq, A.; Gwiazda, R.; Yang, L.; Beal, M.F.; Ratan, R.R.; Andersen, J.K. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: Model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson disease. J. Biol. Chem. 2009, 284, 29065–29076. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, D.I.; Hare, D.J.; Billings, J.L.; Sedjahtera, A.; Nurjono, M.; Arthofer, E.; George, S.; Culvenor, J.G.; Bush, A.I.; Adlard, P.A. Clioquinol Improves Cognitive, Motor Function, and Microanatomy of the Alpha-Synuclein hA53T Transgenic Mice. ACS Chem. Neurosci. 2015, 7, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, W.; Zeng, W.; Cheng, A.; Shi, R.; Zhengli, C. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, Y.; Wang, S.; Pang, Z.; Fan, Y.; Li, J.; Wang, Z.; Guo, C. Lactoferrin ameliorates dopaminergic neurodegeneration and motor deficits in MPTP-treated mice. Redox Biol. 2019, 21, 101090. [Google Scholar] [CrossRef]
- Witten, L.; Sager, T.; Thirstrup, K.; Johansen, J.L.; Larsen, D.B.; Montezinho, L.P.; Mørk, A. HIF prolyl hydroxylase inhibition augments dopamine release in the rat brain in vivo. J. Neurosci. Res. 2009, 87, 1686–1694. [Google Scholar] [CrossRef]
- Johansen, J.L.; Sager, T.N.; Lotharius, J.; Witten, L.; Mørk, A.; Egebjerg, J.; Thirstrup, K. HIF prolyl hydroxylase inhibition increases cell viability and potentiates dopamine release in dopaminergic cells. J. Neurochem. 2010, 115, 209–219. [Google Scholar] [CrossRef]
- Yang, N.; Wei, Y.; Wang, T.; Guo, J.; Sun, Q.; Hu, Y.; Yan, X.; Zhu, X.; Tang, B.; Xu, Q. Genome-wide analysis of DNA methylation during antagonism of DMOG to MnCl2-induced cytotoxicity in the mouse substantia nigra. Sci. Rep. 2016, 6, 28933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Cui, X.; Chen, Y.; Wu, T.; Xu, H.; Yin, H.; Wu, Y. Therapeutic Potential of a Prolyl Hydroxylase Inhibitor FG-4592 for Parkinson’s Diseases in Vitro and in Vivo: Regulation of Redox Biology and Mitochondrial Function. Front. Aging Neurosci. 2018, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Kandil, E.A.; Sayed, R.H.; Ahmed, L.A.; Abd El Fattah, M.A.; El-Sayeh, B.M. Hypoxia-inducible factor 1 alpha and nuclear-related receptor 1 as targets for neuroprotection by albendazole in a rat rotenone model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2019, 46, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, N.; Currò, M.; Giunta, M.L.; Longo, D.; Rizzo, V.; Caccamo, D.; Ientile, R. Up-regulation of HIF-1α is associated with neuroprotective effects of agmatine against rotenone-induced toxicity in differentiated SH-SY5Y cells. Amino Acids 2020, 52, 171–179. [Google Scholar] [CrossRef]
- Mehrabani, M.; Nematollahi, M.H.; Tarzi, M.E.; Juybari, K.B.; Abolhassani, M.; Sharifi, A.M.; Paseban, H.; Saravani, M.; Mirzamohammadi, S. Protective effect of hydralazine on a cellular model of Parkinson’s disease: A possible role of hypoxia-inducible factor (HIF)-1α. Biochem. Cell Biol. 2020, 98, 405–414. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, T.; Li, X.; Liu, Y.; Zhu, X.; Jankovic, J.; Pan, T.; Wu, Y. Neuroprotection by Orexin-A via HIF-1α induction in a cellular model of Parkinson’s disease. Neurosci. Lett. 2014, 579, 35–40. [Google Scholar] [CrossRef]
- Gao, L.; Li, C.; Yang, R.; Lian, W.; Fang, J.; Pang, X.; Qin, X.; Liu, A.; Du, G. Ameliorative effects of baicalein in MPTP-induced mouse model of Parkinson’s disease: A microarray study. Pharmacol. Biochem. Behav. 2015, 133, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Rane, A.; Rajagopalan, S.; Ahuja, M.; Thomas, B.; Chinta, S.J.; Andersen, J.K. Hsp90 Co-chaperone p23 contributes to dopaminergic mitochondrial stress via stabilization of PHD2: Implications for Parkinson’s disease. Neurotoxicology 2018, 65, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Aimé, P.; Karuppagounder, S.S.; Rao, A.; Chen, Y.; Burke, R.E.; Ratan, R.R.; Greene, L.A. The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol. Dis. 2020, 136, 104725. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIF-1α-Target Genes | ||||||
|---|---|---|---|---|---|---|
| Oxygen transport and angiogenesis | ADM | ANGPT1 | ANGPT2 | ANP | BRCP | CP |
| CXCL12 | EDN1 | EPO | FECH | FLK1 | FLT1 | |
| GPI PDGFb | HMOX1 PGF | LEP SERPINE1 | NOS2 TF | NOS3 TFRC | NOX2 VEGF | |
| Stemness/self-renewal | ADM | EDN1 | EPO1 | GPI | ID2 | IGF2 |
| PGM | OCT4 | TERT | TGFA | VEGF | ||
| Proliferation | CD73 | MYC | CTGF | ENG | CDKN1A | CDKN1B |
| IGFBP3 | ITF | MET | NR4A1 | REDD1 | RORα4 | |
| STK15 | TERT | TGFβ3 | WT1 | |||
| Apoptosis | BNIP3/3L | NDRG | NIX | NOXA | PP5 | MCL1 |
| NPM | ||||||
| Oxidative stress | COXAI2 | GPX3 | HMOX1 | LONP1 | NDUFA4L | SOD2 |
| Energy metabolism | ALDOA | ALDOC | CA9 | COXAI2 | ENO1 | GAPDH |
| GLUT1 LONP1 PFKL TPI | GLUT2 MCT4 PGK1 | GPI NDUFA4L PGM | HK1 NHE1 PKM2 | HK2 PDK1 TKT | LDHA PFKFB1-4 TKTL2 | |
| Mitochondrial homeostasis | BHLHE40 | BNIP3/3L | CHCHD4 | HIGD1A | MGARP | MXI1 |
| NIX | PPARGC1 | |||||
| Autophagy | AMPK | BNIP3/3L | NIX | |||
| Dopamine metabolism | DAT | TH | ||||
| Chemical | PD Model | Effects | References |
|---|---|---|---|
| Indirect PHD inhibitors | |||
| Deferoxamine (DFO) | Cell model (SH-SY5Y cells) | ↑HIF-1α expression ↓Cell death ↓cleaved-PARP, cleaved-CASP3 ↑ATP ↓ROS ↑ Autophagolysosomes ↑Cathepsin, Beclin-1 expression | [200] [201] [202] |
| Animal model (6-OHDA rat; α-synuclein rAAV rat) | ↑Striatal DA ↑SN DAergic neurons ↓ROS ↑Motor behaviour ↓α-synuclein inclusions | [203] [204] [205] | |
| Animal model (MPTP mouse) | ↑HIF-1α expression ↑SN DAergic neurons ↑DAT expression ↑Bcl-2/Bax ratio ↑GAP43 expression ↑p-ERK/p-p38 MAPK expression ↓p-JNK1/2 expression ↓astrocyte activation ↑Motor behaviour | [206] | |
| M30 | Animal model (Mouse, MPTP mouse) | ↑HIF-1α expression ↑SN DAergic neurons ↑DA levels ↑TH expression and activity ↑TfR expression ↑Neurotrophic factors ↑Antioxidant enzymes ↑Pro-survival signaling | [207] [208] |
| Clioquinol (CQ) | Animal model (MPTP mouse; α-synuclein hA53T mouse) | ↑HIF-1α expression ↑SN DAergic neurons ↓α-synuclein inclusions | [209] [210] |
| Animal model (MPTP monkey) | ↑Motor behaviour ↓Non-Motor deficits ↑SN DAergic neurons ↑TH, DAT expression ↓ROS ↓SN iron content ↑AKT /mTOR pathway | [211] | |
| Lactoferrin (Lf) | Cell model (SH-SY5Y cells; MN9D cells) | ↑HIF-1α expression ↓Cell death ↓cleaved CASP3 ↑Bcl-2/Bax ratio ↑VEGF, BDNF expression ↑p-ERK expression ↓p-JNK1/2, p-p38 MAPK expression ↑Bcl-2/Bax ratio | [212] |
| Animal model (MPTP mouse) | ↑HIF-1α expression ↑Motor behaviour ↑SN DAergic neurons ↑TH expression ↓α-synuclein expression ↑Bcl-2/Bax ratio ↓cleaved CASP3 ↓glial activation ↓SN iron content ↑GAP43, BDNF, p-ERK expression ↓p-JNK1/2 and p-p38 MAPK expression | [212] | |
| FG-0041 | Cell model (PC12 cells; LUHMES cells; primary rat mesencephalic cells) | ↑HIF-1α expression ↓Cell death ↑TH expression and activity ↑DA release ↑ Mitochondrial membrane potential | [200] [213] [214] |
| Animal model (rat) | ↑TH expression ↑DA levels | [213] | |
| Competitive PHD inhibitors | |||
| Dimethyloxallyl Glycine (DMOG) | Cell model (DJ1-KO MPTP primary cortical neurons; SH-SY5Y cells) | ↑HIF-1α expression ↓Cell death | [61] |
| Animal model (MnCl2 mouse) | ↑SN DAergic neurons ↑Motor behaviour ↓DNA methylation | [215] | |
| FG-4592 | Cell model (SH-SY5Y cells) | ↑HIF-1α expression ↓Cell death ↑Bcl-2/Bax ratio ↑TH expression ↑Mitochondrial respiration ↑Mitochondrial membrane potential ↓p62, Cathepsin, LC3-II expression ↑ PGC-1α expression ↓ROS ↑Antioxidant proteins expression | [216] |
| Animal model (MPTP mouse) | ↑SN DAergic neurons ↑Motor behaviour ↑TH expression ↑DA levels | [216] | |
| JNJ-42041935 | Cell model (SH-SY5Y cells; PC12 cells) | ↑HIF-1α expression ↑ATP ↑DA release | [200] |
| IOX2 | Cell model (iPSC-derived DAergic neurons; primary mouse mesencephalic cells; differentiated SH-SY5Y cells) | ↑HIF-1α expression ↑Mitochondrial membrane potential ↓Iron content ↑ATP13A2 expression | [56] |
| Atypical HIF-1α inducers | |||
| Albendazole (ABZ) | Animal model (rotenone rat) | ↑HIF-1α mRNA ↑VEGF mRNA ↑SN DAergic neurons ↑Motor behaviour ↓α-synuclein expression ↑DA levels ↑TH expression ↑TH mRNA, GDNF mRNA ↓NFκB, TNF-α | [217] |
| Agmantine | Cell model (differentiated SH-SY5Y cells) | ↑HIF-1α expression ↑HIF-1α mRNA ↓Cell death ↓CASP3 activity ↓ROS ↑Mitochondrial membrane potential | [218] |
| Hydralazine | Cell model (SH-SY5Y cells) | ↑HIF-1α expression ↑VEGF, TH, DAT expression ↓Cell death ↑Antioxidant capacity | [219] |
| Orexin-A | Cell model (SH-SY5Y cells) | ↑HIF-1α expression ↑EPO, VEGF expression ↓Cell death ↓cleaved-PARP, cleaved-CASP3 | [220] |
| Baicalein | Animal model (MTPT mouse) | ↑HIF-1α mRNA ↑Motor behaviour | [221] |
| Gedunin | Cell model (iPSC-derived DAergic neurons, N27 cells) | ↓Cell death | [222] |
| Adaptaquin (AQ) | Cell model (PC12 cells; primary rat ventral midbrain DAergic neurons) | ↓Cell death ↓Trib3, ATF4, CHOP expression/ mRNA ↑Parkin expression | [223] |
| Animal model (6-OHDA mouse) | ↓Trib3, CHOP mRNA ↑SN DAergic neurons ↑Motor behaviour | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lestón Pinilla, L.; Ugun-Klusek, A.; Rutella, S.; De Girolamo, L.A. Hypoxia Signaling in Parkinson’s Disease: There Is Use in Asking “What HIF?”. Biology 2021, 10, 723. https://doi.org/10.3390/biology10080723
Lestón Pinilla L, Ugun-Klusek A, Rutella S, De Girolamo LA. Hypoxia Signaling in Parkinson’s Disease: There Is Use in Asking “What HIF?”. Biology. 2021; 10(8):723. https://doi.org/10.3390/biology10080723
Chicago/Turabian StyleLestón Pinilla, Laura, Aslihan Ugun-Klusek, Sergio Rutella, and Luigi A. De Girolamo. 2021. "Hypoxia Signaling in Parkinson’s Disease: There Is Use in Asking “What HIF?”" Biology 10, no. 8: 723. https://doi.org/10.3390/biology10080723
APA StyleLestón Pinilla, L., Ugun-Klusek, A., Rutella, S., & De Girolamo, L. A. (2021). Hypoxia Signaling in Parkinson’s Disease: There Is Use in Asking “What HIF?”. Biology, 10(8), 723. https://doi.org/10.3390/biology10080723