
Comprehensive Genetic Analysis of DGAT2 Mutations and Gene Expression Patterns in Human Cancers

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. DGAT2 Isoforms and Conservation
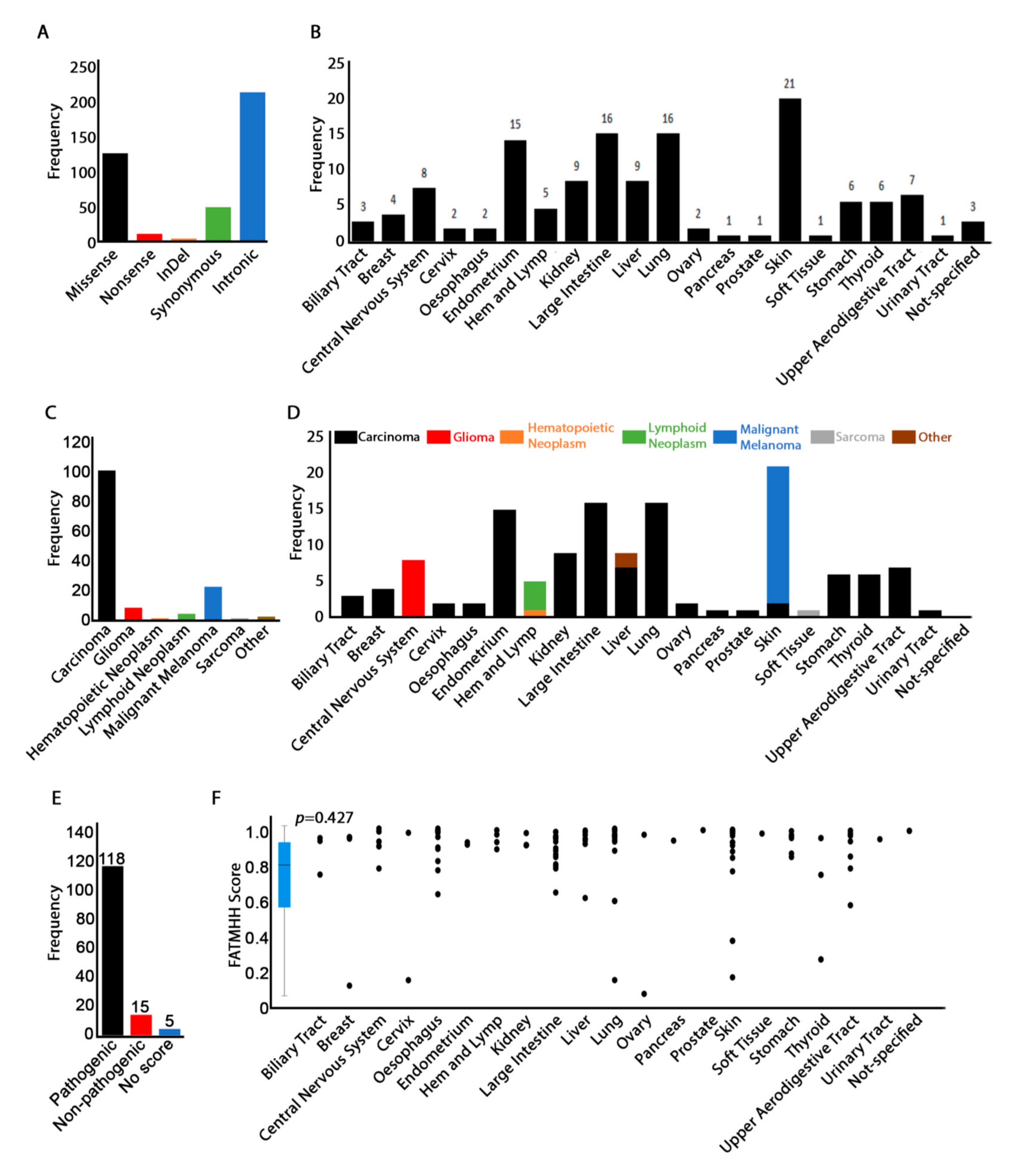
3.2. DGAT2 Mutation Distribution in Human Cancers
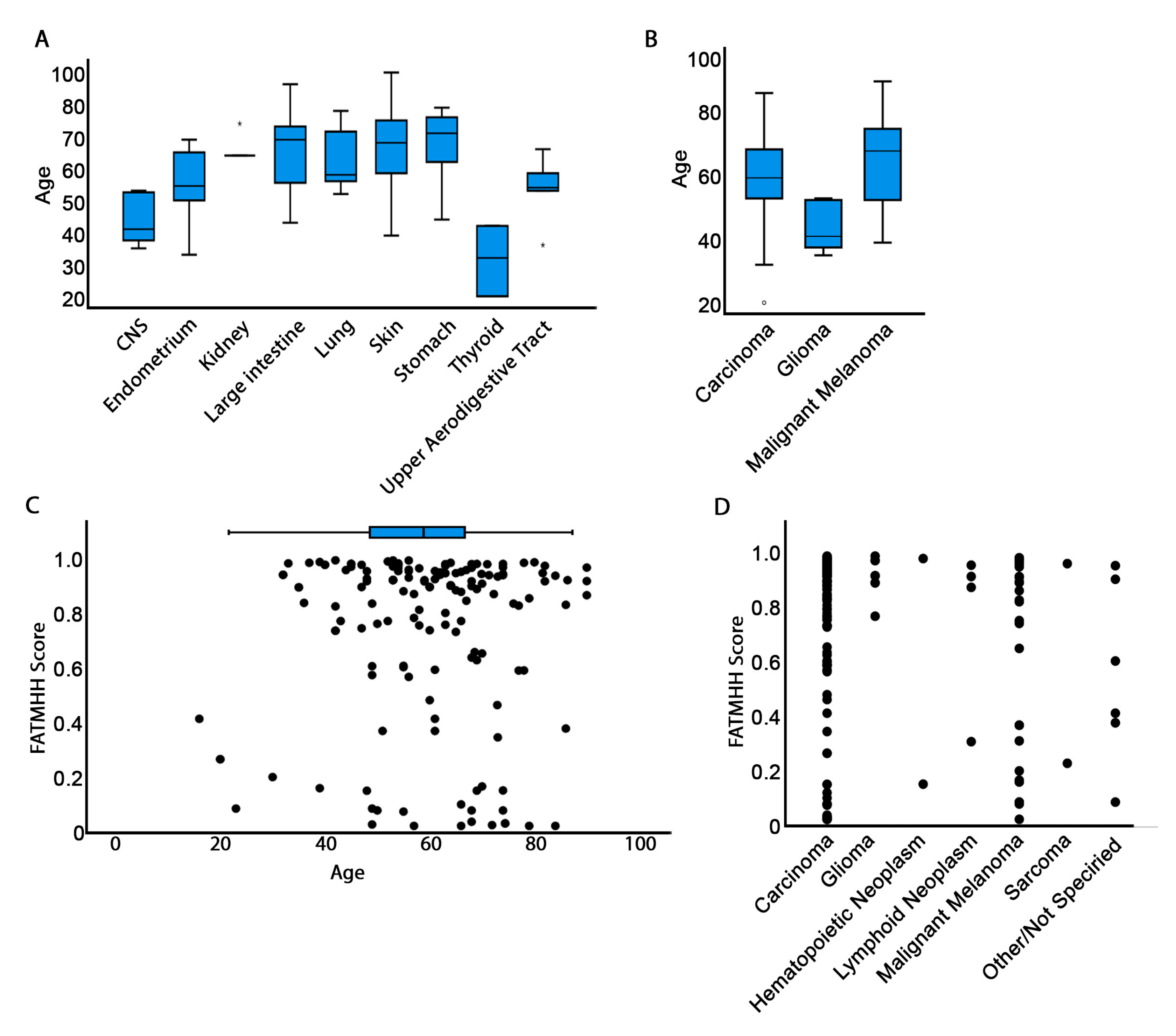
3.3. Mutation Pathogenicity Is Correlated with Older Age
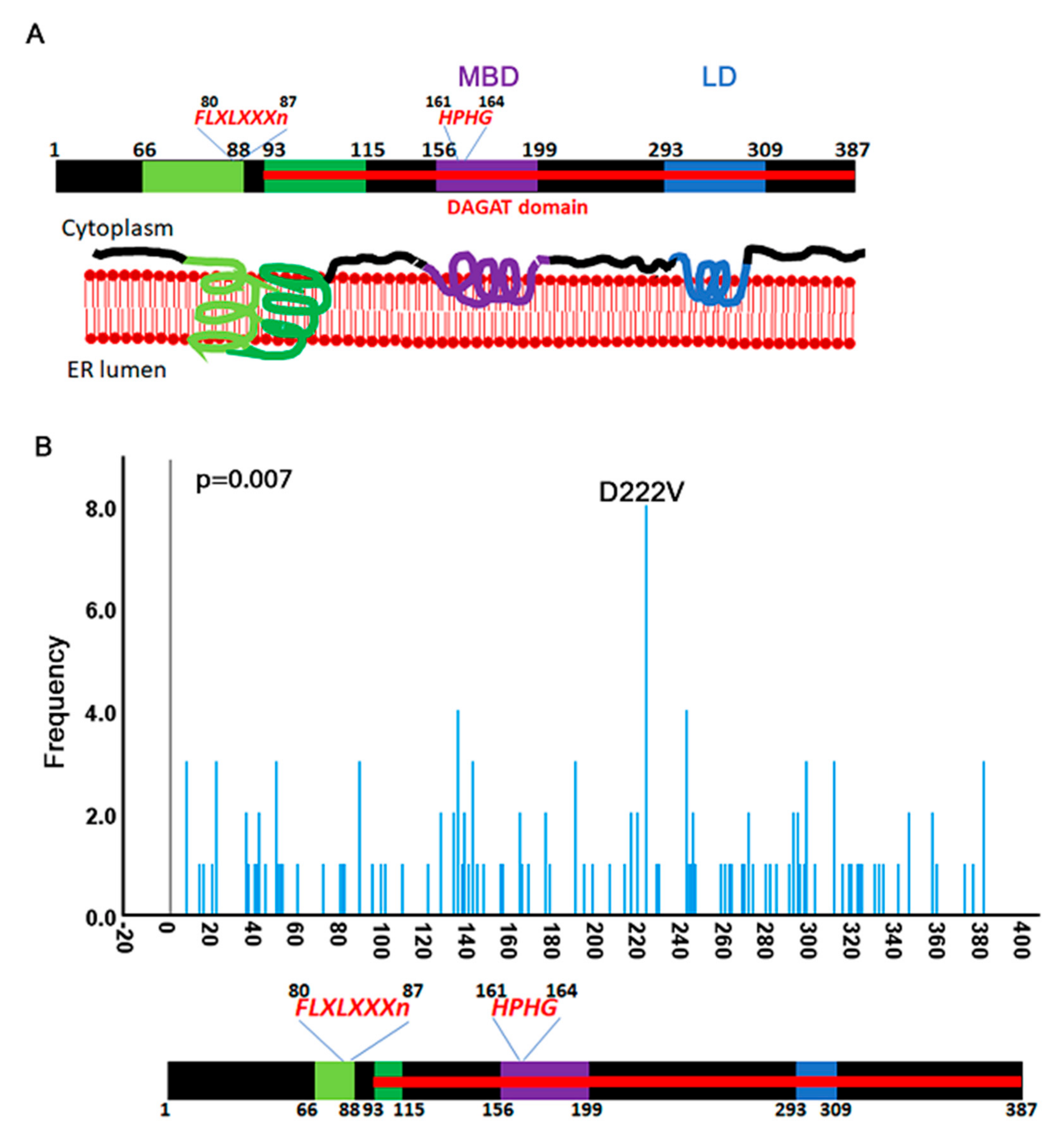
3.4. Identification of a Mutation Hotspot in DGAT2
3.5. Mutational Landscape
3.6. DGAT2 Expression Levels in Cancer Tissues
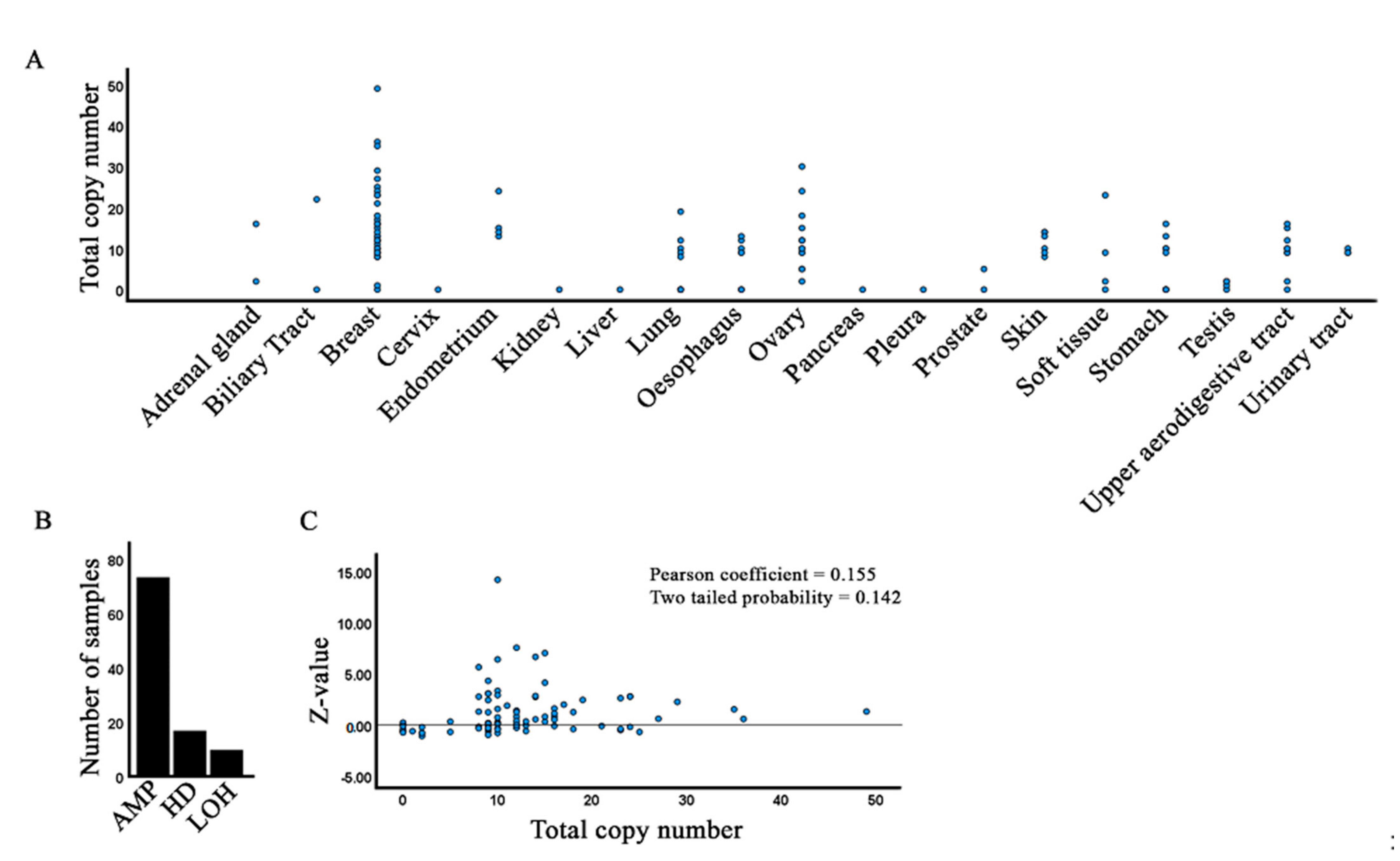
3.7. Copy Number Variations of DGAT2 Alleles
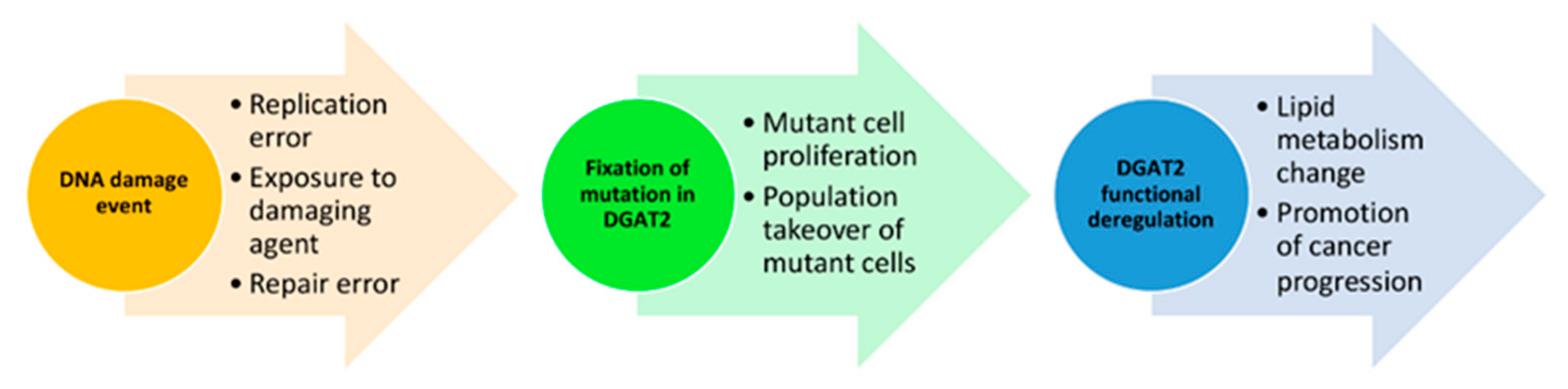
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shyu, P.; Wong, X.F.A.; Crasta, K.; Thibault, G. Dropping in on lipid droplets: Insights into cellular stress and cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bai, L.; Li, W.; Cui, J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Palian, B.M.; Rohira, A.D.; Johnson, S.A.S.; He, L.; Zheng, N.; Dubeau, L.; Stiles, B.L.; Johnson, D.L. Maf1 Is a Novel Target of PTEN and PI3K Signaling That Negatively Regulates Oncogenesis and Lipid Metabolism. PLoS Genet. 2014, 10, e1004789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, M.; Noguchi, C.; Wilson, S.; Martinez, E.; Shiozaki, K.; Sell, C.; Mell, J.C.; Noguchi, E. Maf1-dependent transcriptional regulation of tRNAs prevents genomic instability and is associated with extended lifespan. Aging Cell 2020, 19, e13068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerquist, A.M.; Escorcia, W.; Curran, S.P. Maf1 regulates intracellular lipid homeostasis in response to DNA damage response activation. Mol. Biol. Cell 2021, 32, 1086–1093. [Google Scholar] [CrossRef]
- Bhatt-Wessel, B.; Jordan, T.W.; Miller, J.H.; Peng, L. Role of DGAT enzymes in triacylglycerol metabolism. Arch. Biochem. Biophys. 2018, 655, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitraju, C.; Walther, T.C., Jr.; Farese, R.V. The triglyceride synthesis enzymes DGAT1 and DGAT2 have distinct and overlapping functions in adipocytes. J. Lipid Res. 2019, 60, 1112–1120. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Choi, M.; Choi, K.; Bin Kwon, E.; Kang, M.; Kim, D.-E.; Jeong, H.; Kim, J.; Kim, J.H.; Kim, M.O.; et al. Inactivation of human DGAT2 by oxidative stress on cysteine residues. PLoS ONE 2017, 12, e0181076. [Google Scholar] [CrossRef] [Green Version]
- Bin Hong, Y.; Kang, J.; Kim, J.H.; Lee, J.; Kwak, G.; Hyun, Y.S.; Nam, S.H.; Hong, H.D.; Choi, Y.-R.; Jung, S.-C.; et al. DGAT2Mutation in a Family with Autosomal-Dominant Early-Onset Axonal Charcot-Marie-Tooth Disease. Hum. Mutat. 2016, 37, 473–480. [Google Scholar] [CrossRef]
- Meyers, A.; Weiskittel, T.M.; Dalhaimer, P. Lipid Droplets: Formation to Breakdown. Lipids 2017, 52, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Khor, V.K.; Shen, W.-J.; Kraemer, F. Lipid droplet metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Mol. 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-W. Lipid droplets, lipophagy, and beyond. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2016, 1861, 793–805. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Nelakurti, D.D.; Pappula, A.L.; Rajasekaran, S.; Miles, W.O.; Petreaca, R.C. Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer. Cancers 2020, 12, 2616. [Google Scholar] [CrossRef]
- McPherson, M.T.; Holub, A.S.; Husbands, A.Y.; Petreaca, R.C. Mutation Spectra of the MRN (MRE11, RAD50, NBS1/NBN) Break Sensor in Cancer Cells. Cancers 2020, 12, 3794. [Google Scholar] [CrossRef] [PubMed]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, C.; Zhang, X. Mutplot: An easy-to-use online tool for plotting complex mutation data with flexibility. PLoS ONE 2019, 14, e0215838. [Google Scholar] [CrossRef] [PubMed]
- Douville, C.; Carter, H.; Kim, R.; Niknafs, N.; Diekhans, M.; Stenson, P.D.; Cooper, D.N.; Ryan, M.; Karchin, R. CRAVAT: Cancer-related analysis of variants toolkit. Bioinform. 2013, 29, 647–648. [Google Scholar] [CrossRef]
- Masica, D.L.; Douville, C.; Tokheim, C.; Bhattacharya, R.; Kim, R.; Moad, K.; Ryan, M.C.; Karchin, R. CRAVAT 4: Cancer-Related Analysis of Variants Toolkit. Cancer Res. 2017, 77, e35–e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turchetto-Zolet, A.C.; Maraschin, F.S.; de Morais, G.L.; Cagliari, A.; Andrade, C.M.; Margis-Pinheiro, M.; Margis, R. Evolutionary view of acyl-CoA diacylglycerol acyltransferase (DGAT), a key enzyme in neutral lipid biosynthesis. BMC Evol. Biol. 2011, 11, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turchetto-Zolet, A.C.; Christoff, A.P.; Kulcheski, F.; Loss-Morais, G.; Margis, R.; Margis-Pinheiro, M. Diversity and evolution of plant diacylglycerol acyltransferase (DGATs) unveiled by phylogenetic, gene structure and expression analyses. Genet. Mol. Biol. 2016, 39, 524–538. [Google Scholar] [CrossRef] [Green Version]
- Cao, H. Structure-Function Analysis of Diacylglycerol Acyltransferase Sequences from 70 Organisms. BMC Res. Notes 2011, 4, 249. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.F.; Shihab, H.A.; Mort, M.E.; Cooper, D.N.; Gaunt, T.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinform. 2018, 34, 511–513. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-K.; Choi, Y.-L.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 283–312. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef]
- Li, Y.; Li, T.; Jin, Y.; Shen, J. Dgat2 reduces hepatocellular carcinoma malignancy via downregulation of cell cycle-related gene expression. Biomed. Pharmacother. 2019, 115, 108950. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-Specific High-Throughput Annotation of Somatic Mutations: Computational Prediction of Driver Missense Mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [Green Version]
- Douville, C.; Masica, D.L.; Stenson, P.D.; Cooper, D.N.; Gygax, D.M.; Kim, R.; Ryan, M.; Karchin, R. Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel). Hum. Mutat. 2016, 37, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC Genom. 2013, 14, S3. [Google Scholar] [CrossRef] [Green Version]
- Nurminen, R.; Rantapero, T.; Wong, S.C.; Fischer, D.; Lehtonen, R.J.; Tammela, T.L.; Nykter, M.; Visakorpi, T.; Wahlfors, T.; Schleutker, J. Expressional profiling of prostate cancer risk SNPs at 11q135 identifiesDGAT2as a new target gene. Genes Chromosom. Cancer 2016, 55, 661–673. [Google Scholar] [CrossRef]
- Stone, S.J.; Levin, M.C.; Farese, R.V., Jr. Membrane topology and identification of key functional amino acid residues of murine acyl-CoA:diacylglycerol acyltransferase-2. J. Biol. Chem. 2006, 281, 40273–40282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFie, P.J.; Banman, S.L.; Kary, S.; Stone, S.J. Murine Diacylglycerol Acyltransferase-2 (DGAT2) Can Catalyze Triacylglycerol Synthesis and Promote Lipid Droplet Formation Independent of Its Localization to the Endoplasmic Reticulum. J. Biol. Chem. 2011, 286, 28235–28246. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.; Gilham, D.; Vance, D.E.; Lehner, R. Mutation of F417 but not of L418 or L420 in the lipid binding domain decreases the activity of triacylglycerol hydrolase. J. Lipid Res. 2006, 47, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Anoosha, P.; Sakthivel, R.; Gromiha, M.M. Exploring preferred amino acid mutations in cancer genes: Applications to identify potential drug targets. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Creixell, P.; Schoof, E.M.; Tan, C.S.; Linding, R. Mutational properties of amino acid residues: Implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2584–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brule, C.E.; Grayhack, E.J. Synonymous Codons: Choose Wisely for Expression. Trends Genet. 2017, 33, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of Microarray Data Using Z Score Transformation. J. Mol. Diagn. 2003, 5, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. Faculty Opinions recommendation of The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Fac. Opin.—Post-Publ. Peer Rev. Biomed. Lit. 2015, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational Landscape of Aggressive Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 Transcript | Transcript Length (Nucleotides) | 2 Protein Isoforms | Protein Size (Amino Acids) | UniProt Identifier |
|---|---|---|---|---|
| NM_032564.5 | 2407 | 1 | 388 | Q96PD7-1 |
| NM_001253891.2 | 2278 | 2 | 345 | Q96PD7-2 |
| XM_011545304 | 2156 | X1 | 358 | N/A |
| - | - | 3 | 295 | S4R3S3 |
| - | - | 4 | 160 | S4R383 |
| - | - | 5 | 112 | S4R3Z3 |
| - | - | 6 | 113 | S4R449 |
| 1 Mutation | 2 VEST p-Value | 2 VEST FDR | Tissue | 3 dbSNP |
|---|---|---|---|---|
| G212C | 0.00071000 | 0.05 | CNS | rs777087960 |
| H163R | 0.00071000 | 0.05 | Endometrium | |
| R259H | 0.00091000 | 0.05 | Large Intestine | |
| E243Q | 0.00121000 | 0.05 | Upper Aer Dig | |
| F314S | 0.00152000 | 0.05 | Upper Aer Dig | |
| P329S | 0.00182000 | 0.05 | Endometrium | |
| F262V | 0.00182000 | 0.05 | Pancreas | |
| G261C | 0.00253000 | 0.05 | Lung | |
| G197D | 0.00283000 | 0.05 | Endometrium | rs369680804 |
| S278F | 0.00283000 | 0.05 | Soft Tissue | |
| G318R | 0.00314000 | 0.05 | Kidney | |
| L267R | 0.00314000 | 0.05 | Liver | |
| G164S | 0.00536000 | 0.05 | Haem and Lymph | |
| P215H | 0.00607000 | 0.05 | Endometrium | |
| G120D | 0.008 | 0.10 | Skin | |
| R205K | 0.0082 | 0.10 | Lung | |
| A241V | 0.01002000 | 0.10 | Lung | rs766238005 |
| L245V | 0.01113000 | 0.10 | Lung | |
| W100L | 0.01214000 | 0.10 | Skin | |
| N155S | 0.01295000 | 0.10 | Skin | |
| G270R | 0.01326000 | 0.10 | Lung | |
| G167D | 0.01427000 | 0.10 | Prostate | |
| P141S | 0.01488000 | 0.10 | Lung | |
| S244C | 0.01528000 | 0.10 | Liver | |
| K146N | 0.0164 | 0.10 | Large Intestine | |
| R218W | 0.01791000 | 0.10 | Skin | rs528376420 |
| A310T | 0.01893000 | 0.10 | CNS | rs761761542 |
| W126R | 0.02054000 | 0.10 | Stomach | |
| P215S | 0.02146000 | 0.10 | Skin | |
| D222V | 0.02257000 | 0.10 | Kidney, Large intestine | |
| N228Y | 0.0248 | 0.10 | Stomach | |
| S294F | 0.0249 | 0.10 | Skin | |
| R297Q | 0.02844000 | 0.10 | Haem and Lymph | rs140793537 |
| R297 * | 0.03186000 | 0.10 | Stomach | rs771080849 |
| Y139 * | 0.03273000 | 0.10 | Esophagus | |
| R189L | 0.0339 | 0.10 | Skin | |
| R137 * | 0.03489000 | 0.10 | Skin | rs572486802 |
4 Diagram with mutation distribution 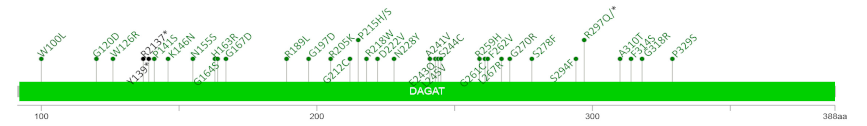 | ||||
| Sample ID 1 | Sample Name | Mutation | Mutation Z-Score | Sample Z-Sore Range 2 |
|---|---|---|---|---|
| COSU540 | Cecum carcinoma | p.G120D | 2.221 | 0.30 ± 4.68 |
| COSU413 3 | Bladder urothelial carcinoma | p.P275= | 2.335 | 0.19 ± 1.19 |
| COSU418 | Lung squamous cell carcinoma | p.P141S | 2.583 | 0.07 ± 1.18 |
| COSU419 | Uterine corpus endometrial carcinoma | p.R189W | 2.706 | 0.22 ± 1.19 |
| COSU540 | Cecum carcinoma | p.R218W | 3.292 | 0.30±4.68 |
| COSU418 | Lung squamous cell carcinoma | p.W40* | 3.368 | 0.07 ± 1.18 |
| COSU435 | Prostate adenocarcinoma | p.G167D | 3.647 | 0.05 ± 1.05 |
| COSU377 | Acute myeloid leukemia | c.*27A>G | 4.978 | 0.0040 ± 0.98 |
| COSU414 | Breast invasive carcinoma | p.I94V | 5.195 | 0.140 ± 2.34 |
| COSU419 | Uterine corpus endometrial carcinoma | c.*556A>T | 6.743 | 0.22 ± 1.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graber, M.; Barta, H.; Wood, R.; Pappula, A.; Vo, M.; Petreaca, R.C.; Escorcia, W. Comprehensive Genetic Analysis of DGAT2 Mutations and Gene Expression Patterns in Human Cancers. Biology 2021, 10, 714. https://doi.org/10.3390/biology10080714
Graber M, Barta H, Wood R, Pappula A, Vo M, Petreaca RC, Escorcia W. Comprehensive Genetic Analysis of DGAT2 Mutations and Gene Expression Patterns in Human Cancers. Biology. 2021; 10(8):714. https://doi.org/10.3390/biology10080714
Chicago/Turabian StyleGraber, Meghan, Hayley Barta, Ryan Wood, Amrit Pappula, Martin Vo, Ruben C. Petreaca, and Wilber Escorcia. 2021. "Comprehensive Genetic Analysis of DGAT2 Mutations and Gene Expression Patterns in Human Cancers" Biology 10, no. 8: 714. https://doi.org/10.3390/biology10080714
APA StyleGraber, M., Barta, H., Wood, R., Pappula, A., Vo, M., Petreaca, R. C., & Escorcia, W. (2021). Comprehensive Genetic Analysis of DGAT2 Mutations and Gene Expression Patterns in Human Cancers. Biology, 10(8), 714. https://doi.org/10.3390/biology10080714