
hnRNP A/B Proteins: An Encyclopedic Assessment of Their Roles in Homeostasis and Disease

 ,
,  ,
, 
Simple Summary
Abstract
1. A Brief History of the hnRNPs


2. hnRNP A/B Proteins: An Overview


3. hnRNP A1 and A2/B1
3.1. DNA Binding: Telomere Regulation, Transcription
3.2. RNA Splicing
3.3. RNA Trafficking
3.4. miRNA Regulation
3.5. mRNA Translation, Stability, and Granules
4. hnRNP A0
4.1. Cellular Proliferation and Cancer
4.2. Intracellular Immune Signaling
4.3. Neurobiology
5. hnRNP A3
5.1. Cellular Proliferation and Senescence
5.2. RNA Metabolism
5.3. Intracellular Immunity
5.4. Neurobiology
6. hnRNP A/B Protein Structural Features
6.1. Structures of Nucleic Acid-Unbound and -Bound RRMs in A1 and A2/B1 Proteins
6.2. Structures of LCD Fibrils of A1 and A2/B1 Proteins
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pagoulatos, G.N.; Yaniv, M. High Resolution Two-Dimensional Electrophoresis of Proteins Bound to Heterogeneous Nuclear RNA. FEBS Lett. 1977, 74, 115–120. [Google Scholar] [CrossRef]
- van Venrooij, W.J.; Janssen, D.B. HnRNP Particles. Mol. Biol. Rep. 1978, 4, 3–8. [Google Scholar] [CrossRef]
- Peters, K.E.; Comings, D.E. Two-Dimensional Gel Electrophoresis of Rat Liver Nuclear Washes, Nuclear Matrix, and HnRNA Proteins. J. Cell Biol. 1980, 86, 135–155. [Google Scholar] [CrossRef]
- Eekelen, C.A.; Riemen, T.; Venrooij, W.J. Specificity in the Interaction of HnRNA and MRNA with Proteins as Revealed by in Vivo Cross Linking. FEBS Lett. 1981, 130, 223–226. [Google Scholar] [CrossRef]
- Upreti, R.K.; Holoubek, V. The Effect of Inhibition of RNA Synthesis by Actinomycin D on the Population of Basic Polypeptides of the 30S Unclear Ribonucleoprotein Particles. Biochimie 1982, 64, 247–254. [Google Scholar] [CrossRef]
- Upreti, R.K.; Holoubek, V. Separation of Proteins of Nuclear Ribonucleoprotein Particles by High-Performance Liquid Chromatography. Anal. Chim. Acta 1981, 131, 239–245. [Google Scholar] [CrossRef]
- Wilk, H.-E.; Werr, H.; Friedrich, D.; Kiltz, H.H.; Schafer, K.P. The Core Proteins of 35 S HnRNP Complexes. Characterization of Nine Different Species. Eur. J. Biochem. 1985, 146, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Mayrand, S.; Setyono, B.; Greenberg, J.R.; Pederson, T. Structure of Nuclear Ribonucleoprotein: Identification of Proteins in Contact with Poly(A)+ Heterogeneous Nuclear RNA in Living HeLa Cells. J. Cell Biol. 1981, 90, 380–384. [Google Scholar] [CrossRef]
- Beyer, A.L.; Christensen, M.E.; Walker, B.W.; LeStourgeon, W.M. Identification and Characterization of the Packaging Proteins of Core 40S HnRNP Particles. Cell 1977, 11, 127–138. [Google Scholar] [CrossRef]
- Piñol-Roma, S.; Choi, Y.D.; Matunis, M.J.; Dreyfuss, G. Immunopurification of Heterogeneous Nuclear Ribonucleoprotein Particles Reveals an Assortment of RNA-Binding Proteins. Genes Dev. 1988, 2, 215–227. [Google Scholar] [CrossRef]
- Valentini, O.; Biamonti, G.; Pandolfo, M.; Morandi, C.; Riva, S. Mammalian Single-Stranded DNA Binding Proteins and Heterogeneous Nuclear RNA Proteins Have Common Antigenic Determinants. Nucleic Acids Res. 1985, 13, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Swanson, M.S.; Dreyfuss, G. RNA Binding Specificity of HnRNP Proteins: A Subset Bind to the 3′ End of Introns. EMBO J. 1988, 7, 3519–3529. [Google Scholar] [CrossRef] [PubMed]
- Karn, J.; Vidali, G.; Boffa, L.C.; Allfrey, V.G. Characterization of the Non-Histone Nuclear Proteins Associated with Rapidly Labeled Heterogeneous Nuclear RNA. J. Biol. Chem. 1977, 252, 7307–7322. [Google Scholar] [CrossRef]
- Pederson, T. Proteins Associated with Heterogeneous Nuclear RNA in Eukaryotic Cells. J. Mol. Biol. 1974, 83, 163–183. [Google Scholar] [CrossRef]
- Jones, R.E.; Okamura, C.S.; Martin, T.E. Immunofluorescent Localization of the Proteins of Nuclear Ribonucleoprotein Complexes. J. Cell Biol. 1980, 86, 235–243. [Google Scholar] [CrossRef]
- Dangli, A.; Plomaritoglou, A.; Boutou, E.; Vassiliadou, N.; Moutsopoulos, H.M.; Guialis, A. Recognition of Subsets of the Mammalian A/B-Type Core Heterogeneous Nuclear Ribonucleoprotein Polypeptides by Novel Autoantibodies. Biochem. J. 1996, 320, 761–767. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Choi, Y.D.; Adam, S.A. Characterization of Heterogeneous Nuclear RNA-Protein Complexes in Vivo with Monoclonal Antibodies. Mol. Cell. Biol. 1984, 4, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G.; Swanson, M.S.; Gorlach, M.; Dreyfuss, G. Primary Structures of the Heterogeneous Nuclear Ribonucleoprotein A2, B1, and C2 Proteins: A Diversity of RNA Binding Proteins Is Generated by Small Peptide Inserts. Proc. Natl. Acad. Sci. USA 1989, 86, 9788–9792. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Matunis, M.J.; Pinol-Roma, S.; Burd, C.G. HnRNP Proteins and the Biogenesis of MRNA. Annu. Rev. Biochem. 1993, 62, 289–321. [Google Scholar] [CrossRef]
- Choi, Y.D.; Dreyfuss, G. Isolation of the Heterogeneous Nuclear RNA-Ribonucleoprotein Complex (HnRNP): A Unique Supramolecular Assembly. Proc. Natl. Acad. Sci. USA 1984, 81, 7471–7475. [Google Scholar] [CrossRef] [PubMed]
- Shishkin, S.; Kovalev, L.; Pashintseva, N.; Kovaleva, M.; Lisitskaya, K. Heterogeneous Nuclear Ribonucleoproteins Involved in the Functioning of Telomeres in Malignant Cells. Int. J. Mol. Sci. 2019, 20, 745. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Ridwan Amode, M.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef]
- Akindahunsi, A.A.; Bandiera, A.; Manzini, G. Vertebrate 2xRBD HnRNP Proteins: A Comparative Analysis of Genome, MRNA and Protein Sequences. Comput. Biol. Chem. 2005, 29, 13–23. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wong, T.K.F.; Kalyaanamoorthy, S.; Meusemann, K.; Yeates, D.K.; Misof, B.; Jermiin, L.S. A Minimum Reporting Standard for Multiple Sequence Alignments. NAR Genom. Bioinform. 2020, 2. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Mayeda, A.; Munroe, S.H.; Cáceres, J.F.; Krainer, A.R. Function of Conserved Domains of HnRNP A1 and Other HnRNP A/B Proteins. EMBO J. 1994, 13, 5483–5495. [Google Scholar] [CrossRef]
- Han, S.P.; Kassahn, K.S.; Skarshewski, A.; Ragan, M.A.; Rothnagel, J.A.; Smith, R. Functional Implications of the Emergence of Alternative Splicing in HnRNP A/B Transcripts. RNA 2010, 16, 1760–1768. [Google Scholar] [CrossRef]
- Biamonti, G.; Ruggiu, M.; Saccone, S.; Della Valle, G.; Riva, S. Two Homologous Genes, Originated by Duplication, Encode the Human HnRNP Proteins A2 and A1. Nucleic Acids Res. 1996, 22. [Google Scholar] [CrossRef] [PubMed]
- Myer, V.E.; Steitz, J.A. Isolation and Characterization of a Novel, Low Abundance HnRNP Protein: A0. RNA 1995, 1, 171–182. [Google Scholar] [PubMed]
- Buvoli, M.; Cobianchi, F.; Bestagno, M.G.; Mangiarotti, A.; Bassi, M.T.; Biamonti, G.; Riva, S. Alternative Splicing in the Human Gene for the Core Protein A1 Generates Another HnRNP Protein. EMBO J. 1990, 9, 1229–1235. [Google Scholar] [CrossRef]
- Biamonti, G.; Buvoli, M.; Bassi, M.T.; Morandi, C.; Cobianchi, F.; Riva, S. Isolation of an Active Gene Encoding Human HnRNP Protein A1. Evidence for Alternative Splicing. J. Mol. Biol. 1989, 207, 491–503. [Google Scholar] [CrossRef]
- Kozu, T.; Henrich, B.; Schäfer, K.P. Structure and Expression of the Gene (HNRPA2B1) Encoding the Human HnRNP Protein A2/B1. Genomics 1995, 25, 365–371. [Google Scholar] [CrossRef]
- Kamma, H.; Horiguchi, H.; Wan, L.; Matsui, M.; Fujiwara, M.; Fujimoto, M.; Yazawa, T.; Dreyfuss, G. Molecular Characterization of the HnRNP A2/B1 Proteins: Tissue-Specific Expression and Novel Isoforms. Exp. Cell Res. 1999, 246, 399–411. [Google Scholar] [CrossRef]
- Hatfield, J.T.; Rothnagel, J.A.; Smith, R. Characterization of the Mouse HnRNP A2/B1/B0 Gene and Identification of Processed Pseudogenes. Gene 2002, 295, 33–42. [Google Scholar] [CrossRef]
- Ma, A.S.W.; Moran-Jones, K.; Shan, J.; Munro, T.P.; Snee, M.J.; Hoek, K.S.; Smith, R. Heterogeneous Nuclear Ribonucleoprotein A3, a Novel RNA Trafficking Response Element-Binding Protein. J. Biol. Chem. 2002, 277, 18010–18020. [Google Scholar] [CrossRef]
- Makeyev, A.V.; Kim, C.B.; Ruddle, F.H.; Enkhmandakh, B.; Erdenechimeg, L.; Bayarsaihan, D. HnRNP A3 Genes and Pseudogenes in the Vertebrate Genomes. J. Exp. Zool. Part A Comp. Exp. Biol. 2005, 303A, 259–271. [Google Scholar] [CrossRef]
- Siapka, S.; Patrinou-Georgoula, M.; Vlachoyiannopoulos, P.G.; Guialis, A. Multiple Specificities of Autoantibodies against HnRNP A/B Proteins in Systemic Rheumatic Diseases and HnRNP L as an Associated Novel Autoantigen. Autoimmunity 2007, 40, 223–233. [Google Scholar] [CrossRef]
- Süleymanoĝlu, E. Molecular Phylogenetics and Functional Evolution of Major RNA Recognition Domains of Recently Cloned and Characterized Autoimmune RNA-Binding Particle. Genom. Proteom. Bioinform. 2003, 1, 310–320. [Google Scholar] [CrossRef]
- Lancaster, A.K.; Nutter-Upham, A.; Lindquist, S.; King, O.D. PLAAC: A Web and Command-Line Application to Identify Proteins with Prion-like Amino Acid Composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef]
- Krecic, A.M.; Swanson, M.S. HnRNP Complexes: Composition, Structure, and Function. Curr. Opin. Cell Biol. 1999, 11, 363–371. [Google Scholar] [CrossRef]
- Friend, L.R.; Han, S.P.; Rothnagel, J.A.; Smith, R. Differential Subnuclear Localisation of HnRNPs A/B Is Dependent on Transcription and Cell Cycle Stage. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1972–1980. [Google Scholar] [CrossRef]
- Buxadé, M.; Parra, J.L.; Rousseau, S.; Shpiro, N.; Marquez, R.; Morrice, N.; Bain, J.; Espel, E.; Proud, C.G. The Mnks Are Novel Components in the Control of TNFα Biosynthesis and Phosphorylate and Regulate HnRNP A1. Immunity 2005, 23, 177–189. [Google Scholar] [CrossRef]
- Rousseau, S.; Morrice, N.; Peggie, M.; Campbell, D.G.; Gaestel, M.; Cohen, P. Inhibition of SAPK2a/P38 Prevents HnRNP A0 Phosphorylation by MAPKAP-K2 and Its Interaction with Cytokine MRNAs. EMBO J. 2002, 21, 6505–6514. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Clements, J.; Finn, R.D. Skylign: A Tool for Creating Informative, Interactive Logos Representing Sequence Alignments and Profile Hidden Markov Models. BMC Bioinform. 2014, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tang, M.L.; Zeng, Z.X.; Wu, R.Y.; Xue, Y.; Hao, Y.H.; Pang, D.W.; Zhao, Y.; Tan, Z. Telomere- and Telomerase-Interacting Protein That Unfolds Telomere G-Quadruplex and Promotes Telomere Extension in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 20413–20418. [Google Scholar] [CrossRef]
- Kamma, H.; Fujimoto, M.; Hamasaki, M.; Fujiwara, M.; Matsui, M.; Horiguchi, H.; Satoh, H. Interaction of HnRNP A2/B1 Isoforms with Telomeric SsDNA and the in Vitro Function. Biochem. Biophys. Res. Commun. 2001, 280, 625–630. [Google Scholar] [CrossRef]
- Clarke, J.P.; Thibault, P.A.; Salapa, H.E.; Levin, M.C. A Comprehensive Analysis of the Role of HnRNP A1 Function and Dysfunction in the Pathogenesis of Neurodegenerative Disease. Front. Mol. Biosci. 2021, 8, 217. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, S. The Roles of HnRNP A2/B1 in RNA Biology and Disease. WIREs RNA 2021, 12, e1612. [Google Scholar] [CrossRef]
- Kamma, H.; Portman, D.S.; Dreyfuss, G. Cell Type-Specific Expression of HnRNP Proteins. Exp. Cell Res. 1995, 221, 187–196. [Google Scholar] [CrossRef]
- Deshaies, J.-E.; Shkreta, L.; Moszczynski, A.J.; Sidibé, H.; Semmler, S.; Fouillen, A.; Bennett, E.R.; Bekenstein, U.; Destroismaisons, L.; Toutant, J.; et al. TDP-43 Regulates the Alternative Splicing of HnRNP A1 to Yield an Aggregation-Prone Variant in Amyotrophic Lateral Sclerosis. Brain 2018, 141, 1320–1333. [Google Scholar] [CrossRef]
- He, Y.; Smith, R. Nuclear Functions of Heterogeneous Nuclear Ribonucleoproteins A/B. Cell. Mol. Life Sci. 2009, 66, 1239–1256. [Google Scholar] [CrossRef]
- Krüger, A.C.; Raarup, M.K.; Nielsen, M.M.; Kristensen, M.; Besenbacher, F.; Kjems, J.; Birkedal, V. Interaction of HnRNP A1 with Telomere DNA G-Quadruplex Structures Studied at the Single Molecule Level. Eur. Biophys. J. 2010, 39, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Hayashi, M.K.; Zhang, Y.; Manche, L.; Krainer, A.R.; Xu, R.M. Crystal Structure of the Two-RRM Domain of HnRNP A1 (UP1) Complexed with Single-Stranded Telomeric DNA. Genes Dev. 1999, 13, 1102–1115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.S.; Manche, L.; Xu, R.M.; Krainer, A.R. HnRNP A1 Associates with Telomere Ends and Stimulates Telomerase Activity. RNA 2006, 12, 1116–1128. [Google Scholar] [CrossRef]
- Mckay, S.J.; Cooke, H. HnRNP A2/B1 Binds Specifically to Single Stranded Vertebrate Telomeric Repeat TTAGGGn. Nucleic Acids Res. 1992, 20, 6461–6464. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-H.; Chen, C.-C.; Hsiao, Y.-C.; Lin, Y.-H.; Pi, W.-C.; Huang, P.-R.; Wang, T.-C.; Chen, C.-Y. Heterogeneous Nuclear Ribonucleoproteins A1 and A2 Function in Telomerase-Dependent Maintenance of Telomeres. Cancers 2019, 11, 334. [Google Scholar] [CrossRef]
- Moran-Jones, K.; Wayman, L.; Kennedy, D.D.; Reddel, R.R.; Sara, S.; Snee, M.J.; Smith, R. HnRNP A2, a Potential SsDNA/RNA Molecular Adapter at the Telomere. Nucleic Acids Res. 2005, 33, 486–496. [Google Scholar] [CrossRef] [PubMed]
- LaBranche, H.; Dupuis, S.; Ben-David, Y.; Bani, M.R.; Wellinger, R.J.; Chabot, B. Telomere Elongation by HnRNP A1 and a Derivative That Interacts with Telomeric Repeats and Telomerase. Nat. Genet. 1998, 19, 199–202. [Google Scholar] [CrossRef]
- Roy, R.; Huang, Y.; Seckl, M.J.; Pardo, O.E. Emerging Roles of HnRNPA1 in Modulating Malignant Transformation. Wiley Interdiscip. Rev. RNA 2017, 8, e1431. [Google Scholar] [CrossRef] [PubMed]
- Patry, C.; Lemieux, B.; Wellinger, R.J.; Chabot, B. Targeting Heterogeneous Nuclear Ribonucleoparticule A1 and A2 Proteins by RNA Interference Promotes Cell Death in Transformed but Not in Normal Mouse Cell Lines. Mol. Cancer Ther. 2004, 3. [Google Scholar]
- Guha, M.; Srinivasan, S.; Johnson, F.B.; Ruthel, G.; Guja, K.; Garcia-Diaz, M.; Kaufman, B.A.; Glineburg, M.R.; Fang, J.; Nakagawa, H.; et al. HnRNPA2 Mediated Acetylation Reduces Telomere Length in Response to Mitochondrial Dysfunction. PLoS ONE 2018, 13, e0206897. [Google Scholar] [CrossRef]
- Chen, H.; Hewison, M.; Hu, B.; Adams, J.S. Heterogeneous Nuclear Ribonucleoprotein (HnRNP) Binding to Hormone Response Elements: A Cause of Vitamin D Resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 6109–6114. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.S.; Baumeister, P.; Kim, E.; Roy, B.; Hsieh, T.-Y.; Lai, M.; Lee, A.S. Heterogeneous Nuclear Ribonucleoproteins as Regulators of Gene Expression through Interactions with the Human Thymidine Kinase Promoter. J. Cell. Biochem. 2000, 79, 395–406. [Google Scholar] [CrossRef]
- Nishikawa, T.; Kuwano, Y.; Takahara, Y.; Nishida, K.; Rokutan, K. HnRNPA1 Interacts with G-Quadruplex in the TRA2B Promoter and Stimulates Its Transcription in Human Colon Cancer Cells. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Paramasivam, M.; Membrino, A.; Cogoi, S.; Fukuda, H.; Nakagama, H.; Xodo, L.E. Protein HnRNP A1 and Its Derivative Up1 Unfold Quadruplex DNA in the Human KRAS Promoter: Implications for Transcription. Nucleic Acids Res. 2009, 37, 2841–2853. [Google Scholar] [CrossRef] [PubMed]
- Scalabrin, M.; Frasson, I.; Ruggiero, E.; Perrone, R.; Tosoni, E.; Lago, S.; Tassinari, M.; Palù, G.; Richter, S.N. The Cellular Protein HnRNP A2/B1 Enhances HIV-1 Transcription by Unfolding LTR Promoter G-Quadruplexes. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Cogoi, S.; Rapozzi, V.; Cauci, S.; Xodo, L.E. Critical Role of HnRNP A1 in Activating KRAS Transcription in Pancreatic Cancer Cells: A Molecular Mechanism Involving G4 DNA. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1389–1398. [Google Scholar] [CrossRef]
- Cui, H.; Wu, F.; Sun, Y.; Fan, G.; Wang, Q. Up-Regulation and Subcellular Localization of HnRNP A2/B1 in the Development of Hepatocellular Carcinoma. BMC Cancer 2010, 10, 356. [Google Scholar] [CrossRef]
- Zech, V.F.E.; Dlaska, M.; Tzankov, A.; Hilbe, W. Prognostic and Diagnostic Relevance of HnRNP A2/B1, HnRNP B1 and S100 A2 in Non-Small Cell Lung Cancer. Cancer Detect. Prev. 2006, 30, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-E.; Li, T.; Shi, S.; Chen, D.-X.; Chen, W.; Chen, H. ESCO2 Promotes Lung Adenocarcinoma Progression by Regulating HnRNPA1 Acetylation. Res. Sq. Prepr. Serv. 2020. [Google Scholar] [CrossRef]
- Han, X.; Xiang, X.; Yang, H.; Zhang, H.; Liang, S.; Wei, J.; Yu, J. P300-Catalyzed Lysine Crotonylation Promotes the Proliferation, Invasion, and Migration of HeLa Cells via Heterogeneous Nuclear Ribonucleoprotein A1. Anal. Cell. Pathol. 2020, 2020. [Google Scholar] [CrossRef]
- Best, A.; Dagliesh, C.; Ehrmann, I.; Kheirollahi-Kouhestani, M.; Tyson-Capper, A.; Elliott, D.J. Expression of Tra2 β in Cancer Cells as a Potential Contributory Factor to Neoplasia and Metastasis. Int. J. Cell Biol. 2013, 2013. [Google Scholar]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Hu, J.; Chen, Z.; Xia, D.; Wu, J.; Xu, H.; Ye, Z.Q. Promoter-Associated Small Double-Stranded RNA Interacts with Heterogeneous Nuclear Ribonucleoprotein A2/B1 to Induce Transcriptional Activation. Biochem. J. 2012, 447, 407–416. [Google Scholar] [CrossRef]
- Meredith, E.K.; Balas, M.M.; Sindy, K.; Haislop, K.; Johnson, A.M. An RNA Matchmaker Protein Regulates the Activity of the Long Noncoding RNA HOTAIR. RNA 2016, 22, 995–1010. [Google Scholar] [CrossRef]
- Nguyen, E.D.; Balas, M.M.; Griffin, A.M.; Roberts, J.T.; Johnson, A.M. Global Profiling of HnRNP A2/B1-RNA Binding on Chromatin Highlights LncRNA Interactions. RNA Biol. 2018, 15, 901–913. [Google Scholar] [CrossRef]
- Carpenter, S.; Aiello, D.; Atianand, M.K.; Ricci, E.P.; Gandhi, P.; Hall, L.L.; Byron, M.; Monks, B.; Henry-Bezy, M.; Lawrence, J.B.; et al. A Long Noncoding RNA Mediates Both Activation and Repression of Immune Response Genes. Science 2013, 341, 789–792. [Google Scholar] [CrossRef]
- Barrandon, C.; Bonnet, F.; Nguyen, V.T.; Labas, V.; Bensaude, O. The Transcription-Dependent Dissociation of P-TEFb-HEXIM1-7SK RNA Relies upon Formation of HnRNP-7SK RNA Complexes. Mol. Cell. Biol. 2007, 27, 6996–7006. [Google Scholar] [CrossRef]
- Lemieux, B.; Blanchette, M.; Monette, A.; Mouland, A.J.; Wellinger, R.J.; Chabot, B. A Function for the HnRNP A1/A2 Proteins in Transcription Elongation. PLoS ONE 2015, 10, e0126654. [Google Scholar] [CrossRef]
- Guha, M.; Pan, H.; Fang, J.-K.; Avadhani, N.G. Heterogeneous Nuclear Ribonucleoprotein A2 Is a Common Transcriptional Coactivator in the Nuclear Transcription Response to Mitochondrial Respiratory Stress. Mol. Biol. Cell 2009, 20, 4107–4119. [Google Scholar] [CrossRef]
- Guha, M.; Fang, J.K.; Monks, R.; Birnbaum, M.J.; Avadhani, N.G. Activation of Akt Is Essential for the Propagation of Mitochondrial Respiratory Stress Signaling and Activation of the Transcriptional Coactivator Heterogeneous Ribonucleoprotein A2. Mol. Biol. Cell 2010, 21, 3578–3589. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Srinivasan, S.; Guja, K.; Mejia, E.; Garcia-Diaz, M.; Johnson, F.B.; Ruthel, G.; Kaufman, B.A.; Rappaport, E.F.; Glineburg, M.R.; et al. HnRNPA2 Is a Novel Histone Acetyltransferase That Mediates Mitochondrial Stress-Induced Nuclear Gene Expression. Cell Discov. 2016, 2, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mili, S.; Shu, H.J.; Zhao, Y.; Piñol-Roma, S. Distinct RNP Complexes of Shuttling HnRNP Proteins with Pre-MRNA and MRNA: Candidate Intermediates in Formation and Export of MRNA. Mol. Cell. Biol. 2001, 21, 7307–7319. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kohlstaedt, L.A.; Damianov, A.; Rio, D.C.; Black, D.L. Polypyrimidine Tract Binding Protein Controls the Transition from Exon Definition to an Intron Defined Spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Bruun, G.H.; Doktor, T.K.; Borch-Jensen, J.; Masuda, A.; Krainer, A.R.; Ohno, K.; Andresen, B.S. Global Identification of HnRNP A1 Binding Sites for SSO-Based Splicing Modulation. BMC Biol. 2016, 14, 1–19. [Google Scholar] [CrossRef]
- Cáceres, J.F.; Stamm, S.; Helfman, D.M.; Krainer, A.R. Regulation of Alternative Splicing in Vivo by Overexpression of Antagonistic Splicing Factors. Science 1994, 265, 1706–1709. [Google Scholar] [CrossRef]
- Olsen, R.K.J.; Brøner, S.; Sabaratnam, R.; Doktor, T.K.; Andersen, H.S.; Bruun, G.H.; Gahrn, B.; Stenbroen, V.; Olpin, S.E.; Dobbie, A.; et al. The ETFDH c.158A>G Variation Disrupts the Balanced Interplay of ESE- and ESS-Binding Proteins Thereby Causing Missplicing and Multiple Acyl-CoA Dehydrogenation Deficiency. Hum. Mutat. 2014, 35, 86–95. [Google Scholar] [CrossRef]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.D.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative Genome-Wide Analysis Reveals Cooperative Regulation of Alternative Splicing by HnRNP Proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef]
- Martinez, F.J.; Pratt, G.A.; Van Nostrand, E.L.; Batra, R.; Huelga, S.C.; Kapeli, K.; Freese, P.; Chun, S.J.; Ling, K.; Gelboin-Burkhart, C.; et al. Protein-RNA Networks Regulated by Normal and ALS-Associated Mutant HNRNPA2B1 in the Nervous System. Neuron 2016, 92, 780–795. [Google Scholar] [CrossRef]
- Van Oordt, W.V.D.H.; Diaz-Meco, M.T.; Lozano, J.; Krainer, A.R.; Moscat, J.; Cáceres, J.F. The MKK(3/6)-P38-Signaling Cascade Alters the Subcellular Distribution of HnRNP A1 and Modulates Alternative Splicing Regulation. J. Cell Biol. 2000, 149, 307–316. [Google Scholar] [CrossRef]
- Guil, S.; Long, J.C.; Cáceres, J.F. HnRNP A1 Relocalization to the Stress Granules Reflects a Role in the Stress Response. Mol. Cell. Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef]
- Ziaei, S.; Shimada, N.; Kucharavy, H.; Hubbard, K. MNK1 Expression Increases during Cellular Senescence and Modulates the Subcellular Localization of HnRNP A1. Exp. Cell Res. 2012, 318, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Bolanos, L.C.; Choi, K.; Liu, X.; Christie, S.; Akunuru, S.; Kumar, R.; Wang, D.; Chen, X.; Greis, K.D.; et al. Ubiquitination of HnRNPA1 by TRAF6 Links Chronic Innate Immune Signaling with Myelodysplasia. Nat. Immunol. 2017, 18, 236–245. [Google Scholar] [CrossRef]
- Wang, F.; Fu, X.; Chen, P.; Wu, P.; Fan, X.; Li, N.; Zhu, H.; Jia, T.T.; Ji, H.; Wang, Z.; et al. SPSB1-Mediated HnRNP A1 Ubiquitylation Regulates Alternative Splicing and Cell Migration in EGF Signaling. Cell Res. 2017, 27, 540–558. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.M.; Lin, H.; Wallace, A.J.; Kim, G.; Draper, J.M.; Haeussler, M.; Katzman, S.; Toloue, M.; Liu, Y.; Sanford, J.R. HNRNPA1 Promotes Recognition of Splice Site Decoys by U2AF2 in Vivo. Genome Res. 2018, 28, 689–698. [Google Scholar] [CrossRef]
- De Araújo, M.M.; Bonnal, S.; Hastings, M.L.; Krainer, A.R.; Valcárcel, J. Differential 3′ Splice Site Recognition of SMN1 and SMN2 Transcripts by U2AF and U2 SnRNP. RNA 2009, 15, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Gupta, S.C.; Peng, W.X.; Zhou, N.; Pochampally, R.; Atfi, A.; Watabe, K.; Lu, Z.; Mo, Y.Y. Regulation of Alternative Splicing of Bcl-x by BC200 Contributes to Breast Cancer Pathogenesis. Cell Death Dis. 2016, 7, e2262. [Google Scholar] [CrossRef] [PubMed]
- Loh, T.J.; Moon, H.; Cho, S.; Jang, H.; Liu, Y.C.; Tai, H.; Jung, D.W.; Williams, D.R.; Kim, H.R.; Shin, M.G.; et al. CD44 Alternative Splicing and HnRNP A1 Expression Are Associated with the Metastasis of Breast Cancer. Oncol. Rep. 2015, 34, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Tauler, J.; Zudaire, E.; Liu, H.; Shih, J.; Mulshine, J.L. HnRNP A2/B1 Modulates Epithelial-Mesenchymal Transition in Lung Cancer Cell Lines. Cancer Res. 2010, 70, 7137–7147. [Google Scholar] [CrossRef]
- Shi, X.; Ran, L.; Liu, Y.; Zhong, S.H.; Zhou, P.P.; Liao, M.X.; Fang, W. Knockdown of HnRNP A2/B1 Inhibits Cell Proliferation, Invasion and Cell Cycle Triggering Apoptosis in Cervical Cancer via PI3K/AKT Signaling Pathway. Oncol. Rep. 2018, 39, 939–950. [Google Scholar] [CrossRef]
- Rajani, D.K.; Walch, M.; Martinvalet, D.; Thomas, M.P.; Lieberman, J. Alterations in RNA Processing during Immune-Mediated Programmed Cell Death. Proc. Natl. Acad. Sci. USA 2012, 109, 8688–8693. [Google Scholar] [CrossRef] [PubMed]
- Doktor, T.K.; Schroeder, L.D.; Vested, A.; Palmfeldt, J.; Andersen, H.S.; Gregersen, N.; Andresen, B.S. SMN2 Exon 7 Splicing Is Inhibited by Binding of HnRNP A1 to a Common ESS Motif That Spans the 3’ Splice Site. Hum. Mutat. 2011, 32, 220–230. [Google Scholar] [CrossRef]
- Kashima, T.; Manley, J.L. A Negative Element in SMN2 Exon 7 Inhibits Splicing in Spinal Muscular Atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef]
- Beusch, I.; Barraud, P.; Moursy, A.; Cléry, A.; Allain, F.H.T. Tandem HnRNP A1 RNA Recognition Motifs Act in Concert to Repress the Splicing of Survival Motor Neuron Exon 7. Elife 2017, 6. [Google Scholar] [CrossRef]
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Barbash, S.; Shaltiel, G.; Goll, Y.; Hanin, G.; Greenberg, D.S.; Ketzef, M.; Becker, A.J.; Friedman, A.; Soreq, H. Cholinergic-associated Loss of HnRNP-A/B in Alzheimer’s Disease Impairs Cortical Splicing and Cognitive Function in Mice. EMBO Mol. Med. 2012, 4, 730–742. [Google Scholar] [CrossRef]
- Kolisnyk, B.; Al-Onaizi, M.A.; Xu, J.; Parfitt, G.M.; Ostapchenko, V.G.; Hanin, G.; Soreq, H.; Prado, M.A.M.; Prado, V.F. Cholinergic Regulation of HnRNPA2/B1 Translation by M1 Muscarinic Receptors. J. Neurosci. 2016, 36, 6287–6296. [Google Scholar] [CrossRef] [PubMed]
- Kolisnyk, B.; Al-Onaizi, M.A.; Hirata, P.H.F.; Guzman, M.S.; Nikolova, S.; Barbash, S.; Soreq, H.; Bartha, R.; Prado, M.A.M.; Prado, V.F. Forebrain Deletion of the Vesicular Acetylcholine Transporter Results in Deficits in Executive Function, Metabolic, and RNA Splicing Abnormalities in the Prefrontal Cortex. J. Neurosci. 2013, 33, 14908–14920. [Google Scholar] [CrossRef] [PubMed]
- Donev, R.; Newall, A.; Thome, J.; Sheer, D. A Role for SC35 and HnRNPA1 in the Determination of Amyloid Precursor Protein Isoforms. Mol. Psychiatry 2007, 12, 681–690. [Google Scholar] [CrossRef]
- Chabot, B.; LeBel, C.; Hutchison, S.; Nasim, F.H.; Simard, M.J. Heterogeneous nuclear ribonucleoprotein particle A/B proteins and the control of alternative splicing of the mammalian heterogeneous nuclear ribonucleoprotein particle A1 pre-mRNA. Prog. Mol. Subcell. Biol. 2003, 31, 59–88. [Google Scholar] [PubMed]
- McGlincy, N.J.; Tan, L.Y.; Paul, N.; Zavolan, M.; Lilley, K.S.; Smith, C.W.J. Expression Proteomics of UPF1 Knockdown in HeLa Cells Reveals Autoregulation of HnRNP A2/B1 Mediated by Alternative Splicing Resulting in Nonsense-Mediated MRNA Decay. BMC Genom. 2010, 11, 1–19. [Google Scholar] [CrossRef]
- Suzuki, H.; Matsuoka, M. HnRNPA1 Autoregulates Its Own MRNA Expression to Remain Non-Cytotoxic. Mol. Cell. Biochem. 2017, 427, 123–131. [Google Scholar] [CrossRef]
- Michael, W.M.; Choi, M.; Dreyfuss, G. A Nuclear Export Signal in HnRNP A1: A Signal-Mediated, Temperature-Dependent Nuclear Protein Export Pathway. Cell 1995, 83, 415–422. [Google Scholar] [CrossRef]
- Siomi, H.; Dreyfuss, G. A Nuclear Localization Domain in the HnRNP A1 Protein. J. Cell Biol. 1995, 129, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Durie, D.; Li, H.; Liu, B.Q.; Skehel, J.M.; Mauri, F.; Cuorvo, L.V.; Barbareschi, M.; Guo, L.; Holcik, M.; et al. HnRNPA1 Couples Nuclear Export and Translation of Specific MRNAs Downstream of FGF-2/S6K2 Signalling. Nucleic Acids Res. 2014, 42, 12483–12497. [Google Scholar] [CrossRef]
- Pollard, V.W.; Michael, W.M.; Nakielny, S.; Siomi, M.C.; Wang, F.; Dreyfuss, G. A Novel Receptor-Mediated Nuclear Protein Import Pathway. Cell 1996, 86, 985–994. [Google Scholar] [CrossRef]
- Siomi, M.C.; Eder, P.S.; Kataoka, N.; Wan, L.; Liu, Q.; Dreyfuss, G. Transportin-Mediated Nuclear Import of Heterogeneous Nuclear RNP Proteins. J. Cell Biol. 1997, 138, 1181–1192. [Google Scholar] [CrossRef]
- Rebane, A.; Aab, A.; Steitz, J.A. Transportins 1 and 2 Are Redundant Nuclear Import Factors for HnRNP A1 and HuR. RNA 2004, 10, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Nakielny, S.; Siomi, M.C.; Siomi, H.; Michael, W.M.; Pollard, V.; Dreyfuss, G. Transportin: Nuclear Transport Receptor of a Novel Nuclear Protein Import Pathway. Exp. Cell Res. 1996, 229, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.; Khalaila, I. The Effect of O-GlcNAcylation on HnRNP A1 Translocation and Interaction with Transportin1. Exp. Cell Res. 2017, 350, 210–217. [Google Scholar] [CrossRef]
- Wang, L.; Wen, M.; Cao, X. Nuclear HnRNPA2B1 Initiates and Amplifies the Innate Immune Response to DNA Viruses. Science 2019, 365. [Google Scholar] [CrossRef]
- Friend, L.R.; Landsberg, M.J.; Nouwens, A.S.; Wei, Y.; Rothnagel, J.A.; Smith, R. Arginine Methylation of HnRNP A2 Does Not Directly Govern Its Subcellular Localization. PLoS ONE 2013, 8, e75669. [Google Scholar] [CrossRef]
- Nichols, R.C.; Wang, X.W.; Tang, J.; Hamilton, B.J.; High, F.A.; Herschman, H.R.; Rigby, W.F.C. The RGG Domain in HnRNP A2 Affects Subcellular Localization. Exp. Cell Res. 2000, 256, 522–532. [Google Scholar] [CrossRef]
- Alexovič, M.; Sabo, J.; Longuespée, R. Microproteomic Sample Preparation. Proteomics 2021, 21, 2000318. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Rathod, R.J.; Rajkumar, S.; Kennedy, D. Nervous Translation, Do You Get the Message? A Review of MRNPs, MRNA-Protein Interactions and Translational Control within Cells of the Nervous System. Cell. Mol. Life Sci. 2014, 71, 3917–3937. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Kidd, G.J.; Carson, J.H.; Smith, R. HnRNP A2 Selectively Binds the Cytoplasmic Transport Sequence of Myelin Basic Protein MRNA. Biochemistry 1998, 37, 7021–7029. [Google Scholar] [CrossRef]
- Smith, R. Moving Molecules: MRNA Trafficking in Mammalian Oligodendrocytes and Neurons. Neuroscientist 2004, 10, 495–500. [Google Scholar] [CrossRef]
- Carson, J.H.; Barbarese, E. Systems Analysis of RNA Trafficking in Neural Cells. Biol. Cell 2005, 97, 51–62. [Google Scholar] [CrossRef]
- Han, S.P.; Friend, L.R.; Carson, J.H.; Korza, G.; Barbarese, E.; Maggipinto, M.; Hatfield, J.T.; Rothnagel, J.A.; Smith, R. Differential Subcellular Distributions and Trafficking Functions of HnRNP A2/B1 Spliceoforms. Traffic 2010, 11, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Muslimov, I.A.; Iacoangeli, A.; Brosius, J.; Tiedge, H. Spatial Codes in Dendritic BC1 RNA. J. Cell Biol. 2006, 175, 427–439. [Google Scholar] [CrossRef]
- Leal, G.; Afonso, P.M.; Duarte, C.B. Neuronal Activity Induces Synaptic Delivery of HnRNP A2/B1 by a BDNF-Dependent Mechanism in Cultured Hippocampal Neurons. PLoS ONE 2014, 9, e108175. [Google Scholar] [CrossRef]
- Kosturko, L.D.; Maggipinto, M.J.; Korza, G.; Lee, J.W.; Carson, J.H.; Barbarese, E. Heterogeneous Nuclear Ribonucleoprotein (HnRNP) E1 Binds to HnRNP A2 and Inhibits Translation of A2 Response Element MRNAs. Mol. Biol. Cell 2006, 17, 3521–3533. [Google Scholar] [CrossRef] [PubMed]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated HnRNPA2B1 Controls the Sorting of MiRNAs into Exosomes through Binding to Specific Motifs. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Pérez-Boza, J.; Boeckx, A.; Lion, M.; Dequiedt, F.; Struman, I. HnRNPA2B1 Inhibits the Exosomal Export of MiR-503 in Endothelial Cells. Cell. Mol. Life Sci. 2020, 77, 4413–4428. [Google Scholar] [CrossRef]
- Tietje, A.; Maron, K.N.; Wei, Y.; Feliciano, D.M. Cerebrospinal Fluid Extracellular Vesicles Undergo Age Dependent Declines and Contain Known and Novel Non-Coding RNAs. PLoS ONE 2014, 9, e113116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, L.; Zou, W.; Chen, X.; Roizman, B.; Zhou, G.G. HnRNPA2B1 Associated with Recruitment of RNA into Exosomes Plays a Key Role in Herpes Simplex Virus 1 Release from Infected Cells. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF Secreted MiR-522 Suppresses Ferroptosis and Promotes Acquired Chemo-Resistance in Gastric Cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Li, S.; Guo, C.; Li, Q.; Zhang, X.; Li, M.; Mi, S. Heterogeneous Nuclear Ribonucleoprotein A1 Loads Batched Tumor-Promoting MicroRNAs Into Small Extracellular Vesicles With the Assist of Caveolin-1 in A549 Cells. Front. Cell Dev. Biol. 2021, 9, 1417. [Google Scholar] [CrossRef]
- O’Carroll, D.; Schaefer, A. General Principals of MiRNA Biogenesis and Regulation in the Brain. Neuropsychopharmacology 2013, 38, 39–54. [Google Scholar] [CrossRef]
- Kooshapur, H.; Choudhury, N.R.; Simon, B.; Mühlbauer, M.; Jussupow, A.; Fernandez, N.; Jones, A.N.; Dallmann, A.; Gabel, F.; Camilloni, C.; et al. Structural Basis for Terminal Loop Recognition and Stimulation of Pri-MiRNA-18a Processing by HnRNP A1. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Michlewski, G.; Guil, S.; Cáceres, J.F. Stimulation of Pri-MiR-18a Processing by HnRNP A1. Adv. Exp. Med. Biol. 2010, 700, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Michlewski, G.; Guil, S.; Semple, C.A.; Cáceres, J.F. Posttranscriptional Regulation of MiRNAs Harboring Conserved Terminal Loops. Mol. Cell 2008, 32, 383–393. [Google Scholar] [CrossRef]
- Guil, S.; Cáceres, J.F. The Multifunctional RNA-Binding Protein HnRNP A1 Is Required for Processing of MiR-18a. Nat. Struct. Mol. Biol. 2007, 14, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Michlewski, G.; Cáceres, J.F. Antagonistic Role of HnRNP A1 and KSRP in the Regulation of Let-7a Biogenesis. Nat. Struct. Mol. Biol. 2010, 17, 1011–1018. [Google Scholar] [CrossRef]
- Klinge, C.M.; Piell, K.M.; Tooley, C.S.; Rouchka, E.C. HNRNPA2/B1 Is Upregulated in Endocrine-Resistant LCC9 Breast Cancer Cells and Alters the MiRNA Transcriptome When Overexpressed in MCF-7 Cells. Sci. Rep. 2019, 9, 1–22. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of M6A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, X.; Lei, T.; Gu, Y.; Gu, J.; Huang, J.; Lu, B.; Yuan, L.; Sun, M.; Wang, Z. Integrative Analysis of NSCLC Identifies LINC01234 as an Oncogenic LncRNA That Interacts with HNRNPA2B1 and Regulates MiR-106b Biogenesis. Mol. Ther. 2020, 28, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, Y.; Jiang, J.; Jiang, H.; Song, J.; Han, T.; Shi, J.; Qiao, H. The Long Noncoding RNA Colon Cancer-Associated Transcript-1/MiR-490 Axis Regulates Gastric Cancer Cell Migration by Targeting HnRNPA1. IUBMB Life 2016, 68, 201–210. [Google Scholar] [CrossRef]
- Sokół, E.; Kędzierska, H.; Czubaty, A.; Rybicka, B.; Rodzik, K.; Tański, Z.; Bogusławska, J.; Piekiełko-Witkowska, A. MicroRNA-Mediated Regulation of Splicing Factors SRSF1, SRSF2 and HnRNP A1 in Context of Their Alternatively Spliced 3′UTRs. Exp. Cell Res. 2018, 363, 208–217. [Google Scholar] [CrossRef]
- Fung, L.; Guzman, H.; Sevrioukov, E.; Idica, A.; Park, E.; Bochnakian, A.; Daugaard, I.; Jury, D.; Mortazavi, A.; Zisoulis, D.G.; et al. MiR-128 Restriction of LINE-1 (L1) Retrotransposition Is Dependent on Targeting HnRNPA1 MRNA. Int. J. Mol. Sci. 2019, 20, 1955. [Google Scholar] [CrossRef]
- Zhang, E.; Li, X. LncRNA SOX2-OT Regulates Proliferation and Metastasis of Nasopharyngeal Carcinoma Cells through MiR-146b-5p/HNRNPA2B1 Pathway. J. Cell. Biochem. 2019, 120, 16575–16588. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liang, L.; Dong, Q.; Huan, L.; He, J.; Li, B.; Yang, C.; Jin, H.; Wei, L.; Yu, C.; et al. Long Noncoding RNA MiR503HG, a Prognostic Indicator, Inhibits Tumor Metastasis by Regulating the HNRNPA2B1/NF-ΚB Pathway in Hepatocellular Carcinoma. Theranostics 2018, 8, 2814–2829. [Google Scholar] [CrossRef]
- Yao, A.; Xiang, Y.; Si, Y.; Fan, L.; Li, J.; Li, H.; Guo, W.; He, H.; Liang, X.; Tan, Y.; et al. PKM2 Promotes Glucose Metabolism through a Let-7a-5p/Stat3/HnRNP-A1 Regulatory Feedback Loop in Breast Cancer Cells. J. Cell. Biochem. 2019, 120, 6542–6554. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Yang, P.; Amin, S.; Li, Z. A Novel MiR-206/HnRNPA1/PKM2 Axis Reshapes the Warburg Effect to Suppress Colon Cancer Growth. Biochem. Biophys. Res. Commun. 2020, 531, 465–471. [Google Scholar] [CrossRef]
- Luan, W.; Wang, Y.; Chen, X.; Shi, Y.; Wang, J.; Zhang, J.; Qian, J.; Li, R.; Tao, T.; Wei, W.; et al. PKM2 Promotes Glucose Metabolism and Cell Growth in Gliomas through a Mechanism Involving a Let-7a/c-Myc/HnRNPA1 Feedback Loop. Oncotarget 2015, 6, 13006–13018. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP Proteins Controlled by C-Myc Deregulate Pyruvate Kinase MRNA Splicing in Cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Hsu, M.-C.; Pan, M.-R.; Chu, P.-Y.; Tsai, Y.-L.; Tsai, C.-H.; Shan, Y.-S.; Chen, L.-T.; Hung, W.-C. Protein Arginine Methyltransferase 3 Enhances Chemoresistance in Pancreatic Cancer by Methylating HnRNPA1 to Increase ABCG2 Expression. Cancers 2018, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Henics, T.; Sanfridson, A.; Hamilton, B.J.; Nagy, E.; Rigby, W.F. Enhanced Stability of Interleukin-2 MRNA in MLA 144 Cells. Possible Role of Cytoplasmic AU-Rich Sequence-Binding Proteins. J. Biol. Chem. 1994, 269, 5377–5383. [Google Scholar] [CrossRef]
- Hamilton, B.J.N.; Burns, C.M.; Nichols, R.C.; Rigby, W.F.C. Modulation of AUUUA Response Element Binding by Heterogeneous Nuclear Ribonucleoprotein A1 in Human T Lymphocytes: The Roles of Cytoplasmic Location, Transcription, and Phosphorylation. J. Biol. Chem. 1997, 272, 28732–28741. [Google Scholar] [CrossRef]
- Hamilton, B.J.; Nagy, E.; Malter, J.S.; Arrick, B.A.; Rigby, W.F. Association of Heterogeneous Nuclear Ribonucleoprotein A1 and C Proteins with Reiterated AUUUA Sequences. J. Biol. Chem. 1993, 268, 8881–8887. [Google Scholar] [CrossRef]
- Brooks, S.A.; Rigby, W.F. Characterization of the MRNA Ligands Bound by the RNA Binding Protein HnRNP A2 Utilizing a Novel in Vivo Technique. Nucleic Acids Res. 2000, 28, 49. [Google Scholar] [CrossRef][Green Version]
- Griffin, M.E.; Hamilton, B.J.N.; Roy, K.M.; Du, M.; Willson, A.M.; Keenan, B.J.; Wang, X.W.; Nichols, R.C. Post-Transcriptional Regulation of Glucose Transporter-1 by an AU-Rich Element in the 3′UTR and by HnRNP A2. Biochem. Biophys. Res. Commun. 2004, 318, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Caput, D.; Beutler, B.; Hartog, K.; Thayer, R.; Brown-Shimer, S.; Cerami, A. Identification of a Common Nucleotide Sequence in the 3’-Untranslated Region of MRNA Molecules Specifying Inflammatory Mediators. Proc. Natl. Acad. Sci. USA 1986, 83, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.A.; Shyu, A. Bin AU-Rich Elements: Characterization and Importance in MRNA Degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Geissler, R.; Simkin, A.; Floss, D.; Patel, R.; Fogarty, E.A.; Scheller, J.; Grimson, A. A Widespread Sequence-Specific MRNA Decay Pathway Mediated by HnRNPs A1 and A2/B1. Genes Dev. 2016, 30, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Seo, J.Y.; Ryu, H.G.; Kim, D.Y.; Lee, K.H.; Kim, K.T. BDNF-Induced Local Translation of GluA1 Is Regulated by HNRNP A2/B1. Sci. Adv. 2020, 6, eabd2163. [Google Scholar] [CrossRef]
- Hung, C.Y.; Wang, Y.C.; Chuang, J.Y.; Young, M.J.; Liaw, H.; Chang, W.C.; Hung, J.J. Nm23-H1-Stabilized HnRNPA2/B1 Promotes Internal Ribosomal Entry Site (IRES)-Mediated Translation of Sp1 in the Lung Cancer Progression. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Lal, S.K. The Multifarious Roles of Heterogeneous Ribonucleoprotein A1 in Viral Infections. Rev. Med. Virol. 2020, 30, e2097. [Google Scholar] [CrossRef]
- Bonnal, S.; Pileur, F.; Orsini, C.; Parker, F.; Pujol, F.; Prats, A.C.; Vagner, S. Heterogeneous Nuclear Ribonucleoprotein A1 Is a Novel Internal Ribosome Entry Site Trans-Acting Factor That Modulates Alternative Initiation of Translation of the Fibroblast Growth Factor 2 MRNA. J. Biol. Chem. 2005, 280, 4144–4153. [Google Scholar] [CrossRef]
- Martin, J.; Masri, J.; Cloninger, C.; Holmes, B.; Artinian, N.; Funk, A.; Ruegg, T.; Anderson, L.; Bashir, T.; Bernath, A.; et al. Phosphomimetic Substitution of Heterogeneous Nuclear Ribonucleoprotein A1 at Serine 199 Abolishes AKT-Dependent Internal Ribosome Entry Site-Transacting Factor (ITAF) Function via Effects on Strand Annealing and Results in Mammalian Target of Rapamycin Co. J. Biol. Chem. 2011, 286, 16402–16413. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Q.; An, W.; Yang, F.; Maguire, E.M.; Chen, D.; Zhang, C.; Wen, G.; Yang, M.; Dai, B.; et al. Novel Pathological Role of HnRNPA1 (Heterogeneous Nuclear Ribonucleoprotein A1) in Vascular Smooth Muscle Cell Function and Neointima Hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2182–2194. [Google Scholar] [CrossRef]
- White, R.; Gonsior, C.; Krämer-Albers, E.M.; Stöhr, N.; Hüttelmaier, S.; Trotter, J. Activation of Oligodendroglial Fyn Kinase Enhances Translation of MRNAs Transported in HnRNP A2-Dependent RNA Granules. J. Cell Biol. 2008, 181, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Liu, F.; Gao, L.-B.; Han, R.; Chen, C.; Ding, X.; Li, S.; Lu, K.; Yang, L.; et al. Heterogeneous Nuclear Ribonucleoprotein A2/B1 Is a Negative Regulator of Human Breast Cancer Metastasis by Maintaining the Balance of Multiple Genes and Pathways. EBioMedicine 2020, 51, 102583. [Google Scholar] [CrossRef]
- Mahboubi, H.; Stochaj, U. Cytoplasmic Stress Granules: Dynamic Modulators of Cell Signaling and Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, S.; Ho, A.; Woo, Y.M.; Kwak, H.; Lee, J.H. Systematic Characterization of Stress-Induced RNA Granulation. Mol. Cell 2018, 70, 175–187.e8. [Google Scholar] [CrossRef]
- Clarke, J.-P.W.E.; Thibault, P.A.; Salapa, H.E.; Kim, D.E.; Hutchinson, C.; Levin, M.C. Multiple Sclerosis-Associated HnRNPA1 Mutations Alter HnRNPA1 Dynamics and Influence Stress Granule Formation. Int. J. Mol. Sci. 2021, 22, 2909. [Google Scholar] [CrossRef]
- Ryan, V.H.; Dignon, G.L.; Zerze, G.H.; Chabata, C.V.; Silva, R.; Conicella, A.E.; Amaya, J.; Burke, K.A.; Mittal, J.; Fawzi, N.L. Mechanistic View of HnRNPA2 Low-Complexity Domain Structure, Interactions, and Phase Separation Altered by Mutation and Arginine Methylation. Mol. Cell 2018, 69, 465–479. [Google Scholar] [CrossRef]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic Analysis of FUS Interacting Proteins Provides Insights into FUS Function and Its Role in ALS. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Bampton, A.; Gittings, L.M.; Fratta, P.; Lashley, T.; Gatt, A. The Role of HnRNPs in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2020, 140, 599–623. [Google Scholar] [CrossRef]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural Basis for Reversible Amyloids of HnRNPA1 Elucidates Their Role in Stress Granule Assembly. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Salapa, H.E.; Johnson, C.; Hutchinson, C.; Popescu, B.F.; Levin, M.C. Dysfunctional RNA Binding Proteins and Stress Granules in Multiple Sclerosis. J. Neuroimmunol. 2018, 324, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Levin, M. Novel Somatic Single Nucleotide Variants within the RNA Binding Protein HnRNP A1 in Multiple Sclerosis Patients. F1000Research 2014, 3, 132. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in Prion-like Domains in HnRNPA2B1 and HnRNPA1 Cause Multisystem Proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic Mutations in RNA-Binding Proteins and Their Roles in ALS. Hum. Genet. 2017, 136, 1193–1214. [Google Scholar] [CrossRef]
- Villa, C.; Fenoglio, C.; De Riz, M.; Clerici, F.; Marcone, A.; Benussi, L.; Ghidoni, R.; Gallone, S.; Cortini, F.; Serpente, M.; et al. Role of HnRNP-A1 and MiR-590-3p in Neuronal Death: Genetics and Expression Analysis in Patients with Alzheimer Disease and Frontotemporal Lobar Degeneration. Rejuvenation Res. 2011, 14, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct Stages in Stress Granule Assembly and Disassembly. Elife 2016, 5. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, K.; Xia, W.; Feng, G.; Gu, J.; Ma, Y.; Gui, X.; Zhang, X.; Fang, Y.; Sun, B.; et al. The Nuclear Localization Sequence Mediates HnRNPA1 Amyloid Fibril Formation Revealed by CryoEM Structure. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cipolat Mis, M.S.; Brajkovic, S.; Frattini, E.; Di Fonzo, A.; Corti, S. Autophagy in Motor Neuron Disease: Key Pathogenetic Mechanisms and Therapeutic Targets. Mol. Cell. Neurosci. 2016, 72, 84–90. [Google Scholar] [CrossRef]
- Purice, M.D.; Taylor, J.P. Linking HnRNP Function to ALS and FTD Pathology. Front. Neurosci. 2018, 12, 326. [Google Scholar] [CrossRef]
- Salapa, H.E.; Hutchinson, C.; Popescu, B.F.; Levin, M.C. Neuronal RNA-binding Protein Dysfunction in Multiple Sclerosis Cortex. Ann. Clin. Transl. Neurol. 2020, 7, 1214–1224. [Google Scholar] [CrossRef]
- Salapa, H.E.; Lee, S.; Shin, Y.; Levin, M.C. Contribution of the Degeneration of the Neuro-Axonal Unit to the Pathogenesis of Multiple Sclerosis. Brain Sci. 2017, 7, 69. [Google Scholar] [CrossRef]
- Salapa, H.E.; Libner, C.D.; Levin, M.C. Dysfunctional RNA-Binding Protein Biology and Neurodegeneration in Experimental Autoimmune Encephalomyelitis in Female Mice. J. Neurosci. Res. 2020, 98, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Libner, C.D.; Salapa, H.E.; Hutchinson, C.; Lee, S.; Levin, M.C. Antibodies to the RNA Binding Protein Heterogeneous Nuclear Ribonucleoprotein A1 Contribute to Neuronal Cell Loss in an Animal Model of Multiple Sclerosis. J. Comp. Neurol. 2020, 528, 1704–1724. [Google Scholar] [CrossRef]
- Libner, C.D.; Salapa, H.E.; Levin, M.C. The Potential Contribution of Dysfunctional RNA-Binding Proteins to the Pathogenesis of Neurodegeneration in Multiple Sclerosis and Relevant Models. Int. J. Mol. Sci. 2020, 21, 4571. [Google Scholar] [CrossRef]
- Wesselingh, R.; Butzkueven, H.; Buzzard, K.; Tarlinton, D.; O’Brien, T.J.; Monif, M. Innate Immunity in the Central Nervous System: A Missing Piece of the Autoimmune Encephalitis Puzzle? Front. Immunol. 2019, 10, 2066. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Hamasaki, H.; Wakamiya, T.; Koyama, S.; Suzuki, S.O.; Fujii, N.; Iwaki, T. Loss of HnRNPA1 in ALS Spinal Cord Motor Neurons with TDP-43-Positive Inclusions. Neuropathology 2015, 35, 37–43. [Google Scholar] [CrossRef]
- Mohagheghi, F.; Prudencio, M.; Stuani, C.; Cook, C.; Jansen-West, K.; Dickson, D.W.; Petrucelli, L.; Buratti, E. TDP-43 Functions within a Network of HnRNP Proteins to Inhibit the Production of a Truncated Human SORT1 Receptor. Hum. Mol. Genet. 2016, 25, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Maslyanskiy, A.; Lazareva, N.; Olinek, P.; Schierack, P.; Hentschel, C.; Cuccato, J.; Bogdanos, D.P.; Lapin, S.V.; Roggenbuck, D. Anti-HnRNP B1 (RA33) Autoantibodies Are Associated with the Clinical Phenotype in Russian Patients with Rheumatoid Arthritis and Systemic Sclerosis. J. Immunol. Res. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Hayer, S.; Tohidast-Akrad, M.; Haralambous, S.; Jahn-Schmid, B.; Skriner, K.; Trembleau, S.; Dumortier, H.; Pinol-Roma, S.; Redlich, K.; Schett, G.; et al. Aberrant Expression of the Autoantigen Heterogeneous Nuclear Ribonucleoprotein-A2 (RA33) and Spontaneous Formation of Rheumatoid Arthritis-Associated Anti-RA33 Autoantibodies in TNF-α Transgenic Mice. J. Immunol. 2005, 175, 8327–8336. [Google Scholar] [CrossRef]
- Sueoka, E.; Yukitake, M.; Iwanaga, K.; Sueoka, N.; Aihara, T.; Kuroda, Y. Autoantibodies against Heterogeneous Nuclear Ribonucleoprotein B1 in CSF of MS Patients. Ann. Neurol. 2004, 56, 778–786. [Google Scholar] [CrossRef]
- Caporali, R.; Bugatti, S.; Bruschi, E.; Cavagna, L.; Montecucco, C. Autoantibodies to Heterogeneous Nuclear Ribonucleoproteins. Autoimmunity 2005, 38, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, G.C.B.A.; Bestagno, M.; Cerino, A.; Negri, C.; Caporali, R.; Cobianchi, F.; Longhi, M.; Montecucco, C.M. Antibodies to HnRNP Core Protein A1 in Connective Tissue Diseases. J. Cell. Biochem. 1989, 40, 43–47. [Google Scholar] [CrossRef]
- Op De Beéck, K.; Maes, L.; Van den Bergh, K.; Derua, R.; Waelkens, E.; Van Steen, K.; Vermeersch, P.; Westhovens, R.; De Vlam, K.; Verschueren, P.; et al. Heterogeneous Nuclear RNPs as Targets of Autoantibodies in Systemic Rheumatic Diseases. Arthritis Rheum. 2012, 64, 213–221. [Google Scholar] [CrossRef]
- Lee, S.; Xu, L.; Shin, Y.; Gardner, L.; Hartzes, A.; Dohan, F.C.; Raine, C.; Homayouni, R.; Levin, M.C. A Potential Link between Autoimmunity and Neurodegeneration in Immune-Mediated Neurological Disease. J. Neuroimmunol. 2011, 235, 56–69. [Google Scholar] [CrossRef]
- Hassfeld, W.; Steiner, G.; Studnicka-Benke, A.; Skriner, K.; Graninger, W.; Fischer, I.; Smolen, J.S. Autoimmune Response to the Spliceosome. Arthritis Rheum. 1995, 38, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Yukitake, M.; Sueoka, E.; Sueoka-Aragane, N.; Sato, A.; Ohashi, H.; Yakushiji, Y.; Saito, M.; Osame, M.; Izumo, S.; Kuroda, Y. Significantly Increased Antibody Response to Heterogeneous Nuclear Ribonucleoproteins in Cerebrospinal Fluid of Multiple Sclerosis Patients but Not in Patients with Human T-Lymphotropic Virus Type I - Associated Myelopathy/Tropical Spastic Paraparesis. J. Neurovirol. 2008, 14, 130–135. [Google Scholar] [CrossRef]
- Nell, V.P.K.; Machold, K.P.; Stamm, T.A.; Eberl, G.; Heinzl, H.; Uffmann, M.; Smolen, J.S.; Steiner, G. Autoantibody Profiling as Early Diagnostic and Prognostic Tool for Rheumatoid Arthritis. Ann. Rheum. Dis. 2005, 64, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Steiner, G.; Skriner, K.; Smolen, J.S. Autoantibodies to the a/b Proteins of the Heterogeneous Nuclear Ribonucleoprotein Complex: Novel Tools for the Diagnosis of Rheumatic Diseases. Int. Arch. Allergy Immunol. 1996, 111, 314–319. [Google Scholar] [CrossRef]
- Steiner, G.; Skriner, K.; Hassfeld, W.; Smolen, J.S. Clinical and Immunological Aspects of Autoantibodies to RA33/HnRNP-A/B Proteins-A Link between RA, SLE and MCTD. Mol. Biol. Rep. 1996, 23, 167–171. [Google Scholar] [CrossRef]
- Schett, G.; Dumortier, H.; Hoefler, E.; Muller, S.; Steiner, G. B Cell Epitopes of the Heterogeneous Nuclear Ribonucleoprotein A2: Identification of a New Specific Antibody Marker for Active Lupus Disease. Ann. Rheum. Dis. 2009, 68, 729–735. [Google Scholar] [CrossRef]
- Sun, K.-H.; Tang, S.-J.; Wang, Y.-S.; Lin, W.-J.; You, R.-I. Autoantibodies to DsDNA Cross-React with the Arginine-Glycine-Rich Domain of Heterogeneous Nuclear Ribonucleoprotein A2 (HnRNP A2) and Promote Methylation of HnRNP A2. Rheumatology 2003, 42, 154–161. [Google Scholar] [CrossRef][Green Version]
- Muslimov, I.A.; Iacoangeli, A.; Eom, T.; Ruiz, A.; Lee, M.; Stephenson, S.; Ginzler, E.M.; Tiedge, H. Neuronal BC RNA Transport Impairments Caused by Systemic Lupus Erythematosus Autoantibodies. J. Neurosci. 2019, 39, 7759–7777. [Google Scholar] [CrossRef]
- Douglas, J.N.; Gardner, L.A.; Levin, M.C. Antibodies to an Intracellular Antigen Penetrate Neuronal Cells and Cause Deleterious Effects. J. Clin. Cell. Immunol. 2013, 4, 134. [Google Scholar] [CrossRef]
- Ong, S.E.; Mittler, G.; Mann, M. Identifying and Quantifying in Vivo Methylation Sites by Heavy Methyl SILAC. Nat. Methods 2004, 1, 119–126. [Google Scholar] [CrossRef]
- Thiede, B.; Dimmler, C.; Siejak, F.; Rudel, T. Predominant Identification of RNA-Binding Proteins in Fas-Induced Apoptosis by Proteome Analysis. J. Biol. Chem. 2001, 276, 26044–26050. [Google Scholar] [CrossRef]
- Gueroussov, S.; Weatheritt, R.J.; O’Hanlon, D.; Lin, Z.Y.; Narula, A.; Gingras, A.C.; Blencowe, B.J. Regulatory Expansion in Mammals of Multivalent HnRNP Assemblies That Globally Control Alternative Splicing. Cell 2017, 170, 324–339.e23. [Google Scholar] [CrossRef] [PubMed]
- Belhadj, S.; Terradas, M.; Munoz-Torres, P.M.; Aiza, G.; Navarro, M.; Capellá, G.; Valle, L. Candidate Genes for Hereditary Colorectal Cancer: Mutational Screening and Systematic Review. Hum. Mutat. 2020, 41, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Peng, B.; Han, Y.; Chen, W.V.; Rother, J.; Tomlinson, G.E.; Boland, C.R.; Chaussabel, M.; Frazier, M.L.; Amos, C.I. Mutations of HNRNPA0 and WIF1 Predispose Members of a Large Family to Multiple Cancers. Fam. Cancer 2015, 14, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Sun, S.; Zhang, L.; Tian, S.; Xu, K.; Zhang, G.; Chen, M. Integrative Analysis of DNA Methylation Identified 12 Signature Genes Specific to Metastatic CcRCC. Front. Oncol. 2020, 10, 556018. [Google Scholar] [CrossRef]
- La Thangue, N.B. DRTF1/E2F: An Expanding Family of Heterodimeric Transcription Factors Implicated in Cell-Cycle Control. Trends Biochem. Sci. 1994, 19, 108–114. [Google Scholar] [CrossRef]
- Li, Z.; Kreutzer, M.; Mikkat, S.; Miše, N.; Glocker, M.O.; Pützer, B.M. Proteomic Analysis of the E2F1 Response in P53-Negative Cancer Cells: New Aspects in the Regulation of Cell Survival and Death. Proteomics 2006, 6, 5735–5745. [Google Scholar] [CrossRef]
- Zhang, T.; Cheng, G.; Deng, L.; Yang, Y.; Sun, L.; Chen, P.; He, X.; Su, D.; Bi, N.; Qiu, B. Silence of S1 RNA Binding Domain 1 Represses Cell Growth and Promotes Apoptosis in Human Non-Small Cell Lung Cancer Cells. Transl. Lung Cancer Res. 2019, 8, 760–774. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Hasskamp, P.; Schmedding, I.; Morandell, S.; van Vugt, M.A.T.M.; Wang, X.Z.; Linding, R.; Ong, S.E.; Weaver, D.; Carr, S.A.; et al. DNA Damage Activates a Spatially Distinct Late Cytoplasmic Cell-Cycle Checkpoint Network Controlled by MK2-Mediated RNA Stabilization. Mol. Cell 2010, 40, 34–49. [Google Scholar] [CrossRef]
- Cannell, I.G.; Merrick, K.A.; Morandell, S.; Zhu, C.Q.; Braun, C.J.; Grant, R.A.; Cameron, E.R.; Tsao, M.S.; Hemann, M.T.; Yaffe, M.B. A Pleiotropic RNA-Binding Protein Controls Distinct Cell Cycle Checkpoints to Drive Resistance of P53-Defective Tumors to Chemotherapy. Cancer Cell 2015, 28, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Fujiya, M.; Kashima, S.; Sakatani, A.; Dokoshi, T.; Ando, K.; Ueno, N.; Iwama, T.; Moriichi, K.; Tanaka, H.; et al. A Tumor-Specific Modulation of Heterogeneous Ribonucleoprotein A0 Promotes Excessive Mitosis and Growth in Colorectal Cancer Cells. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Zhang, J.; Kong, L.; Guo, S.; Bu, M.; Guo, Q.; Xiong, Y.; Zhu, N.; Qiu, C.; Yan, X.; Chen, Q.; et al. HnRNPs and ELAVL1 Cooperate with UORFs to Inhibit Protein Translation. Nucleic Acids Res. 2017, 45, 2849–2864. [Google Scholar] [CrossRef]
- Young, D.J.; Stoddart, A.; Nakitandwe, J.; Chen, S.C.; Qian, Z.; Downing, J.R.; Le Beau, M.M. Knockdown of Hnrnpa0, a Del(5q) Gene, Alters Myeloid Cell Fate in Murine Cells through Regulation of AU-Rich Transcripts. Haematologica 2014, 99, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Bollig, F.; Winzen, R.; Gaestel, M.; Kostka, S.; Resch, K.; Holtmann, H. Affinity Purification of ARE-Binding Proteins Identifies Poly(A)-Binding Protein 1 as a Potential Substrate in MK2-Induced MRNA Stabilization. Biochem. Biophys. Res. Commun. 2003, 301, 665–670. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Zhang, Y.; Kern, S.; Danner, R.L. Nitric Oxide-P38 MAPK Signaling Stabilizes MRNA through AU-Rich Element-Dependent and -Independent Mechanisms. J. Leukoc. Biol. 2008, 83, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Galán, C.; Sola, I.; Nogales, A.; Thomas, B.; Akoulitchev, A.; Enjuanes, L.; Almazán, F. Host Cell Proteins Interacting with the 3′ End of TGEV Coronavirus Genome Influence Virus Replication. Virology 2009, 391, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Marco, P.; Romero-López, C.; Berzal-Herranz, A. The Cis-Acting Replication Element of the Hepatitis C Virus Genome Recruits Host Factors That Influence Viral Replication and Translation. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Lu, H.; Li, W.; Noble, W.S.; Payan, D.; Anderson, D.C. Riboproteomics of the Hepatitis C Virus Internal Ribosomal Entry Site. J. Proteome Res. 2004, 3, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Iliev, D.B.; Skjæveland, I.; Jørgensen, J.B. CpG Oligonucleotides Bind TLR9 and RRM-Containing Proteins in Atlantic Salmon (Salmo Salar). BMC Immunol. 2013, 14, 12. [Google Scholar] [CrossRef]
- Zhang, Z.; Weinschenk, T.; Schluesener, H.J. Uptake, Intracellular Distribution, and Novel Binding Proteins of Immunostimulatory CpG Oligodeoxynucleotides in Microglial Cells. J. Neuroimmunol. 2005, 160, 32–40. [Google Scholar] [CrossRef]
- Dammer, E.B.; Duong, D.M.; Diner, I.; Gearing, M.; Feng, Y.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Neuron Enriched Nuclear Proteome Isolated from Human Brain. J. Proteome Res. 2013, 12, 3193–3206. [Google Scholar] [CrossRef]
- Martins-De-Souza, D.; Gattaz, W.F.; Schmitt, A.; Rewerts, C.; Maccarrone, G.; Dias-Neto, E.; Turck, C.W. Prefrontal Cortex Shotgun Proteome Analysis Reveals Altered Calcium Homeostasis and Immune System Imbalance in Schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Pilling, L.C.; Bandinelli, S.; Ferrucci, L.; Melzer, D.; Harries, L.W. The Transcript Expression Levels of HNRNPM, HNRNPA0 and AKAP17A Splicing Factors May Be Predictively Associated with Ageing Phenotypes in Human Peripheral Blood. Biogerontology 2019, 20, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Darlington, T.M.; Pimentel, R.; Smith, K.; Bakian, A.V.; Jerominski, L.; Cardon, J.; Camp, N.J.; Callor, W.B.; Grey, T.; Singleton, M.; et al. Identifying Rare Variants for Genetic Risk through a Combined Pedigree and Phenotype Approach: Application to Suicide and Asthma. Transl. Psychiatry 2014, 4, e471. [Google Scholar] [CrossRef] [PubMed]
- Maziuk, B.F.; Apicco, D.J.; Cruz, A.L.; Jiang, L.; Ash, P.E.A.; da Rocha, E.L.; Zhang, C.; Yu, W.H.; Leszyk, J.; Abisambra, J.F.; et al. RNA Binding Proteins Co-Localize with Small Tau Inclusions in Tauopathy. Acta Neuropathol. Commun. 2018, 6, 71. [Google Scholar] [CrossRef]
- Han, S.P.; Tang, Y.H.; Smith, R. Functional Diversity of the HnRNPs: Past, Present and Perspectives. Biochem. J. 2010, 430, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Plomaritoglou, A.; Choli-Papadopoulou, T.; Guialis, A. Molecular Characterization of a Murine, Major A/B Type HnRNP Protein: MBx. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1490, 54–62. [Google Scholar] [CrossRef]
- Good, P.J.; Rebbert, M.L.; Dawid, L.B. Three New Members of the RNP Protein Family in Xenopus. Nucleic Acids Res. 1993, 21, 999–1006. [Google Scholar] [CrossRef][Green Version]
- Dangli, A.; Guialis, A.; Vretou, E.; Sekeris, C.E. Autoantibodies to the Core Proteins of HnRNPs. FEBS Lett. 1988, 231, 118–124. [Google Scholar] [CrossRef]
- Yamaoka, K.; Imajoh-Ohmi, S.; Fukuda, H.; Akita, Y.; Kurosawa, K.; Yamamoto, Y.; Sanai, Y. Identification of Phosphoproteins Associated with Maintenance of Transformed State in Temperature-Sensitive Rous Sarcoma-Virus Infected Cells by Proteomic Analysis. Biochem. Biophys. Res. Commun. 2006, 345, 1240–1246. [Google Scholar] [CrossRef]
- Allemand, E.; Guil, S.; Myers, M.; Moscat, J.; Cáceres, J.F.; Krainer, A.R. Regulation of Heterogenous Nuclear Ribonucleoprotein A1 Transport by Phosphorylation in Cells Stressed by Osmotic Shock. Proc. Natl. Acad. Sci. USA 2005, 102, 3605–3610. [Google Scholar] [CrossRef]
- Li, T.; Evdokimov, E.; Shen, R.F.; Chao, C.C.; Tekle, E.; Wang, T.; Stadtman, E.R.; Yang, D.C.H.; Chock, P.B. Sumoylation of Heterogeneous Nuclear Ribonucleoproteins, Zinc Finger Proteins, and Nuclear Pore Complex Proteins: A Proteomic Analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 8551–8556. [Google Scholar] [CrossRef]
- Guo, H.; Wang, R.; Zheng, W.; Chen, Y.; Blum, G.; Deng, H.; Luo, M. Profiling Substrates of Protein Arginine N-Methyltransferase 3 with S-Adenosyl-L-Methionine Analogues. ACS Chem. Biol. 2014, 9, 476–484. [Google Scholar] [CrossRef]
- Boukakis, G.; Patrinou-Georgoula, M.; Lekarakou, M.; Valavanis, C.; Guialis, A. Deregulated Expression of HnRNP A/B Proteins in Human Non-Small Cell Lung Cancer: Parallel Assessment of Protein and MRNA Levels in Paired Tumour/Non-Tumour Tissues. BMC Cancer 2010, 10, 434. [Google Scholar] [CrossRef] [PubMed]
- Demasi, M.A.A.; Montor, W.R.; Ferreira, G.B.; Pimenta, D.C.; Labriola, L.; Sogayar, M.C. Differential Proteomic Analysis of the Anti-Proliferative Effect of Glucocorticoid Hormones in ST1 Rat Glioma Cells. J. Steroid Biochem. Mol. Biol. 2007, 103, 137–148. [Google Scholar] [CrossRef]
- Chapman, K.M.; Powell, H.M.; Chaudhary, J.; Shelton, J.M.; Richardson, J.A.; Richardson, T.E.; Hamra, F.K. Linking Spermatid Ribonucleic Acid (RNA) Binding Protein and Retrogene Diversity to Reproductive Success. Mol. Cell. Proteom. 2013, 12, 3221–3236. [Google Scholar] [CrossRef]
- Comegna, M.; Succoio, M.; Napolitano, M.; Vitale, M.; D’Ambrosio, C.; Scaloni, A.; Passaro, F.; Zambrano, N.; Cimino, F.; Faraonio, R. Identification of MiR-494 Direct Targets Involved in Senescence of Human Diploid Fibroblasts. FASEB J. 2014, 28, 3720–3733. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yoon, J.-G.; Li, L.; Tsai, Y.S.; Zheng, S.; Hood, L.; Goodlett, D.R.; Foltz, G.; Lin, B. Landscape of the SOX2 Protein-Protein Interactome. Proteomics 2011, 11, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Wu, C.C.; Huang, K.Y.; Chuang, W.Y.; Hsueh, C.; Li, H.J.; Chen, C.Y. Profiling of Subcellular EGFR Interactome Reveals HnRNP A3 Modulates Nuclear EGFR Localization. Oncogenesis 2020, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Paasch, A.; Huber, S.; Fehrenbacher, B.; Schaller, M.; Rodemann, H.P. Nuclear EGFR Renders Cells Radio-Resistant by Binding MRNA Species and Triggering a Metabolic Switch to Increase Lactate Production. Radiother. Oncol. 2015, 116, 431–437. [Google Scholar]
- Chen, X.; Lloyd, S.M.; Kweon, J.; Gamalong, G.M.; Bao, X. Epidermal Progenitors Suppress GRHL3-Mediated Differentiation through Intronic Polyadenylation Promoted by CPSF-HNRNPA3 Collaboration. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and Telomerase: Three Decades of Progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Tanaka, E.; Fukuda, H.; Nakashima, K.; Tsuchiya, N.; Seimiya, H.; Nakagama, H. HnRNP A3 Binds to and Protects Mammalian Telomeric Repeats in Vitro. Biochem. Biophys. Res. Commun. 2007, 358, 608–614. [Google Scholar] [CrossRef]
- Huang, P.R.; Tsai, S.T.; Hsieh, K.H.; Wang, T.C.V. Heterogeneous Nuclear Ribonucleoprotein A3 Binds Single-Stranded Telomeric DNA and Inhibits Telomerase Extension in Vitro. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 193–202. [Google Scholar] [CrossRef]
- Huang, P.R.; Hung, S.C.; Wang, T.C.V. Telomeric DNA-Binding Activities of Heterogeneous Nuclear Ribonucleoprotein A3 in Vitro and in Vivo. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 1164–1174. [Google Scholar] [CrossRef]
- Nakagama, H.; Higuchi, K.; Tanaka, E.; Tsuchiya, N.; Nakashima, K.; Katahira, M.; Fukuda, H. Molecular Mechanisms for Maintenance of G-Rich Short Tandem Repeats Capable of Adopting G4 DNA Structures. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2006, 598, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Travina, A.O.; Ilicheva, N.V.; Mittenberg, A.G.; Shabelnikov, S.V.; Kotova, A.V.; Podgornaya, O.I. The Long Linker Region of Telomere-Binding Protein TRF2 Is Responsible for Interactions with Lamins. Int. J. Mol. Sci. 2021, 22, 3293. [Google Scholar] [CrossRef]
- Soeno, Y.; Taya, Y.; Stasyk, T.; Huber, L.A.; Aoba, T.; Hüttenhofer, A. Identification of Novel Ribonucleo-Protein Complexes from the Brain-Specific SnoRNA MBII-52. RNA 2010, 16, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Kishore, S.; Stamm, S. The SnoRNA HBII-52 Regulates Alternative Splicing of the Serotonin Receptor 2C. Science 2006, 311, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Kishore, S.; Khanna, A.; Zhang, Z.; Hui, J.; Balwierz, P.J.; Stefan, M.; Beach, C.; Nicholls, R.D.; Zavolan, M.; Stamm, S. The SnoRNA MBII-52 (SNORD 115) Is Processed into Smaller RNAs and Regulates Alternative Splicing. Hum. Mol. Genet. 2010, 19, 1153–1164. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative Polyadenylation of MRNA Precursors. Nat. Rev. Mol. Cell Biol. 2016, 18, 18–30. [Google Scholar] [CrossRef]
- Fukuda, N.; Fukuda, T.; Sinnamon, J.; Hernandez-Hernandez, A.; Izadi, M.; Raju, C.S.; Czaplinski, K.; Percipalle, P. The Transacting Factor CBF-A/Hnrnpab Binds to the A2RE/RTS Element of Protamine 2 MRNA and Contributes to Its Translational Regulation during Mouse Spermatogenesis. PLoS Genet. 2013, 9, e1003858. [Google Scholar] [CrossRef] [PubMed]
- Srikantan, S.; Gorospe, M. HuR Function in Disease. Front. Biosci. 2012, 17, 189–205. [Google Scholar] [CrossRef]
- Papadopoulou, C.; Patrinou-Georgoula, M.; Guialis, A. Extensive Association of HuR with HnRNP Proteins within Immunoselected HnRNP and MRNP Complexes. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Chaumet, A.; Castella, S.; Gasmi, L.; Fradin, A.; Clodic, G.; Bolbach, G.; Poulhe, R.; Denoulet, P.; Larcher, J.C. Proteomic Analysis of Interleukin Enhancer Binding Factor 3 (Ilf3) and Nuclear Factor 90 (NF90) Interactome. Biochimie 2013, 95, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J.; Miki, T.; Takano, K.; Maruhashi, M.; Uchikawa, M.; Tachibana, T.; Yoneda, Y. Nuclear RNA Export Factor 7 Is Localized in Processing Bodies and Neuronal RNA Granules through Interactions with Shuttling HnRNPs. Nucleic Acids Res. 2008, 36, 616–628. [Google Scholar] [CrossRef]
- Cok, S.J.; Acton, S.J.; Sexton, A.E.; Morrison, A.R. Identification of RNA-Binding Proteins in RAW 264.7 Cells That Recognize a Lipopolysaccharide-Responsive Element in the 3-Untranslated Region of the Murine Cyclooxygenase-2 MRNA. J. Biol. Chem. 2004, 279, 8196–8205. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.T.; Yu, G.-Y.; Lai, M.M.C. Multiple Type A/B Heterogeneous Nuclear Ribonucleoproteins (HnRNPs) Can Replace HnRNP A1 in Mouse Hepatitis Virus RNA Synthesis. J. Virol. 2003, 77, 10584–10593. [Google Scholar] [CrossRef]
- Bogunovic, D.; Boisson-Dupuis, S.; Casanova, J.L. ISG15: Leading a Double Life as a Secreted Molecule. Exp. Mol. Med. 2013, 45, 45. [Google Scholar] [CrossRef]
- Real, C.I.; Megger, D.A.; Sitek, B.; Jahn-Hofmann, K.; Ickenstein, L.M.; John, M.J.; Walker, A.; Timm, J.; Kuhlmann, K.; Eisenacher, M.; et al. Identification of Proteins That Mediate the Pro-Viral Functions of the Interferon Stimulated Gene 15 in Hepatitis C Virus Replication. Antiviral Res. 2013, 100, 654–661. [Google Scholar] [CrossRef]
- Mishra, N.; Reddy, K.S.; Timilsina, U.; Gaur, D.; Gaur, R. Human APOBEC3B Interacts with the Heterogenous Nuclear Ribonucleoprotein A3 in Cancer Cells. J. Cell. Biochem. 2018, 119, 6695–6703. [Google Scholar] [CrossRef]
- Lopes, G.S.; Brusco, J.; Rosa, J.C.; Larson, R.E.; Lico, D.T.P. Selectively RNA Interaction by a HnRNPA/B-like Protein at Presynaptic Terminal of Squid Neuron. Invertebr. Neurosci. 2020, 20, 14. [Google Scholar] [CrossRef]
- Ou, M.Y.; Ju, X.C.; Cai, Y.J.; Sun, X.Y.; Wang, J.F.; Fu, X.Q.; Sun, Q.; Luo, Z.G. Heterogeneous Nuclear Ribonucleoprotein A3 Controls Mitotic Progression of Neural Progenitors via Interaction with Cohesin. Development 2020, 147. [Google Scholar] [CrossRef]
- Mori, K.; Lammich, S.; Mackenzie, I.R.A.; Forné, I.; Zilow, S.; Kretzschmar, H.; Edbauer, D.; Janssens, J.; Kleinberger, G.; Cruts, M.; et al. HnRNP A3 Binds to GGGGCC Repeats and Is a Constituent of P62-Positive/TDP43-Negative Inclusions in the Hippocampus of Patients with C9orf72 Mutations. Acta Neuropathol. 2013, 125, 413–423. [Google Scholar] [CrossRef]
- Nihei, Y.; Mori, K.; Werner, G.; Arzberger, T.; Zhou, Q.; Khosravi, B.; Japtok, J.; Hermann, A.; Sommacal, A.; Weber, M.; et al. Poly-Glycine–Alanine Exacerbates C9orf72 Repeat Expansion-Mediated DNA Damage via Sequestration of Phosphorylated ATM and Loss of Nuclear HnRNPA3. Acta Neuropathol. 2020, 139, 99–118. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The Expanding Biology of the C9orf72 Nucleotide Repeat Expansion in Neurodegenerative Disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef]
- Davidson, Y.S.; Flood, L.; Robinson, A.C.; Nihei, Y.; Mori, K.; Rollinson, S.; Richardson, A.; Benson, B.C.; Jones, M.; Snowden, J.S.; et al. Heterogeneous Ribonuclear Protein A3 (HnRNP A3) Is Present in Dipeptide Repeat Protein Containing Inclusions in Frontotemporal Lobar Degeneration and Motor Neurone Disease Associated with Expansions in C9orf72 Gene. Acta Neuropathol. Commun. 2017, 5, 31. [Google Scholar] [CrossRef]
- Fifita, J.A.; Zhang, K.Y.; Galper, J.; Williams, K.L.; McCann, E.P.; Hogan, A.L.; Saunders, N.; Bauer, D.; Tarr, I.S.; Pamphlett, R.; et al. Genetic and Pathological Assessment of HnRNPA1, HnRNPA2/B1, and HnRNPA3 in Familial and Sporadic Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2017, 17, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Nihei, Y.; Arzberger, T.; Zhou, Q.; Mackenzie, I.R.; Hermann, A.; Hanisch, F.; Kamp, F.; Nuscher, B.; Orozco, D.; et al. Reduced HnRNPA3 Increases C9orf72 Repeat RNA Levels and Dipeptide-repeat Protein Deposition. EMBO Rep. 2016, 17, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 Binds Heterogeneous Nuclear Ribonucleoprotein A/B through Its C-Terminal Tail: An Important Region for the Inhibition of Cystic Fibrosis Transmembrane Conductance Regulator Exon 9 Splicing. J. Biol. Chem. 2005, 280, 37572–37584. [Google Scholar] [CrossRef] [PubMed]
- Prpar Mihevc, S.; Baralle, M.; Buratti, E.; Rogelj, B. TDP-43 Aggregation Mirrors TDP-43 Knockdown, Affecting the Expression Levels of a Common Set of Proteins. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/Dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Gilpin, K.M.; Chang, L.; Monteiro, M.J. ALS-Linked Mutations in Ubiquilin-2 or HnRNPA1 Reduce Interaction between Ubiquilin-2 and HnRNPA1. Hum. Mol. Genet. 2015, 24, 2565–2577. [Google Scholar] [CrossRef][Green Version]
- Van Acker, Z.P.; Van Raemdonck, G.A.; Logie, E.; Van Acker, S.I.; Baggerman, G.; Vanden Berghe, W.; Ponsaerts, P.; Dewilde, S. Connecting the Dots in the Neuroglobin-Protein Interaction Network of an Unstressed and Ferroptotic Cell Death Neuroblastoma Model. Cells 2019, 8, 873. [Google Scholar] [CrossRef]
- Tunca, C.; Şeker, T.; Akçimen, F.; Coşkun, C.; Bayraktar, E.; Palvadeau, R.; Zor, S.; Koçoğlu, C.; Kartal, E.; Şen, N.E.; et al. Revisiting the Complex Architecture of ALS in Turkey: Expanding Genotypes, Shared Phenotypes, Molecular Networks, and a Public Variant Database. Hum. Mutat. 2020, 41, e7–e45. [Google Scholar] [CrossRef] [PubMed]
- Calini, D.; Corrado, L.; Del Bo, R.; Gagliardi, S.; Pensato, V.; Verde, F.; Corti, S.; Mazzini, L.; Milani, P.; Castellotti, B.; et al. Analysis of HnRNPA1, A2/B1, and A3 Genes in Patients with Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2013, 34, 2695.e11–2695.e12. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Cao, Q.; Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Cascio, D.; Eisenberg, D.S. CryoEM Structure of the Low-Complexity Domain of HnRNPA2 and Its Conversion to Pathogenic Amyloid. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Goldschmidt, L.; Rodriguez, J.A.; Cascio, D.; Chong, L.; Gonen, T.; Eisenberg, D.S. Atomic Structures of Low-Complexity Protein Segments Reveal Kinked b Sheets That Assemble Networks. Science 2018, 359, 698–701. [Google Scholar] [CrossRef]
- Barraud, P.; Allain, F.H.T. Solution Structure of the Two RNA Recognition Motifs of HnRNP A1 Using Segmental Isotope Labeling: How the Relative Orientation between RRMs Influences the Nucleic Acid Binding Topology. J. Biomol. NMR 2013, 55, 119–138. [Google Scholar] [CrossRef]
- Vitali, J.; Ding, J.; Jiang, J.; Zhang, Y.; Krainer, A.R.; Xu, R.M. Correlated Alternative Side Chain Conformations in the RNA-Recognition Motif of Heterogeneous Nuclear Ribonucleoprotein A1. Nucleic Acids Res. 2002, 30, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Shamoo, Y.; Krueger, U.; Rice, L.M.; Williams, K.R.; Steitz, T.A. Crystal Structure of the Two RNA Binding Domains of Human HnRNP A1 at 1.75 A Resolution. Nat. Struct. Biol. 1997, 4, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.M.; Jokhan, L.; Cheng, X.; Mayeda, A.; Krainer, A.R. Crystal Structure of Human UP1, the Domain of HnRNP A1 That Contains Two RNA-Recognition Motifs. Structure 1997, 5, 559–570. [Google Scholar] [CrossRef]
- Myers, J.C.; Shamoo, Y. Human UP1 as a Model for Understanding Purine Recognition in the Family of Proteins Containing the RNA Recognition Motif (RRM). J. Mol. Biol. 2004, 342, 743–756. [Google Scholar] [CrossRef]
- Myers, J.C.; Moore, S.A.; Shamoo, Y. Structure-Based Incorporation of 6-Methyl-8-(2-Deoxy-β -Ribofuranosyl)Isoxanthopteridine into the Human Telomeric Repeat DNA as a Probe for UP1 Binding and Destabilization of G-Tetrad Structures. J. Biol. Chem. 2003, 278, 42300–42306. [Google Scholar] [CrossRef]
- Morgan, C.E.; Meagher, J.L.; Levengood, J.D.; Delproposto, J.; Rollins, C.; Stuckey, J.A.; Tolbert, B.S. The First Crystal Structure of the UP1 Domain of HnRNP A1 Bound to RNA Reveals a New Look for an Old RNA Binding Protein. J. Mol. Biol. 2015, 427, 3241–3257. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Su, S.; Patil, D.P.; Liu, H.; Gan, J.; Jaffrey, S.R.; Ma, J. Molecular Basis for the Specific and Multivariant Recognitions of RNA Substrates by Human HnRNP A2/B1. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mayeda, A.; Munroe, S.H.; Xu, R.M.; Krainer, A.R. Distinct Functions of the Closely Related Tandem RNA-Recognition Motifs of HnRNP A1. RNA 1998, 4, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Shamoo, Y.; Abdul-manan, N.; Williams, K.R. Multiple RNA Binding Domains (RBDs) Just Don’t Add Up. Nucleic Acids Res. 1995, 23, 725–728. [Google Scholar] [CrossRef]
- Afroz, T.; Cienikova, Z.; Cléry, A.; Allain, F.H.T. One, two, three, four! How multiple RRMs read the genome sequence. Methods Enzymol. 2015, 558, 235–278. [Google Scholar]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the Cross-β Spine of Amyloid-like Fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and Patterning of Aromatic Residues Determine the Phase Behavior of Prion-like Domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Martin, E.W.; Thomasen, F.E.; Milkovic, N.M.; Cuneo, M.J.; Grace, C.R.; Nourse, A.; Lindorff-Larsen, K.; Mittag, T. Interplay of Folded Domains and the Disordered Low-Complexity Domain in Mediating HnRNPA1 Phase Separation. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
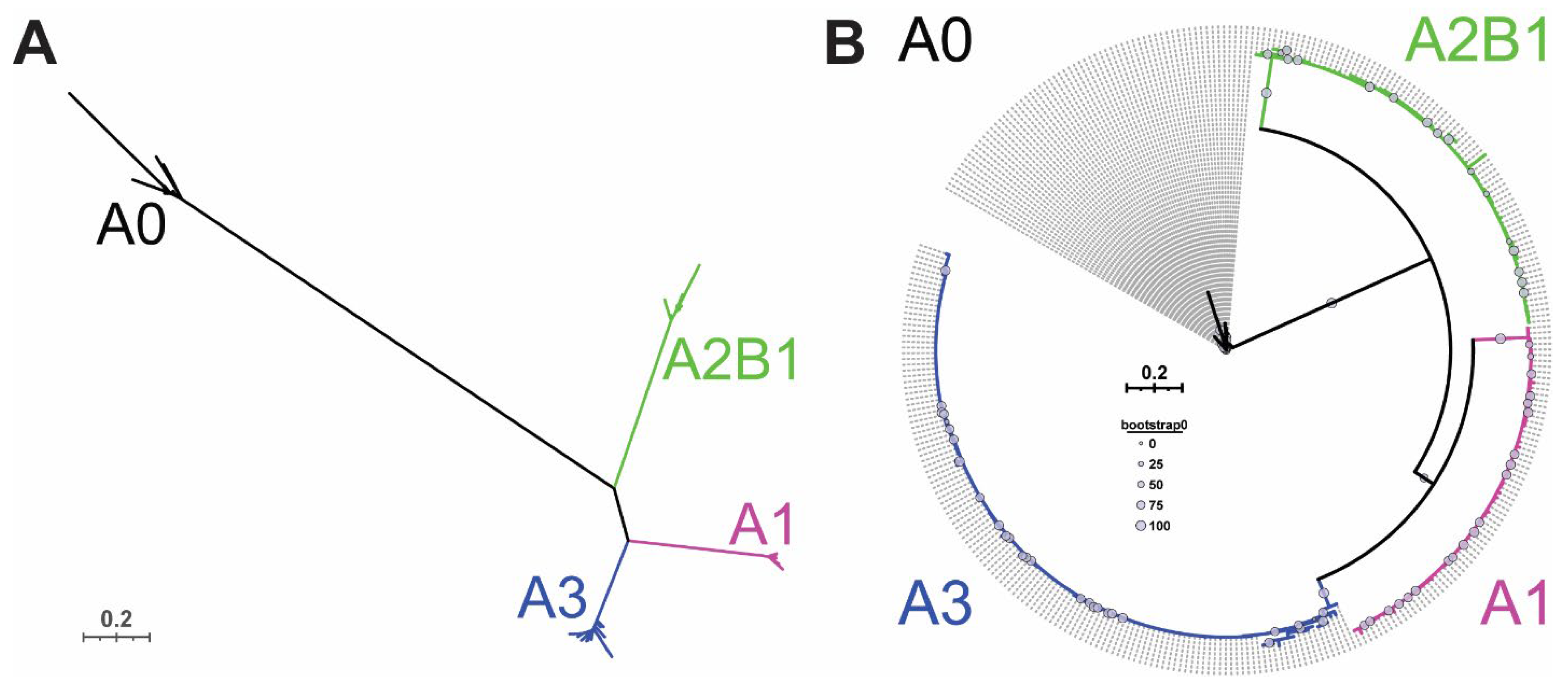{kind=link}
{kind=link}
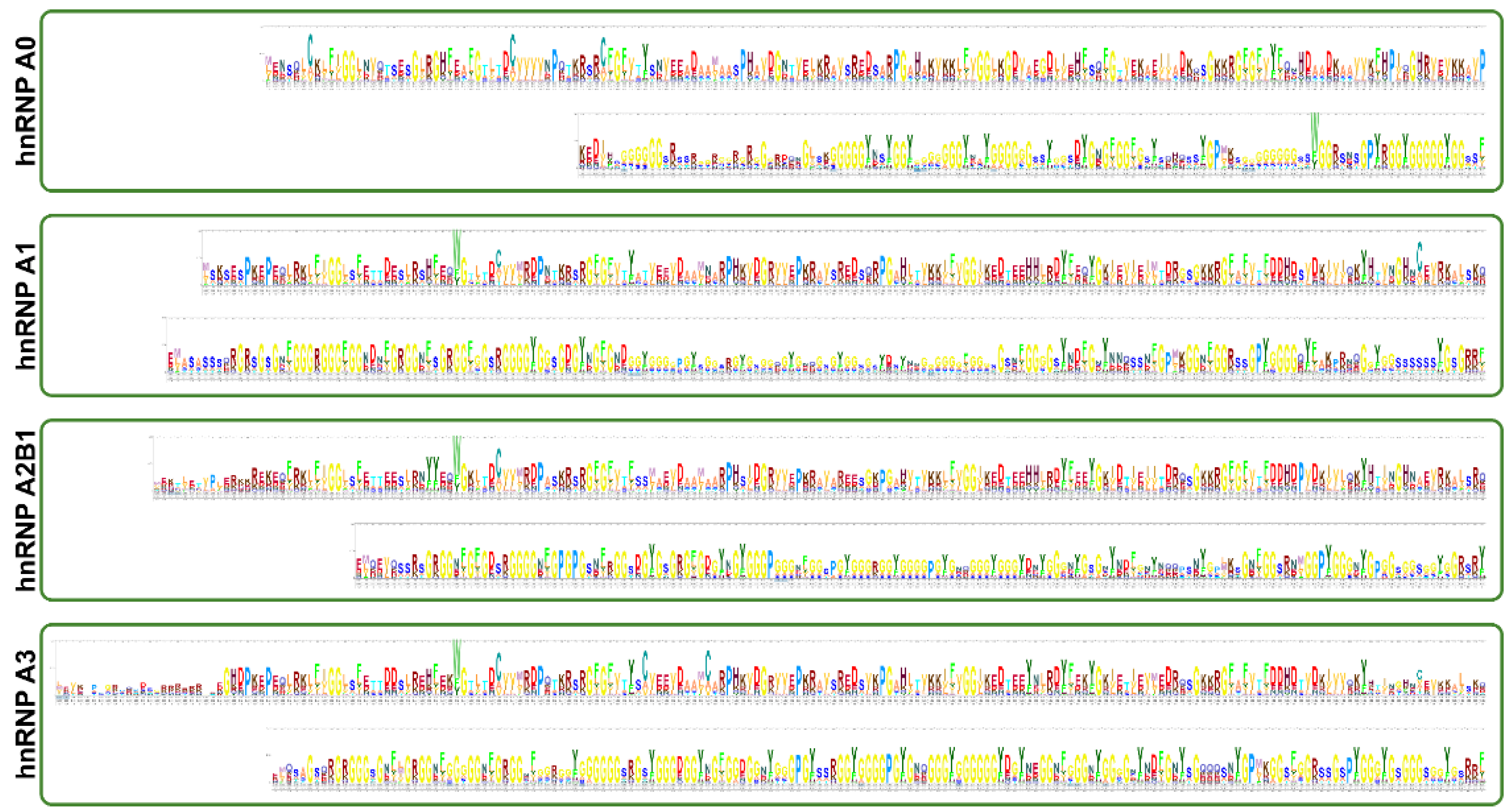{kind=link}
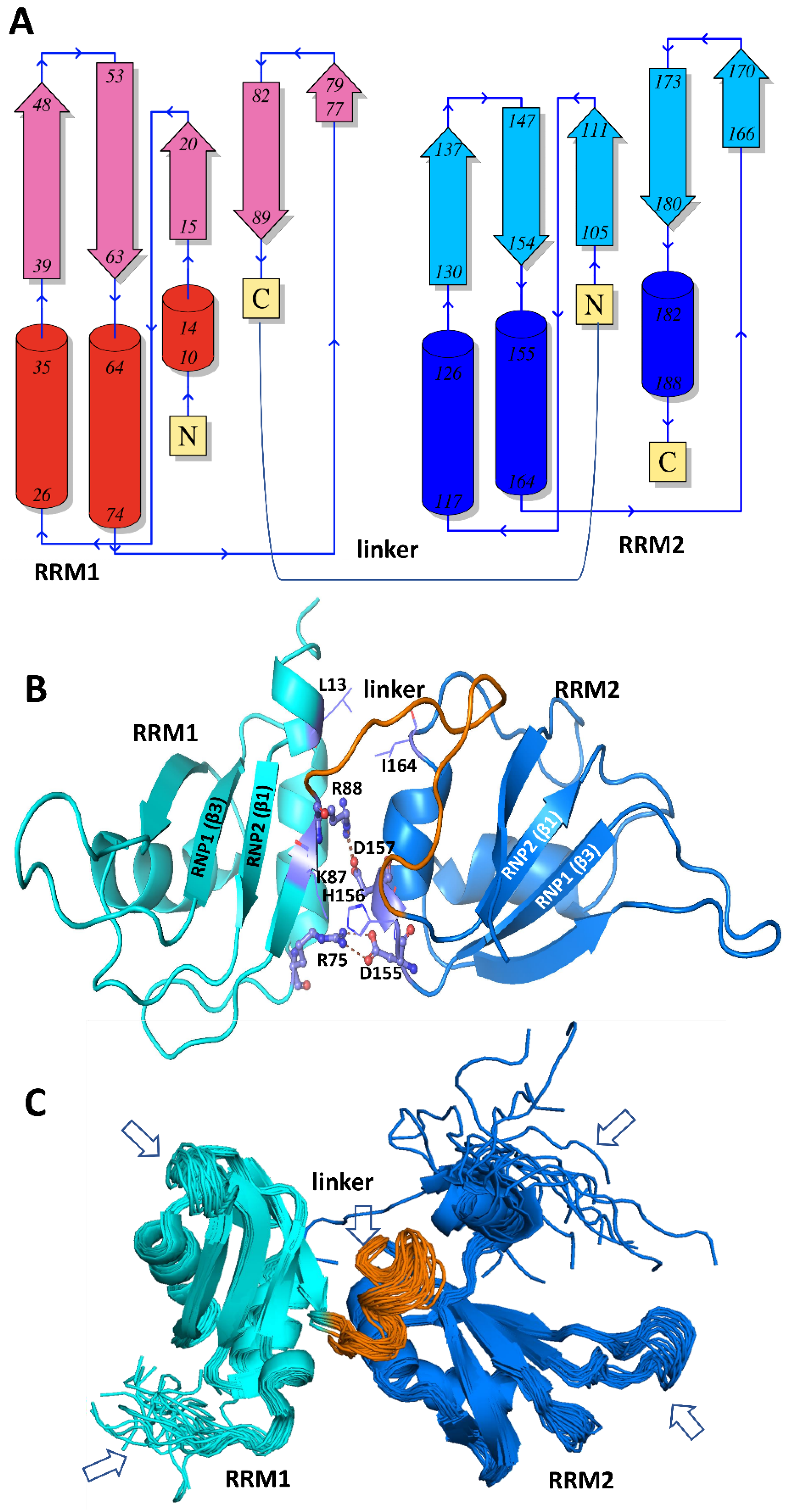{kind=link}
{kind=link}
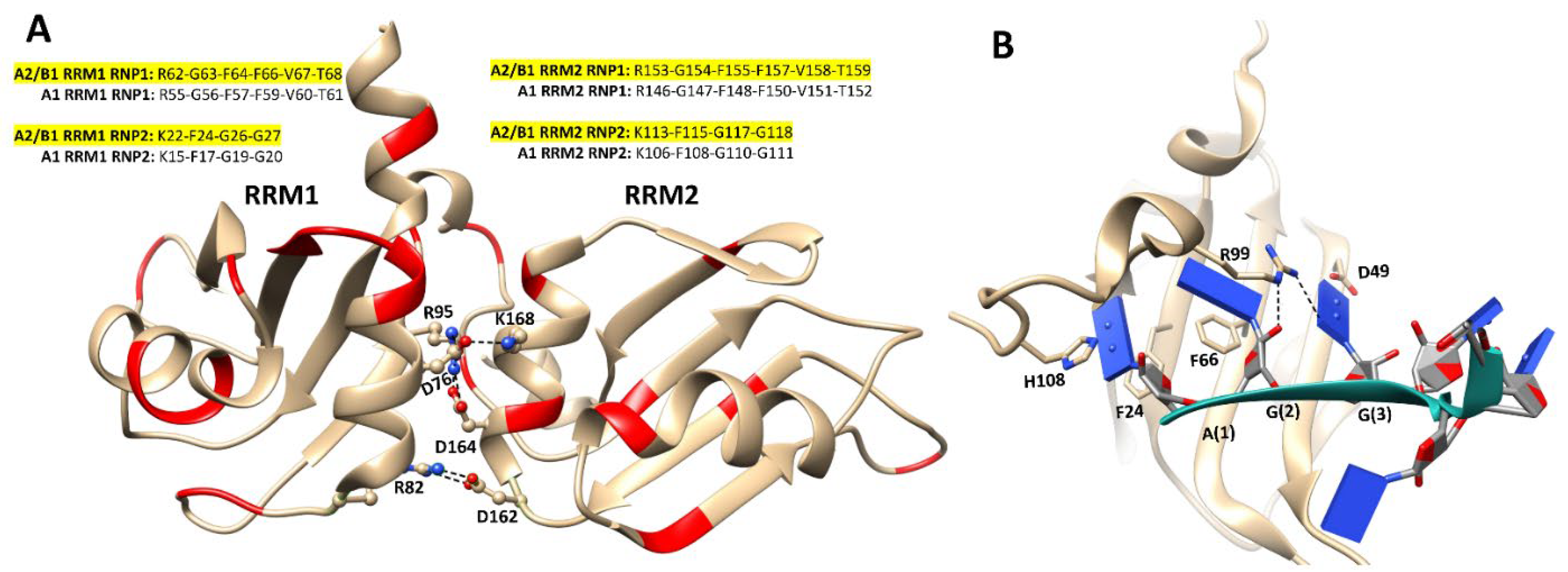{kind=link}
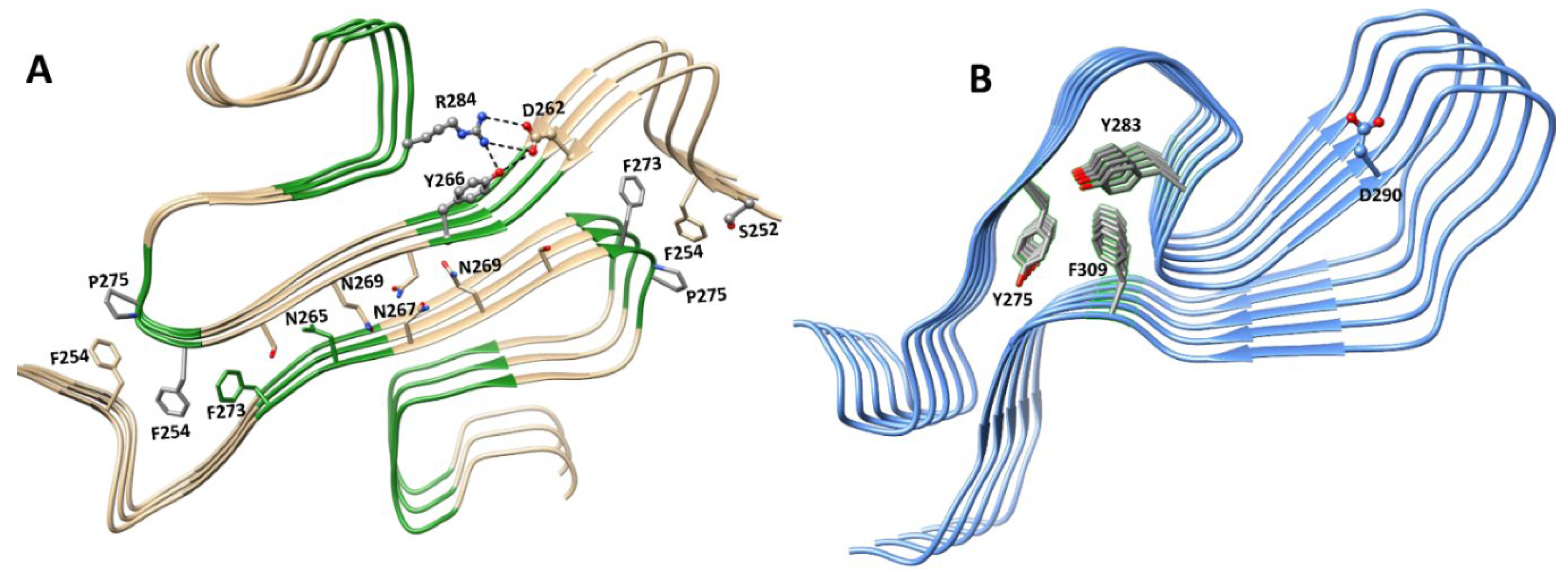{kind=link}
{kind=link}
| Common Protein Name | Gene Name a (Ensembl ID) | Chromosomal Location | Isoforms b (in Order of Abundance) | Protein Length (aa) | References |
|---|---|---|---|---|---|
| hnRNP A0 | HNRNPA0 | Ch5q31.2, rev | n/a | 305 | [33] |
| (ENSG00000177733) | |||||
| hnRNP A1 | HNRNPA1 | Ch12q13.13, for | hnRNP A1 | 320 | [34,35] |
| (ENSG00000135486) | hnRNP A1-B | 372 | |||
| hnRNP A2/B1 | HNRNPA2B1 | Ch7p15.2, rev | hnRNP A2 | 341 | [18,36,49,50] |
| hnRNP B1 | 353 | ||||
| (ENSG00000122566) | hnRNP A2b | 301 | |||
| hnRNP B1b | 313 | ||||
| hnRNP A2* | 253 | ||||
| hnRNP A3 | HNRNPA3 | Ch2q31.2, for | hnRNP A3 var 1 | 378 | [31,39,40] |
| hnRNP A3 var 2 | 356 | ||||
| (ENSG00000170144) | hnRNP A3 var 3 | 331 | |||
| hnRNP A3 var 4 | 309 |
| PDB Code | Structure Description | Technique (Resolution in Å) | Refs | Year |
|---|---|---|---|---|
| hnRNP A1 | ||||
| 7BX7 | A1 LCD (residues 251–295) | Cryo-EM (2.8) | [193] | 2020 |
| 6BXX | A1 LCD (residues 243–248) | X-ray diffraction (1.1) | [298] | 2018 |
| 5ZGL | A1 LCD (residues 234–240) | X-ray diffraction (0.95) | [185] | 2019 |
| 6J60 | A1 LCD (residues 209–217) | Electron crystallography (0.96) | [185] | 2019 |
| 2LYV | A1 RRMs | Solution NMR | [299] | 2013 |
| 1L3K | A1 RRMs | X-ray diffraction (1.1) | [300] | 2002 |
| 1HA1 | A1 RRMs | X-ray diffraction (1.75) | [301] | 1997 |
| 1UP1 | A1 RRMs | X-ray diffraction (1.9) | [302] | 1997 |
| 1U1L/1U1K/ 1U1R/1U1Q/ 1U1P/1U1O/ 1U1N/1U1M | A1 RRMs complexed with modified telomeric DNA repeats | X-ray diffraction (1.8–2.1) | [303] | 2004 |
| 1PO6/ 1PGZ | A1 RRMs complexed with DNA | X-ray diffraction (2.1/2.6) | [304] | 2003 |
| 2UP1 | A1 RRM-single-stranded telomeric DNA complex | X-ray diffraction | [57] | 1999 |
| 6DCL | A1 RRMs-bound to pri-miRNA-18a | X-ray diffraction (2.5) | [144] | 2018 |
| 5MPG | A1 RRM1 bound to 5′-UUAGGUC-3′ | Solution NMR | [108] | 2017 |
| 5MPL | A1 RRM2 bound to 5′-UCAGUU-3′ | Solution NMR | [108] | 2017 |
| 4YOE | A1 RRMs bound to 5’-AGU-3’ RNA | X-ray diffraction (1.92) | [305] | 2015 |
| hnRNP A2/B1 | ||||
| 6WQK | A2 LCD (residues 263–319) | Cryo-EM (3.1) | [297] | 2020 |
| 6WPQ | D290V mutant A2 LCD (residues 286–291) | X-ray diffraction (1.10) | [297] | 2020 |
| 5WWE/ 5WWF/5WWG | A2/B1 RRMs in complex with 10-mer RNA | X-ray diffraction (2.03–2.15) | [306] | 2018 |
| 5HO4 | A2/B1 RRMs in complex with 10-mer RNA | X-ray diffraction (1.85) | [306] | 2018 |
| 5EN1 | A2/B1 RRMs in complex with 8-mer RNA | X-ray diffraction (2.58) | [306] | 2018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thibault, P.A.; Ganesan, A.; Kalyaanamoorthy, S.; Clarke, J.-P.W.E.; Salapa, H.E.; Levin, M.C. hnRNP A/B Proteins: An Encyclopedic Assessment of Their Roles in Homeostasis and Disease. Biology 2021, 10, 712. https://doi.org/10.3390/biology10080712
Thibault PA, Ganesan A, Kalyaanamoorthy S, Clarke J-PWE, Salapa HE, Levin MC. hnRNP A/B Proteins: An Encyclopedic Assessment of Their Roles in Homeostasis and Disease. Biology. 2021; 10(8):712. https://doi.org/10.3390/biology10080712
Chicago/Turabian StyleThibault, Patricia A., Aravindhan Ganesan, Subha Kalyaanamoorthy, Joseph-Patrick W. E. Clarke, Hannah E. Salapa, and Michael C. Levin. 2021. "hnRNP A/B Proteins: An Encyclopedic Assessment of Their Roles in Homeostasis and Disease" Biology 10, no. 8: 712. https://doi.org/10.3390/biology10080712
APA StyleThibault, P. A., Ganesan, A., Kalyaanamoorthy, S., Clarke, J.-P. W. E., Salapa, H. E., & Levin, M. C. (2021). hnRNP A/B Proteins: An Encyclopedic Assessment of Their Roles in Homeostasis and Disease. Biology, 10(8), 712. https://doi.org/10.3390/biology10080712