
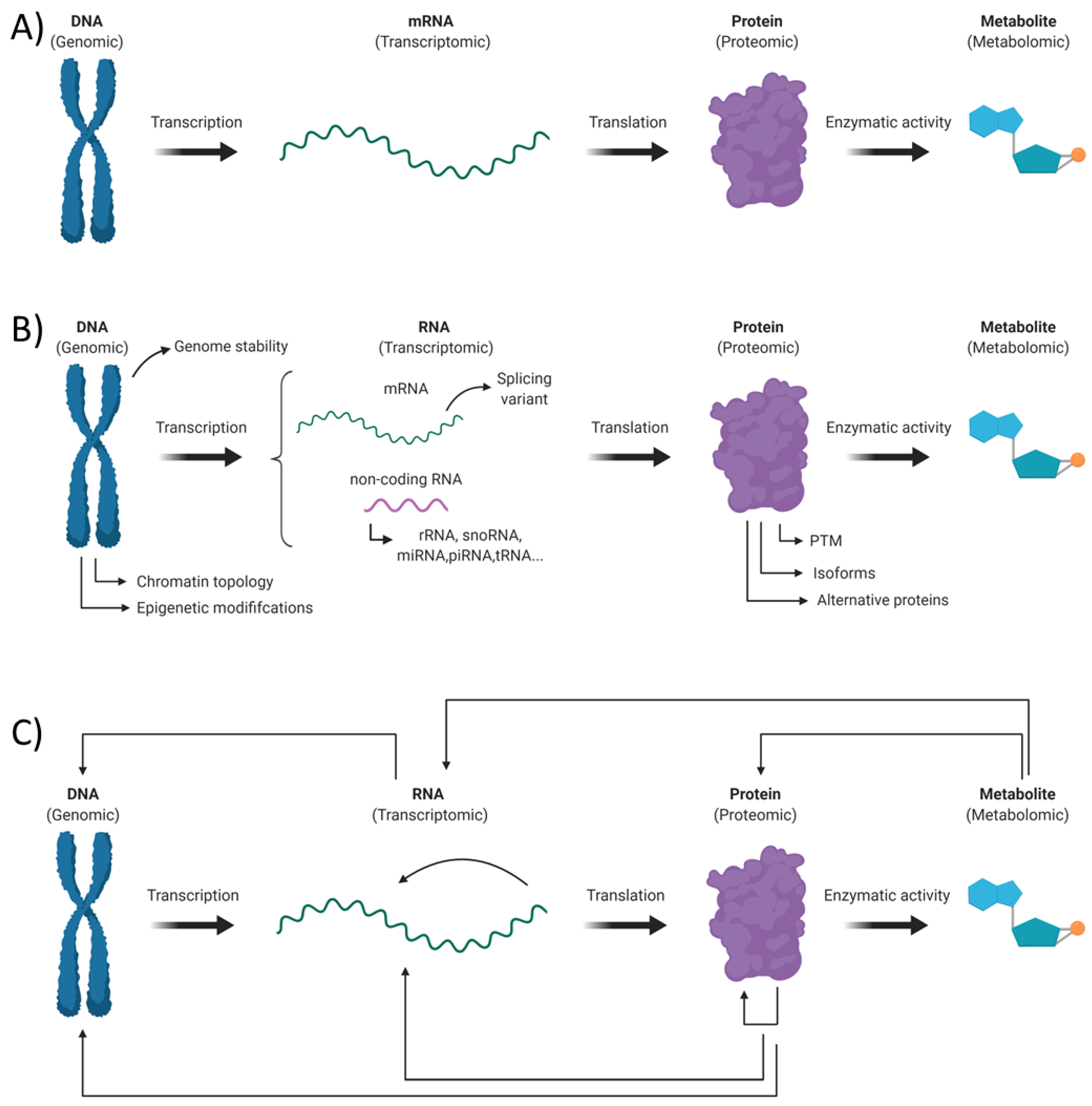
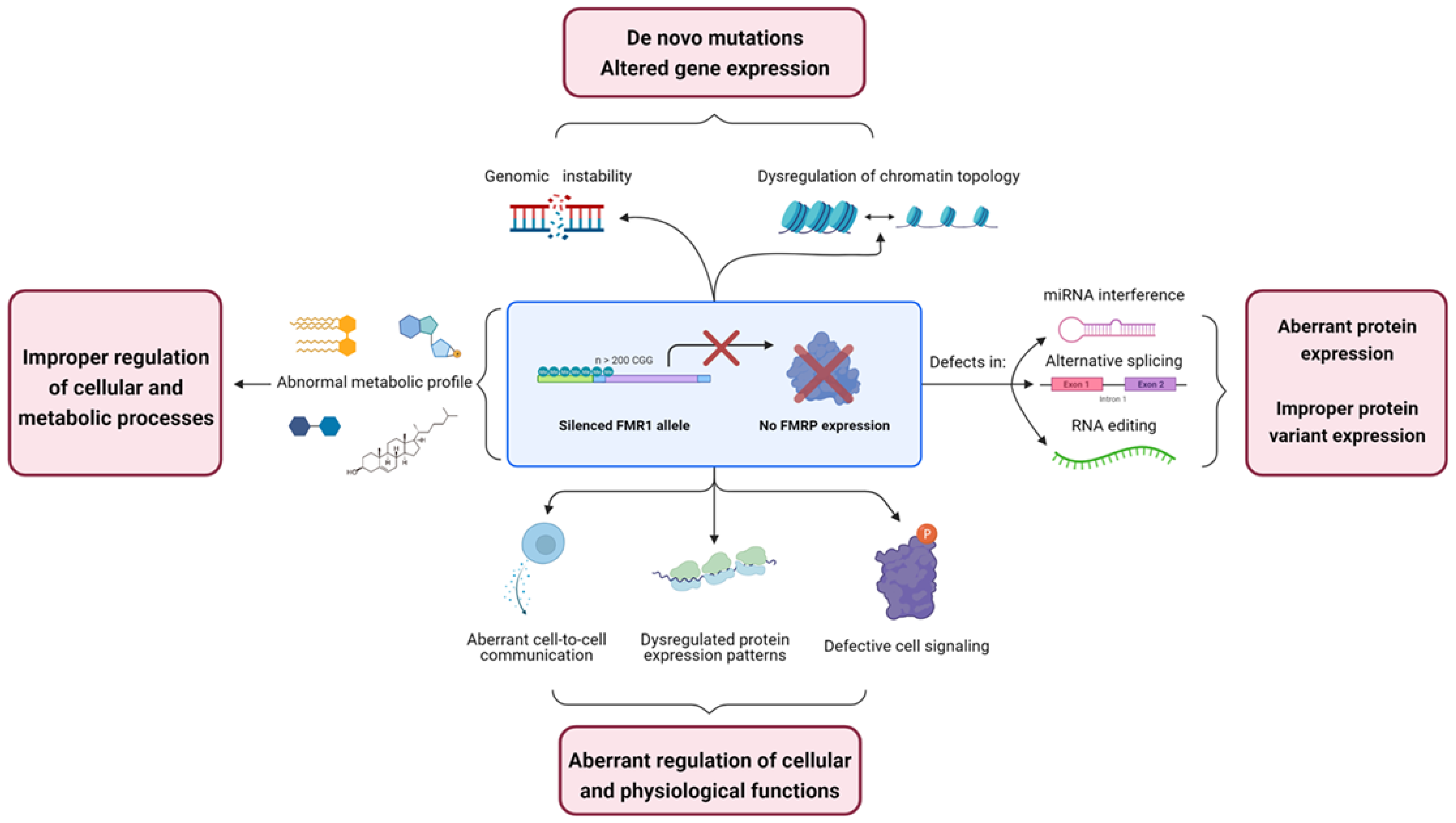
An “Omic” Overview of Fragile X Syndrome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Genomic Alterations in Fragile X Syndrome
2.1. FMRP Is Involved in Genomic Stability Maintenance
2.2. Alteration of Chromatin Topology in FXS
3. Transcriptomic Alterations in Fragile X Syndrome
3.1. FMRP Mediates Micro RNA-Related Interference
3.2. Alternative Splicing and FMRP
3.3. FMRP Absence May Lead to Defect in RNA Editing
4. Proteomics Alterations in Fragile X Syndrome
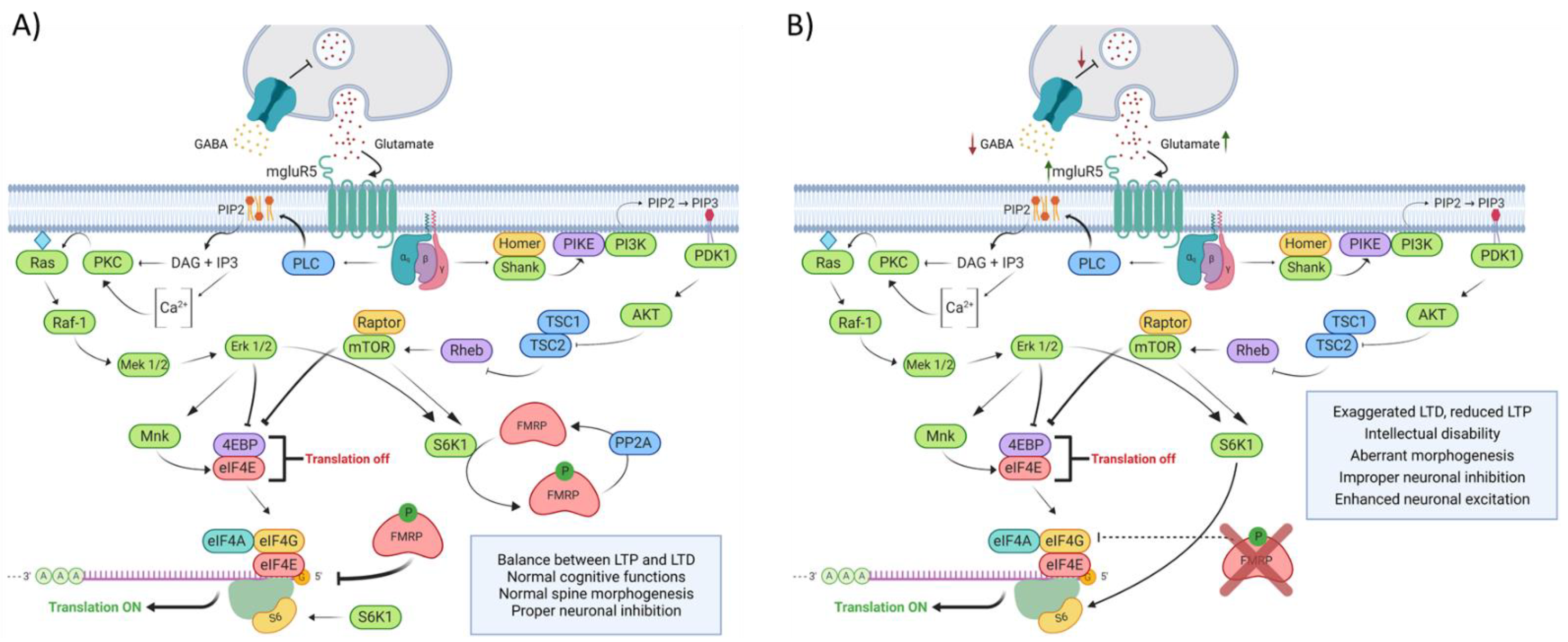
4.1. Rate of Protein Synthesis Alteration: A Still Misunderstood Hallmark of FXS
4.2. Cell Signaling Defects in FXS
4.3. The Matrix Metalloproteinase-9 Involvement in FXS Physiopathology
4.4. Aberrant Cytokines Profile: A Sign of Immune Dysfunction in FXS?
5. Metabolomic Alterations in Fragile X Syndrome
5.1. The Hypocholesterolemic Phenotype of FXS
5.2. Cyclic AMP Metabolism Is Defective in Fragile X
5.3. The Amyloid-β Precursor Protein and Its Secreted Metabolites
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peprah, E. Fragile X Syndrome: The FMR1 CGG Repeat Distribution among World Populations. Ann. Hum. Genet. 2012, 76, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Azarang, A.; Wilaisakditipakorn, T.; Hagerman, R.J. Fragile X Syndrome: A Review of Clinical Management. Intractable Rare Dis. Res. 2016, 5, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X Syndrome: A Review of Clinical and Molecular Diagnoses. Ital. J. Pediatr. 2017, 43. [Google Scholar] [CrossRef]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X Syndrome: A Review of Associated Medical Problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Saldarriaga, W.; Tassone, F.; González-Teshima, L.Y.; Forero-Forero, J.V.; Ayala-Zapata, S.; Hagerman, R. Fragile X Syndrome. Colomb Med. 2014, 45, 190–198. [Google Scholar] [CrossRef]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Pieretti, M.; Warren, T.; Thomas, C.; Nelson’, D.L. Absence of Expression of the HIM-7 Gene in Fragile X Syndrome. Cell 1997, 66, 817–822. [Google Scholar] [CrossRef]
- Rousseau, F.; Heitz, D.; Tarleton, J.; MacPherson, J.; Malmgren, H.; Dahl, N.; Barnicoat, A.; Mathew, C.; Mornet, E.; Tejada, I. A Multicenter Study on Genotype-Phenotype Correlations in the Fragile X Syndrome, Using Direct Diagnosis with Probe StB12.3: The First 2,253 Cases. Am. J. Hum. Genet. 1994, 55, 225–237. [Google Scholar]
- Nolin, S.L.; Glicksman, A.; Houck, G.E.; Brown, W.T.; Dobkin, C.S. Mosaicism in Fragile X Affected Males. Am. J. Med. Genet. 1994, 51, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.; Yrigollen, C.M.; Tang, H.-T.; Williamson, J.; Espinal, G.; Iwahashi, C.K.; Durbin-Johnson, B.; Hagerman, R.J.; Hagerman, P.J.; Tassone, F. Clinical and Molecular Implications of Mosaicism in FMR1 Full Mutations. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Lessard, M.; Chouiali, A.; Drouin, R.; Sébire, G.; Corbin, F. Quantitative Measurement of FMRP in Blood Platelets as a New Screening Test for Fragile X Syndrome. Clin. Genet. 2012, 82, 472–477. [Google Scholar] [CrossRef]
- Willemsen, R. Predictive Testing for Cognitive Functioning in Female Carriers of the Fragile X Syndrome Using Hair Root Analysis. J. Med. Genet. 2003, 40, 377–379. [Google Scholar] [CrossRef]
- Kim, K.; Hessl, D.; Randol, J.L.; Espinal, G.M.; Schneider, A.; Protic, D.; Aydin, E.Y.; Hagerman, R.J.; Hagerman, P.J. Association between IQ and FMR1 Protein (FMRP) across the Spectrum of CGG Repeat Expansions. PLoS ONE 2019, 14, e0226811. [Google Scholar] [CrossRef]
- Hinds, H.L.; Ashley, C.T.; Sutcliffe, J.S.; Nelson, D.L.; Warren, S.T.; Housman, D.E.; Schalling, M. Tissue Specific Expression of FMR–1 Provides Evidence for a Functional Role in Fragile X Syndrome. Nat. Genet. 1993, 3, 36–43. [Google Scholar] [CrossRef]
- Bakker, C.E.; de Diego Otero, Y.; Bontekoe, C.; Raghoe, P.; Luteijn, T.; Hoogeveen, A.T.; Oostra, B.A.; Willemsen, R. Immunocytochemical and Biochemical Characterization of FMRP, FXR1P, and FXR2P in the Mouse. Exp. Cell Res. 2000, 258, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Patzlaff, N.E.; Shen, M.; Zhao, X. Regulation of Adult Neurogenesis by the Fragile X Family of RNA Binding Proteins. Brain Plast. 2018, 3, 205–223. [Google Scholar] [CrossRef]
- Darnell, J.C.; Fraser, C.E.; Mostovetsky, O.; Stefani, G.; Jones, T.A.; Eddy, S.R.; Darnell, R.B. Kissing Complex RNAs Mediate Interaction between the Fragile-X Mental Retardation Protein KH2 Domain and Brain Polyribosomes. Genes Dev. 2005, 19, 903–918. [Google Scholar] [CrossRef]
- Darnell, J.C.; Jensen, K.B.; Jin, P.; Brown, V.; Warren, S.T.; Darnell, R.B. Fragile X Mental Retardation Protein Targets G Quartet MRNAs Important for Neuronal Function. Cell 2001, 107, 489–499. [Google Scholar] [CrossRef]
- Bechara, E.G.; Didiot, M.C.; Melko, M.; Davidovic, L.; Bensaid, M.; Martin, P.; Castets, M.; Pognonec, P.; Khandjian, E.W.; Moine, H.; et al. A Novel Function for Fragile X Mental Retardation Protein in Translational Activation. PLoS Biol. 2009, 7, e1000016. [Google Scholar] [CrossRef]
- Chen, E.; Joseph, S. Fragile X Mental Retardation Protein: A Paradigm for Translational Control by RNA-Binding Proteins. Biochimie 2015, 114, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Choi, M.; Siomi, M.C.; Nussbaum, R.L.; Dreyfuss, G. Essential Role for KH Domains in RNA Binding: Impaired RNA Binding by a Mutation in the KH Domain of FMR1 That Causes Fragile X Syndrome. Cell 1994, 77, 33–39. [Google Scholar] [CrossRef]
- Myrick, L.K.; Nakamoto-Kinoshita, M.; Lindor, N.M.; Kirmani, S.; Cheng, X.; Warren, S.T. Fragile X Syndrome Due to a Missense Mutation. Eur. J. Hum. Genet. 2014, 22, 1185–1189. [Google Scholar] [CrossRef]
- Corbin, F.; Bouillon, M.; Fortin, A.; Morin, S.; Rousseau, F.; Khandjian, E.W. The Fragile X Mental Retardation Protein Is Associated with Poly(A)+ MRNA in Actively Translating Polyribosomes. Hum. Mol. Genet. 1997, 6, 1465–1472. [Google Scholar] [CrossRef]
- Darnell, J.C.; Klann, E. The Translation of Translational Control by FMRP: Therapeutic Targets for FXS. Nat. Neurosci. 2013, 16, 1530–1536. [Google Scholar] [CrossRef]
- Maurin, T.; Bardoni, B. Fragile X Mental Retardation Protein: To Be or Not to Be a Translational Enhancer. Front. Mol. Biosci. 2018, 5, 113. [Google Scholar] [CrossRef]
- Sandoval, G.M.; Shim, S.; Hong, D.S.; Garrett, A.S.; Quintin, E.-M.; Marzelli, M.J.; Patnaik, S.; Lightbody, A.A.; Reiss, A.L. Neuroanatomical Abnormalities in Fragile X Syndrome during the Adolescent and Young Adult Years. J. Psychiatr. Res. 2018, 107, 138–144. [Google Scholar] [CrossRef]
- Hallahan, B.P.; Craig, M.C.; Toal, F.; Daly, E.M.; Moore, C.J.; Ambikapathy, A.; Robertson, D.; Murphy, K.C.; Murphy, D.G.M. In Vivo Brain Anatomy of Adult Males with Fragile X Syndrome: An MRI Study. Neuroimage 2011, 54, 16–24. [Google Scholar] [CrossRef]
- Hoeft, F.; Carter, J.C.; Lightbody, A.A.; Cody Hazlett, H.; Piven, J.; Reiss, A.L. Region-Specific Alterations in Brain Development in One- to Three-Year-Old Boys with Fragile X Syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 9335–9339. [Google Scholar] [CrossRef]
- Lombroso, P.J. Genetics of Childhood Disorders: XLVIII. Learning and Memory, Part 1: Fragile X Syndrome Update. J. Am. Acad. Child. Adolesc. Psychiatry 2003, 42, 372–375. [Google Scholar] [CrossRef][Green Version]
- He, C.X.; Portera-Cailliau, C. The Trouble with Spines in Fragile X Syndrome: Density, Maturity and Plasticity. Neuroscience 2013, 251, 120–128. [Google Scholar] [CrossRef]
- Contractor, A.; Klyachko, V.A.; Portera-Cailliau, C. Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 2015, 87, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.R.; Bartley, A.F.; Hays, S.A.; Huber, K.M. Imbalance of Neocortical Excitation and Inhibition and Altered UP States Reflect Network Hyperexcitability in the Mouse Model of Fragile X Syndrome. J. Neurophysiol. 2008, 100, 2615–2626. [Google Scholar] [CrossRef] [PubMed]
- Morin-Parent, F.; Champigny, C.; Lacroix, A.; Corbin, F.; Lepage, J.-F. Hyperexcitability and Impaired Intracortical Inhibition in Patients with Fragile-X Syndrome. Transl. Psychiatry 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, M.S.; Auerbach, B.D.; Bear, M.F. Fragile X Mental Retardation Protein and Synaptic Plasticity. Mol. Brain 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.C.; Hagerman, R.J. Serotonin Dysregulation in Fragile X Syndrome: Implications for Treatment. Intractable Rare Dis. Res. 2014, 3, 110–117. [Google Scholar] [CrossRef][Green Version]
- Protic, D.; Salcedo-Arellano, M.J.; Dy, J.B.; Potter, L.A.; Hagerman, R.J. New Targeted Treatments for Fragile X Syndrome. Curr. Pediatr. Rev. 2019, 15, 251–258. [Google Scholar] [CrossRef]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The MGluR Theory of Fragile X Mental Retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef]
- Dölen, G.; Bear, M.F. Role for Metabotropic Glutamate Receptor 5 (MGluR5) in the Pathogenesis of Fragile X Syndrome: Pathogenesis of Fragile X Syndrome. J. Physiol. 2008, 586, 1503–1508. [Google Scholar] [CrossRef]
- Braat, S.; Kooy, R.F. Insights into GABAAergic System Deficits in Fragile X Syndrome Lead to Clinical Trials. Neuropharmacology 2015, 88, 48–54. [Google Scholar] [CrossRef]
- Van der Aa, N.; Kooy, R.F. GABAergic Abnormalities in the Fragile X Syndrome. Eur. J. Paediatr. Neurol. 2020, 24, 100–104. [Google Scholar] [CrossRef]
- Hagerman, R.; Lozano, R.; Hare, E. Modulation of the GABAergic Pathway for the Treatment of Fragile X Syndrome. Neuropsychiatr. Dis. Treat. 2014, 1769. [Google Scholar] [CrossRef]
- D’Hulst, C.; Heulens, I.; Brouwer, J.R.; Willemsen, R.; De Geest, N.; Reeve, S.P.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Expression of the GABAergic System in Animal Models for Fragile X Syndrome and Fragile X Associated Tremor/Ataxia Syndrome (FXTAS). Brain Res. 2009, 1253, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic Pharmacological MGlu5 Inhibition Corrects Fragile X in Adult Mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef]
- Osterweil, E.K.; Chuang, S.-C.; Chubykin, A.A.; Sidorov, M.; Bianchi, R.; Wong, R.K.S.; Bear, M.F. Lovastatin Corrects Excess Protein Synthesis and Prevents Epileptogenesis in a Mouse Model of Fragile X Syndrome. Neuron 2013, 77, 243–250. [Google Scholar] [CrossRef]
- de Vrij, F.M.S.; Levenga, J.; van der Linde, H.C.; Koekkoek, S.K.; De Zeeuw, C.I.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Rescue of Behavioral Phenotype and Neuronal Protrusion Morphology in Fmr1 KO Mice. Neurobiol. Dis. 2008, 31, 127–132. [Google Scholar] [CrossRef]
- Henderson, C.; Wijetunge, L.; Kinoshita, M.N.; Shumway, M.; Hammond, R.S.; Postma, F.R.; Brynczka, C.; Rush, R.; Thomas, A.; Paylor, R.; et al. Reversal of Disease-Related Pathologies in the Fragile X Mouse Model by Selective Activation of GABAB Receptors with Arbaclofen. Sci. Transl. Med. 2012, 4, 152ra128. [Google Scholar] [CrossRef]
- Heulens, I.; D’Hulst, C.; Van Dam, D.; De Deyn, P.P.; Kooy, R.F. Pharmacological Treatment of Fragile X Syndrome with GABAergic Drugs in a Knockout Mouse Model. Behav. Brain Res. 2012, 229, 244–249. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Corbin, J.G.; Burns, M.P. The GABA(A) Receptor Agonist THIP Ameliorates Specific Behavioral Deficits in the Mouse Model of Fragile X Syndrome. Dev. Neurosci. 2011, 33, 395–403. [Google Scholar] [CrossRef]
- Qin, M.; Huang, T.; Kader, M.; Krych, L.; Xia, Z.; Burlin, T.; Zeidler, Z.; Zhao, T.; Smith, C.B. R-Baclofen Reverses a Social Behavior Deficit and Elevated Protein Synthesis in a Mouse Model of Fragile X Syndrome. Int. J. Neuropsychopharmacol. 2015, 18, pyv034. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hessl, D.; Coffey, S.; Hervey, C.; Schneider, A.; Yuhas, J.; Hutchison, J.; Snape, M.; Tranfaglia, M.; Nguyen, D.V.; et al. A Pilot Open Label, Single Dose Trial of Fenobam in Adults with Fragile X Syndrome. J. Med. Genet. 2009, 46, 266–271. [Google Scholar] [CrossRef]
- Youssef, E.A.; Berry-Kravis, E.; Czech, C.; Hagerman, R.J.; Hessl, D.; Wong, C.Y.; Rabbia, M.; Deptula, D.; John, A.; Kinch, R.; et al. Effect of the MGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 2018, 43, 503–512. [Google Scholar] [CrossRef]
- Hagerman, R.; Jacquemont, S.; Berry-Kravis, E.; Des Portes, V.; Stanfield, A.; Koumaras, B.; Rosenkranz, G.; Murgia, A.; Wolf, C.; Apostol, G.; et al. Mavoglurant in Fragile X Syndrome: Results of Two Open-Label, Extension Trials in Adults and Adolescents. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Jacquemont, S.; Curie, A.; des Portes, V.; Torrioli, M.G.; Berry-Kravis, E.; Hagerman, R.J.; Ramos, F.J.; Cornish, K.; He, Y.; Paulding, C.; et al. Epigenetic Modification of the FMR1 Gene in Fragile X Syndrome Is Associated with Differential Response to the MGluR5 Antagonist AFQ056. Sci. Transl. Med. 2011, 3, 64ra1. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Weng, N.; Weiler, I.J.; Greenough, W.T.; Stigler, K.A.; Wink, L.K.; McDougle, C.J. Open-Label Riluzole in Fragile X Syndrome. Brain Res. 2011, 1380, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Ligsay, A.; Van Dijck, A.; Nguyen, D.V.; Lozano, R.; Chen, Y.; Bickel, E.S.; Hessl, D.; Schneider, A.; Angkustsiri, K.; Tassone, F.; et al. A Randomized Double-Blind, Placebo-Controlled Trial of Ganaxolone in Children and Adolescents with Fragile X Syndrome. J. Neurodev. Disord. 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.M.; Hessl, D.; Rathmell, B.; Zarevics, P.; Cherubini, M.; Walton-Bowen, K.; Mu, Y.; Nguyen, D.V.; Gonzalez-Heydrich, J.; Wang, P.P.; et al. Effects of STX209 (Arbaclofen) on Neurobehavioral Function in Children and Adults with Fragile X Syndrome: A Randomized, Controlled, Phase 2 Trial. Sci. Transl. Med. 2012, 4, 152ra127. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hagerman, R.; Visootsak, J.; Budimirovic, D.; Kaufmann, W.E.; Cherubini, M.; Zarevics, P.; Walton-Bowen, K.; Wang, P.; Bear, M.F.; et al. Arbaclofen in Fragile X Syndrome: Results of Phase 3 Trials. J. Neurodev. Disord. 2017, 9. [Google Scholar] [CrossRef]
- Erickson, C.A.; Wink, L.K.; Ray, B.; Early, M.C.; Stiegelmeyer, E.; Mathieu-Frasier, L.; Patrick, V.; Lahiri, D.K.; McDougle, C.J. Impact of Acamprosate on Behavior and Brain-Derived Neurotrophic Factor: An Open-Label Study in Youth with Fragile X Syndrome. Psychopharmacology 2013, 228, 75–84. [Google Scholar] [CrossRef]
- Dahlhaus, R. Of Men and Mice: Modeling the Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Zhao, X.; Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef] [PubMed]
- Sittler, A.; Devys, D.; Weber, C.; Mandel, J.L. Alternative Splicing of Exon 14 Determines Nuclear or Cytoplasmic Localisation of Fmr1 Protein Isoforms. Hum. Mol. Genet. 1996, 5, 95–102. [Google Scholar] [CrossRef]
- Feng, Y.; Gutekunst, C.-A.; Eberhart, D.E.; Yi, H.; Warren, S.T.; Hersch, S.M. Fragile X Mental Retardation Protein: Nucleocytoplasmic Shuttling and Association with Somatodendritic Ribosomes. J. Neurosci. 1997, 17, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Dury, A.Y.; Fatimy, R.E.; Tremblay, S.; Rose, T.M.; Côté, J.; Koninck, P.D.; Khandjian, E.W. Nuclear Fragile X Mental Retardation Protein Is Localized to Cajal Bodies. PLoS Genet. 2013, 9, e1003890. [Google Scholar] [CrossRef] [PubMed]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stützer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A Chromatin-Dependent Role of the Fragile X Mental Retardation Protein FMRP in the DNA Damage Response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Bozzetti, M.P.; Specchia, V.; Cattenoz, P.B.; Laneve, P.; Geusa, A.; Sahin, H.B.; Di Tommaso, S.; Friscini, A.; Massari, S.; Diebold, C.; et al. The Drosophila Fragile X Mental Retardation Protein Participates in the PiRNA Pathway. J. Cell. Sci. 2015, 128, 2070–2084. [Google Scholar] [CrossRef]
- Jiang, F.; Lu, F.; Li, P.; Liu, W.; Zhao, L.; Wang, Q.; Cao, X.; Zhang, L.; Zhang, Y.Q. Drosophila Homolog of FMRP Maintains Genome Integrity by Interacting with Piwi. J. Genet. Genom. 2016, 43, 11–24. [Google Scholar] [CrossRef]
- Tóth, K.F.; Pezic, D.; Stuwe, E.; Webster, A. The PiRNA Pathway Guards the Germline Genome Against Transposable Elements. Adv. Exp. Med. Biol. 2016, 886, 51–77. [Google Scholar] [CrossRef]
- Fyodorov, D.V.; Zhou, B.-R.; Skoultchi, A.I.; Bai, Y. Emerging Roles of Linker Histones in Regulating Chromatin Structure and Function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP Stalls Ribosomal Translocation on MRNAs Linked to Synaptic Function and Autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Gresack, J.; Allis, C.D.; Darnell, R.B. Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition. Cell 2017, 170, 1209–1223.e20. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond Transcriptional Regulation. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Molinaro, G.; Liu, B.; Wang, R.; Huber, K.M.; Richter, J.D. FMRP Control of Ribosome Translocation Promotes Chromatin Modifications and Alternative Splicing of Neuronal Genes Linked to Autism. Cell Rep. 2020, 30, 4459–4472.e6. [Google Scholar] [CrossRef]
- He, Q.; Ge, W. FMRP: A New Chapter with Chromatin. Protein Cell 2014, 5, 885–888. [Google Scholar] [CrossRef][Green Version]
- Goodman, J.V.; Bonni, A. Regulation of Neuronal Connectivity in the Mammalian Brain by Chromatin Remodeling. Curr. Opin. Neurobiol. 2019, 59, 59–68. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, N.; Zhang, Y.; Du, Y.; Zhang, T.; Li, Z.; Wu, J.; Wang, X. Prioritized High-Confidence Risk Genes for Intellectual Disability Reveal Molecular Convergence During Brain Development. Front. Genet. 2018, 9, 349. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Cao, T.; Zhen, X. Dysregulation of MiRNA and Its Potential Therapeutic Application in Schizophrenia. CNS Neurosci. Ther. 2018, 24, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. In MicroRNA Profiling: Methods and Protocols; Rani, S., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 1–10. ISBN 978-1-4939-6524-3. [Google Scholar]
- Caudy, A.A.; Myers, M.; Hannon, G.J.; Hammond, S.M. Fragile X-Related Protein and VIG Associate with the RNA Interference Machinery. Genes Dev. 2002, 16, 2491–2496. [Google Scholar] [CrossRef]
- Jin, P.; Zarnescu, D.C.; Ceman, S.; Nakamoto, M.; Mowrey, J.; Jongens, T.A.; Nelson, D.L.; Moses, K.; Warren, S.T. Biochemical and Genetic Interaction between the Fragile X Mental Retardation Protein and the MicroRNA Pathway. Nat. Neurosci. 2004, 7, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Plante, I.; Davidovic, L.; Ouellet, D.L.; Gobeil, L.-A.; Tremblay, S.; Khandjian, E.W.; Provost, P. Dicer-Derived MicroRNAs Are Utilized by the Fragile X Mental Retardation Protein for Assembly on Target RNAs. J. Biomed. Biotechnol. 2006, 2006, 64347. [Google Scholar] [CrossRef]
- Cheever, A.; Ceman, S. Phosphorylation of FMRP Inhibits Association with Dicer. RNA 2009, 15, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Ishizuka, A.; Siomi, H.; Siomi, M.C. Distinct Roles for Argonaute Proteins in Small RNA-Directed RNA Cleavage Pathways. Genes Dev. 2004, 18, 1655–1666. [Google Scholar] [CrossRef]
- Kenny, P.J.; Zhou, H.; Kim, M.; Skariah, G.; Khetani, R.S.; Drnevich, J.; Arcila, M.L.; Kosik, K.S.; Ceman, S. MOV10 and FMRP Regulate AGO2 Association with MicroRNA Recognition Elements. Cell Rep. 2014, 9, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of Synaptic Structure and Function by FMRP-Associated MicroRNAs MiR-125b and MiR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef]
- Halevy, T.; Czech, C.; Benvenisty, N. Molecular Mechanisms Regulating the Defects in Fragile X Syndrome Neurons Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 37–46. [Google Scholar] [CrossRef]
- Muddashetty, R.S.; Nalavadi, V.C.; Gross, C.; Yao, X.; Xing, L.; Laur, O.; Warren, S.T.; Bassell, G.J. Reversible Inhibition of PSD-95 MRNA Translation by MiR-125a, FMRP Phosphorylation, and MGluR Signaling. Mol. Cell 2011, 42, 673–688. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, B.; Stefanovic, S.; Williams, A.; Moss, K.R.; Anderson, B.R.; Bassell, G.J.; Mihailescu, M.R. FMRP—G-Quadruplex MRNA—MiR-125a Interactions: Implications for MiR-125a Mediated Translation Regulation of PSD-95 MRNA. PLoS ONE 2019, 14, e0217275. [Google Scholar] [CrossRef] [PubMed]
- Putkonen, N.; Laiho, A.; Ethell, D.; Pursiheimo, J.; Anttonen, A.-K.; Pitkonen, J.; Gentile, A.M.; de Diego-Otero, Y.; Castrén, M.L. Urine MicroRNA Profiling Displays MiR-125a Dysregulation in Children with Fragile X Syndrome. Cells 2020, 9, 289. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Ooi, J.Y.; Lin, R.C.; McMullen, J.R. MiRNA Therapeutics: A New Class of Drugs with Potential Therapeutic Applications in the Heart. Future Med. Chem. 2015, 7, 1771–1792. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Meese, E.; Keller, A. Specific MiRNA Disease Biomarkers in Blood, Serum and Plasma: Challenges and Prospects. Mol. Diagn. Ther. 2016, 20, 509–518. [Google Scholar] [CrossRef]
- Didiot, M.-C.; Tian, Z.; Schaeffer, C.; Subramanian, M.; Mandel, J.-L.; Moine, H. The G-Quartet Containing FMRP Binding Site in FMR1 MRNA Is a Potent Exonic Splicing Enhancer. Nucleic Acids Res. 2008, 36, 4902–4912. [Google Scholar] [CrossRef]
- Brooks, A.N.; Duff, M.O.; May, G.; Yang, L.; Bolisetty, M.; Landolin, J.; Wan, K.; Sandler, J.; Booth, B.W.; Celniker, S.E.; et al. Regulation of Alternative Splicing in Drosophila by 56 RNA Binding Proteins. Genome Res. 2015, 25, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, M.H.; Olson, S.; May, G.E.; Duff, M.O.; Manent, J.; Obar, R.; Guruharsha, K.G.; Bickel, P.J.; Artavanis-Tsakonas, S.; Brown, J.B.; et al. Extensive Cross-Regulation of Post-Transcriptional Regulatory Networks in Drosophila. Genome Res. 2015, 25, 1692–1702. [Google Scholar] [CrossRef]
- Pasciuto, E.; Bagni, C. SnapShot: FMRP Interacting Proteins. Cell 2014, 159, 218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-T.; Ye, S.-H.; Yang, H.-X.; Zhou, Y.-T.; Zhao, Q.-H.; Sun, W.-W.; Gao, M.-M.; Yi, Y.-H.; Long, Y.-S. A Novel Role of Fragile X Mental Retardation Protein in Pre-MRNA Alternative Splicing through RNA-Binding Protein 14. Neuroscience 2017, 349, 64–75. [Google Scholar] [CrossRef]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef]
- Krestel, H.; Meier, J.C. RNA Editing and Retrotransposons in Neurology. Front. Mol. Neurosci. 2018, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Jepson, J.E.; Savva, Y.A.; Pepper, A.S.-R.; Reenan, R.A.; Jongens, T.A. Modulation of DADAR-Dependent RNA Editing by the Drosophila Fragile X Mental Retardation Protein. Nat. Neurosci. 2011, 14, 1517–1524. [Google Scholar] [CrossRef]
- Shamay-Ramot, A.; Khermesh, K.; Porath, H.T.; Barak, M.; Pinto, Y.; Wachtel, C.; Zilberberg, A.; Lerer-Goldshtein, T.; Efroni, S.; Levanon, E.Y.; et al. Fmrp Interacts with Adar and Regulates RNA Editing, Synaptic Density and Locomotor Activity in Zebrafish. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Bonini, D.; Lacoux, C.; Pacini, L.; Zingariello, M.; Sancillo, L.; Bosisio, D.; Salvi, V.; Mingardi, J.; La Via, L.; et al. Absence of the Fragile X Mental Retardation Protein Results in Defects of RNA Editing of Neuronal MRNAs in Mouse. RNA Biol. 2017, 14, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Laggerbauer, B.; Ostareck, D.; Keidel, E.M.; Ostareck-Lederer, A.; Fischer, U. Evidence That Fragile X Mental Retardation Protein Is a Negative Regulator of Translation. Hum. Mol. Genet. 2001, 10, 329–338. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Ku, L.; Wilkinson, K.D.; Warren, S.T.; Feng, Y. The Fragile X Mental Retardation Protein Inhibits Translation via Interacting with MRNA. Nucleic Acids Res. 2001, 29, 2276–2283. [Google Scholar] [CrossRef]
- Bolduc, F.V.; Bell, K.; Cox, H.; Broadie, K.S.; Tully, T. Excess Protein Synthesis in Drosophila Fragile X Mutants Impairs Long-Term Memory. Nat. Neurosci. 2008, 11, 1143–1145. [Google Scholar] [CrossRef]
- Qin, M.; Kang, J.; Burlin, T.V.; Jiang, C.; Smith, C.B. Postadolescent Changes in Regional Cerebral Protein Synthesis: An in Vivo Study in the FMR1 Null Mouse. J. Neurosci. 2005, 25, 5087–5095. [Google Scholar] [CrossRef]
- Till, S.M.; Asiminas, A.; Jackson, A.D.; Katsanevaki, D.; Barnes, S.A.; Osterweil, E.K.; Bear, M.F.; Chattarji, S.; Wood, E.R.; Wyllie, D.J.A.; et al. Conserved Hippocampal Cellular Pathophysiology but Distinct Behavioural Deficits in a New Rat Model of FXS. Hum. Mol. Genet. 2015, 24, 5977–5984. [Google Scholar] [CrossRef]
- Osterweil, E.K.; Krueger, D.D.; Reinhold, K.; Bear, M.F. Hypersensitivity to MGluR5 and ERK1/2 Leads to Excessive Protein Synthesis in the Hippocampus of a Mouse Model of Fragile X Syndrome. J. Neurosci. 2010, 30, 15616–15627. [Google Scholar] [CrossRef]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin Ameliorates Core Deficits in a Mouse Model of Fragile X Syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef] [PubMed]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective Inhibition of Glycogen Synthase Kinase 3α Corrects Pathophysiology in a Mouse Model of Fragile X Syndrome. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Kumari, D.; Bhattacharya, A.; Nadel, J.; Moulton, K.; Zeak, N.M.; Glicksman, A.; Dobkin, C.; Brick, D.J.; Schwartz, P.H.; Smith, C.B.; et al. Identification of Fragile X Syndrome Specific Molecular Markers in Human Fibroblasts: A Useful Model to Test the Efficacy of Therapeutic Drugs. Hum. Mutat. 2014, 35, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Pacini, L.; Jønch, A.E.; Cencelli, G.; Rozenberg, I.; He, Y.; D’Andrea, L.; Pedini, G.; Eldeeb, M.; Willemsen, R.; et al. Protein Synthesis Levels Are Increased in a Subset of Individuals with Fragile X Syndrome. Hum. Mol. Genet. 2018, 27, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Schmidt, K.C.; Zametkin, A.J.; Bishu, S.; Horowitz, L.M.; Burlin, T.V.; Xia, Z.; Huang, T.; Quezado, Z.M.; Smith, C.B. Altered Cerebral Protein Synthesis in Fragile X Syndrome: Studies in Human Subjects and Knockout Mice. J. Cereb. Blood Flow Metab. 2013, 33, 499–507. [Google Scholar] [CrossRef]
- Schmidt, K.C.; Loutaev, I.; Quezado, Z.; Sheeler, C.; Smith, C.B. Regional Rates of Brain Protein Synthesis Are Unaltered in Dexmedetomidine Sedated Young Men with Fragile X Syndrome: A L-[1-11C]Leucine PET Study. Neurobiol. Dis. 2020, 143, 104978. [Google Scholar] [CrossRef] [PubMed]
- Bowling, H.; Bhattacharya, A.; Zhang, G.; Alam, D.; Lebowitz, J.Z.; Bohm-Levine, N.; Lin, D.; Singha, P.; Mamcarz, M.; Puckett, R.; et al. Altered Steady State and Activity-Dependent de Novo Protein Expression in Fragile X Syndrome. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Gross, C.; Nakamoto, M.; Yao, X.; Chan, C.-B.; Yim, S.Y.; Ye, K.; Warren, S.T.; Bassell, G.J. Excess Phosphoinositide 3-Kinase Subunit Synthesis and Activity as a Novel Therapeutic Target in Fragile X Syndrome. J. Neurosci. 2010, 30, 10624–10638. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hessl, D.; Abbeduto, L.; Reiss, A.L.; Beckel-Mitchener, A.; Urv, T.K. Outcome Measures for Clinical Trials in Fragile X Syndrome. J. Dev. Behav. Pediatr. 2013, 34, 508–522. [Google Scholar] [CrossRef]
- Fayard, E. Protein Kinase B/Akt at a Glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef]
- Buscà, R.; Pouysségur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front. Cell Dev. Biol. 2016, 4. [Google Scholar] [CrossRef]
- Ascano, M.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP Targets Distinct MRNA Sequence Elements to Regulate Protein Expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Hou, L.; Antion, M.D.; Hu, D.; Spencer, C.M.; Paylor, R.; Klann, E. Dynamic Translational and Proteasomal Regulation of Fragile X Mental Retardation Protein Controls MGluR-Dependent Long-Term Depression. Neuron 2006, 51, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Rashid, M.H.; Millecamps, M.; Sanoja, R.; Entrena, J.M.; Cervero, F. Decreased Nociceptive Sensitization in Mice Lacking the Fragile X Mental Retardation Protein: Role of MGluR1/5 and MTOR. J. Neurosci. 2007, 27, 13958–13967. [Google Scholar] [CrossRef]
- Weng, N.; Weiler, I.J.; Sumis, A.; Berry-Kravis, E.; Greenough, W.T. Early-Phase ERK Activation as a Biomarker for Metabolic Status in Fragile X Syndrome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147B, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of MTOR Signaling in Fragile X Syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kaphzan, H.; Alvarez-Dieppa, A.C.; Murphy, J.P.; Pierre, P.; Klann, E. Genetic Removal of P70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice. Neuron 2012, 76, 325–337. [Google Scholar] [CrossRef]
- Hoeffer, C.A.; Sanchez, E.; Hagerman, R.J.; Mu, Y.; Nguyen, D.V.; Wong, H.; Whelan, A.M.; Zukin, R.S.; Klann, E.; Tassone, F. Altered MTOR Signaling and Enhanced CYFIP2 Expression Levels in Subjects with Fragile X Syndrome. Genes Brain Behav. 2012, 11, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Snape, M.; Klann, E.; Stone, J.G.; Singh, A.; Petersen, R.B.; Castellani, R.J.; Casadesus, G.; Smith, M.A.; Zhu, X. Activation of the Extracellular Signal-Regulated Kinase Pathway Contributes to the Behavioral Deficit of Fragile x-Syndrome. J. Neurochem. 2012, 121, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, D.; Çaku, A.; Fradet, M.; Bouvier, P.; Dubé, J.; Corbin, F. Lovastatin Corrects ERK Pathway Hyperactivation in Fragile X Syndrome: Potential of Platelet’s Signaling Cascades as New Outcome Measures in Clinical Trials. Biomarkers 2016, 21, 497–508. [Google Scholar] [CrossRef]
- Çaku, A.; Pellerin, D.; Bouvier, P.; Riou, E.; Corbin, F. Effect of Lovastatin on Behavior in Children and Adults with Fragile X Syndrome: An Open-Label Study. Am. J. Med. Genet. Part A 2014, 164, 2834–2842. [Google Scholar] [CrossRef]
- Wlodarczyk, J.; Mukhina, I.; Kaczmarek, L.; Dityatev, A. Extracellular Matrix Molecules, Their Receptors, and Secreted Proteases in Synaptic Plasticity. Dev. Neurobiol. 2011, 71, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Dansie, L.E.; Ethell, I.M. Casting a Net on Dendritic Spines: The Extracellular Matrix and Its Receptors. Dev. Neurobiol. 2011, 71, 956–981. [Google Scholar] [CrossRef] [PubMed]
- Michaluk, P.; Wawrzyniak, M.; Alot, P.; Szczot, M.; Wyrembek, P.; Mercik, K.; Medvedev, N.; Wilczek, E.; De Roo, M.; Zuschratter, W.; et al. Influence of Matrix Metalloproteinase MMP-9 on Dendritic Spine Morphology. J. Cell Sci. 2011, 124, 3369–3380. [Google Scholar] [CrossRef]
- Reinhard, S.M.; Razak, K.; Ethell, I.M. A Delicate Balance: Role of MMP-9 in Brain Development and Pathophysiology of Neurodevelopmental Disorders. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Nagy, V.; Bozdagi, O.; Matynia, A.; Balcerzyk, M.; Okulski, P.; Dzwonek, J.; Costa, R.M.; Silva, A.J.; Kaczmarek, L.; Huntley, G.W. Matrix Metalloproteinase-9 Is Required for Hippocampal Late-Phase Long-Term Potentiation and Memory. J. Neurosci. 2006, 26, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bozdagi, O.; Nikitczuk, J.S.; Zhai, Z.W.; Zhou, Q.; Huntley, G.W. Extracellular Proteolysis by Matrix Metalloproteinase-9 Drives Dendritic Spine Enlargement and Long-Term Potentiation Coordinately. Proc. Natl. Acad. Sci. USA 2008, 105, 19520–19525. [Google Scholar] [CrossRef]
- Janusz, A.; Milek, J.; Perycz, M.; Pacini, L.; Bagni, C.; Kaczmarek, L.; Dziembowska, M. The Fragile X Mental Retardation Protein Regulates Matrix Metalloproteinase 9 MRNA at Synapses. J. Neurosci. 2013, 33, 18234–18241. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, H.; Dansie, L.E.; Hickmott, P.W.; Ethell, D.W.; Ethell, I.M. Genetic Removal of Matrix Metalloproteinase 9 Rescues the Symptoms of Fragile X Syndrome in a Mouse Model. J. Neurosci. 2014, 34, 9867–9879. [Google Scholar] [CrossRef]
- Vandooren, J.; Knoops, S.; Aldinucci Buzzo, J.L.; Boon, L.; Martens, E.; Opdenakker, G.; Kolaczkowska, E. Differential Inhibition of Activity, Activation and Gene Expression of MMP-9 in THP-1 Cells by Azithromycin and Minocycline versus Bortezomib: A Comparative Study. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Dansie, L.E.; Phommahaxay, K.; Okusanya, A.G.; Uwadia, J.; Huang, M.; Rotschafer, S.E.; Razak, K.A.; Ethell, D.W.; Ethell, I.M. Long-Lasting Effects of Minocycline on Behavior in Young but Not Adult Fragile X Mice. Neuroscience 2013, 246, 186–198. [Google Scholar] [CrossRef]
- Bilousova, T.V.; Dansie, L.; Ngo, M.; Aye, J.; Charles, J.R.; Ethell, D.W.; Ethell, I.M. Minocycline Promotes Dendritic Spine Maturation and Improves Behavioural Performance in the Fragile X Mouse Model. J. Med. Genet. 2009, 46, 94–102. [Google Scholar] [CrossRef]
- Leigh, M.J.S.; Nguyen, D.V.; Mu, Y.; Winarni, T.I.; Schneider, A.; Chechi, T.; Polussa, J.; Doucet, P.; Tassone, F.; Rivera, S.M.; et al. A Randomized Double-Blind, Placebo-Controlled Trial of Minocycline in Children and Adolescents with Fragile x Syndrome. J. Dev. Behav. Pediatr. 2013, 34, 147–155. [Google Scholar] [CrossRef]
- Paribello, C.; Tao, L.; Folino, A.; Berry-Kravis, E.; Tranfaglia, M.; Ethell, I.M.; Ethell, D.W. Open-Label Add-on Treatment Trial of Minocycline in Fragile X Syndrome. BMC Neurol. 2010, 10, 91. [Google Scholar] [CrossRef]
- Dziembowska, M.; Pretto, D.I.; Janusz, A.; Kaczmarek, L.; Leigh, M.J.; Gabriel, N.; Durbin-Johnson, B.; Hagerman, R.J.; Tassone, F. High MMP-9 Activity Levels in Fragile X Syndrome Are Lowered by Minocycline. Am. J. Med. Genet. Part A 2013, 161, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef] [PubMed]
- Prilutsky, D.; Kho, A.T.; Palmer, N.P.; Bhakar, A.L.; Smedemark-Margulies, N.; Kong, S.W.; Margulies, D.M.; Bear, M.F.; Kohane, I.S. Gene Expression Analysis in Fmr1KO Mice Identifies an Immunological Signature in Brain Tissue and MGluR5-Related Signaling in Primary Neuronal Cultures. Mol. Autism 2015, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Hodges, S.L.; Nolan, S.O.; Taube, J.H.; Lugo, J.N. Adult Fmr1 Knockout Mice Present with Deficiencies in Hippocampal Interleukin-6 and Tumor Necrosis Factor-α Expression. Neuroreport 2017, 28, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; Noyon, T.; Basuta, K.; Van de Water, J.; Tassone, F.; Hagerman, R.J.; Ashwood, P. Group I Metabotropic Glutamate Receptor Mediated Dynamic Immune Dysfunction in Children with Fragile X Syndrome. J. Neuroinflamm. 2014, 11, 110. [Google Scholar] [CrossRef]
- Van Dijck, A.; Barbosa, S.; Bermudez-Martin, P.; Khalfallah, O.; Gilet, C.; Martinuzzi, E.; Elinck, E.; Kooy, R.F.; Glaichenhaus, N.; Davidovic, L. Reduced Serum Levels of Pro-Inflammatory Chemokines in Fragile X Syndrome. BMC Neurol. 2020, 20, 138. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Nguyen, D.V.; Hessl, D.; Hagerman, R.J.; Tassone, F. Plasma Cytokine Profiles in Fragile X Subjects: Is There a Role for Cytokines in the Pathogenesis? Brain Behav. Immun. 2010, 24, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-H.; Palmer, N.; Fox, K.; Prock, L.; Mandl, K.D.; Kohane, I.S.; Prilutsky, D. The Phenotypical Implications of Immune Dysregulation in Fragile X Syndrome. Eur. J. Neurol. 2020, 27, 590–593. [Google Scholar] [CrossRef]
- Leboucher, A.; Pisani, D.F.; Martinez-Gili, L.; Chilloux, J.; Bermudez-Martin, P.; Van Dijck, A.; Ganief, T.; Macek, B.; Becker, J.A.J.; Le Merrer, J.; et al. The Translational Regulator FMRP Controls Lipid and Glucose Metabolism in Mice and Humans. Mol. Metab. 2019, 21, 22–35. [Google Scholar] [CrossRef]
- Davidovic, L.; Navratil, V.; Bonaccorso, C.M.; Catania, M.V.; Bardoni, B.; Dumas, M.-E. A Metabolomic and Systems Biology Perspective on the Brain of the Fragile X Syndrome Mouse Model. Genome Res. 2011, 21, 2190–2202. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Thematic Review Series: Brain Lipids. Cholesterol Metabolism in the Central Nervous System during Early Development and in the Mature Animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Martín, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in Brain Disease: Sometimes Determinant and Frequently Implicated. EMBO Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef]
- Diaz-Stransky, A.; Tierney, E. Cognitive and Behavioral Aspects of Smith–Lemli–Opitz Syndrome. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2012, 160C, 295–300. [Google Scholar] [CrossRef]
- Boland, M.R.; Tatonetti, N.P. Investigation of 7-Dehydrocholesterol Reductase Pathway to Elucidate off-Target Prenatal Effects of Pharmaceuticals: A Systematic Review. Pharm. J. 2016, 16, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Tierney, E.; Bukelis, I.; Thompson, R.E.; Ahmed, K.; Aneja, A.; Kratz, L.; Kelley, R.I. Abnormalities of Cholesterol Metabolism in Autism Spectrum Disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-K.; Neggers, Y.H.; Shin, C.-S.; Kim, E.; Kim, E.M. Alterations in Lipid Profile of Autistic Boys: A Case Control Study. Nutr. Res. 2010, 30, 255–260. [Google Scholar] [CrossRef]
- Benachenhou, S.; Etcheverry, A.; Galarneau, L.; Dubé, J.; Çaku, A. Implication of Hypocholesterolemia in Autism Spectrum Disorder and Its Associated Comorbidities: A Retrospective Case–Control Study. Autism Res. 2019, 12, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Levin, R.; Shah, H.; Mathur, S.; Darnell, J.C.; Ouyang, B. Cholesterol Levels in Fragile X Syndrome. Am. J. Med. Genet. 2015, 167, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Çaku, A.; Seidah, N.G.; Lortie, A.; Gagné, N.; Perron, P.; Dubé, J.; Corbin, F. New Insights of Altered Lipid Profile in Fragile X Syndrome. PLoS ONE 2017, 12, e0174301. [Google Scholar] [CrossRef]
- Ben Djoudi Ouadda, A.; Gauthier, M.-S.; Susan-Resiga, D.; Girard, E.; Essalmani, R.; Black, M.; Marcinkiewicz, J.; Forget, D.; Hamelin, J.; Evagelidis, A.; et al. Ser-Phosphorylation of PCSK9 (Proprotein Convertase Subtilisin-Kexin 9) by Fam20C (Family with Sequence Similarity 20, Member C) Kinase Enhances Its Ability to Degrade the LDLR (Low-Density Lipoprotein Receptor). ATVB 2019, 39, 1996–2013. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Bloyd, M.; Stratakis, C.A. PKA Functions in Metabolism and Resistance to Obesity: Lessons from Mouse and Human Studies. J. Endocrinol. 2020, 246, R51–R64. [Google Scholar] [CrossRef] [PubMed]
- Conti, M. Phosphodiesterases and Cyclic Nucleotide Signaling in Endocrine Cells. Mol. Endocrinol. 2000, 14, 1317–1327. [Google Scholar] [CrossRef]
- Goldsmith, Z.G.; Dhanasekaran, D.N. G Protein Regulation of MAPK Networks. Oncogene 2007, 26, 3122–3142. [Google Scholar] [CrossRef]
- Dityatev, A.; El-Husseini, A. (Eds.) Molecular Mechanisms of Synaptogenesis; Springer: New York, NY, USA, 2006; ISBN 978-0-387-32560-6. [Google Scholar]
- Crawford, D.C.; Mennerick, S. Presynaptically Silent Synapses: Dormancy and Awakening of Presynaptic Vesicle Release. Neuroscientist 2012, 18, 216–223. [Google Scholar] [CrossRef]
- Beavo, J.A.; Brunton, L.L. Cyclic Nucleotide Research -- Still Expanding after Half a Century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Sklena, P. Demonstration of Abnormal Cyclic AMP Production in Platelets from Patients with Fragile X Syndrome. Am. J. Med. Genet. 1993, 45, 81–87. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Huttenlocher, P.R. Cyclic AMP Metabolism in Fragile X Syndrome. Ann. Neurol. 1992, 31, 22–26. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Hicar, M.; Ciurlionis, R. Reduced cyclic AMP production in fragile X syndrome: Cytogenetic and molecular correlations. Pediatr. Res. 1995, 38, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Ciurlionis, R. Overexpression of Fragile X Gene (FMR-1) Transcripts Increases CAMP Production in Neural Cells. J. Neurosci. Res. 1998, 51, 41–48. [Google Scholar] [CrossRef]
- Kelley, D.J.; Davidson, R.J.; Elliott, J.L.; Lahvis, G.P.; Yin, J.C.P.; Bhattacharyya, A. The Cyclic AMP Cascade Is Altered in the Fragile X Nervous System. PLoS ONE 2007, 2, e931. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Bhattacharyya, A.; Lahvis, G.; Yin, J.; Malter, J.; Davidson, R. The Cyclic AMP Phenotype of Fragile X and Autism. Neurosci. Biobehav. Rev. 2008, 32, 1533–1543. [Google Scholar] [CrossRef]
- Costa, L.; Sardone, L.M.; Bonaccorso, C.M.; D’Antoni, S.; Spatuzza, M.; Gulisano, W.; Tropea, M.R.; Puzzo, D.; Leopoldo, M.; Lacivita, E.; et al. Activation of Serotonin 5-HT7 Receptors Modulates Hippocampal Synaptic Plasticity by Stimulation of Adenylate Cyclases and Rescues Learning and Behavior in a Mouse Model of Fragile X Syndrome. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Maurin, T.; Melancia, F.; Jarjat, M.; Castro, L.; Costa, L.; Delhaye, S.; Khayachi, A.; Castagnola, S.; Mota, E.; Di Giorgio, A.; et al. Involvement of Phosphodiesterase 2A Activity in the Pathophysiology of Fragile X Syndrome. Cereb. Cortex 2019, 29, 3241–3252. [Google Scholar] [CrossRef]
- Gurney, M.E.; Cogram, P.; Deacon, R.M.; Rex, C.; Tranfaglia, M. Multiple Behavior Phenotypes of the Fragile-X Syndrome Mouse Model Respond to Chronic Inhibition of Phosphodiesterase-4D (PDE4D). Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Schoenfeld, B.P.; Bell, A.J.; Hinchey, J.; Rosenfelt, C.; Gertner, M.J.; Campbell, S.R.; Emerson, D.; Hinchey, P.; Kollaros, M.; et al. Multiple Drug Treatments That Increase CAMP Signaling Restore Long-Term Memory and Aberrant Signaling in Fragile X Syndrome Models. Front. Behav. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Baumkötter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid Precursor Protein Dimerization and Synaptogenic Function Depend on Copper Binding to the Growth Factor-like Domain. J. Neurosci. 2014, 34, 11159–11172. [Google Scholar] [CrossRef]
- Nguyen, K.V. β-Amyloid Precursor Protein (APP) and the Human Diseases. Aims Neurosci. 2019, 6, 273–281. [Google Scholar] [CrossRef]
- Lee, E.K.; Kim, H.H.; Kuwano, Y.; Abdelmohsen, K.; Srikantan, S.; Subaran, S.S.; Gleichmann, M.; Mughal, M.R.; Martindale, J.L.; Yang, X.; et al. HnRNP C Promotes APP Translation by Competing with FMRP for APP MRNA Recruitment to P Bodies. Nat. Struct. Mol. Biol. 2010, 17, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Westmark, C.J.; Malter, J.S. FMRP Mediates MGluR5-Dependent Translation of Amyloid Precursor Protein. PLoS Biol. 2007, 5. [Google Scholar] [CrossRef] [PubMed]
- Westmark, C.J.; Westmark, P.R.; O’Riordan, K.J.; Ray, B.C.; Hervey, C.M.; Salamat, M.S.; Abozeid, S.H.; Stein, K.M.; Stodola, L.A.; Tranfaglia, M.; et al. Reversal of Fragile X Phenotypes by Manipulation of AβPP/Aβ Levels in Fmr1KO Mice. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Sokol, D.K.; Chen, D.; Farlow, M.R.; Dunn, D.W.; Maloney, B.; Zimmer, J.A.; Lahiri, D.K. High Levels of Alzheimer Beta-Amyloid Precursor Protein (APP) in Children With Severely Autistic Behavior and Aggression. J. Child Neurol. 2006, 21, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Long, J.M.; Sokol, D.K.; Lahiri, D.K. Increased Secreted Amyloid Precursor Protein-α (SAPPα) in Severe Autism: Proposal of a Specific, Anabolic Pathway and Putative Biomarker. PLoS ONE 2011, 6, e20405. [Google Scholar] [CrossRef]
- Ray, B.; Sokol, D.K.; Maloney, B.; Lahiri, D.K. Finding Novel Distinctions between the SAPPα-Mediated Anabolic Biochemical Pathways in Autism Spectrum Disorder and Fragile X Syndrome Plasma and Brain Tissue. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- McLane, R.D.; Schmitt, L.M.; Pedapati, E.V.; Shaffer, R.C.; Dominick, K.C.; Horn, P.S.; Gross, C.; Erickson, C.A. Peripheral Amyloid Precursor Protein Derivative Expression in Fragile X Syndrome. Front. Integr. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef]
- Erickson, C.A.; Ray, B.; Maloney, B.; Wink, L.K.; Bowers, K.; Schaefer, T.L.; McDougle, C.J.; Sokol, D.K.; Lahiri, D.K. Impact of Acamprosate on Plasma Amyloid-β Precursor Protein in Youth: A Pilot Analysis in Fragile X Syndrome-Associated and Idiopathic Autism Spectrum Disorder Suggests a Pharmacodynamic Protein Marker. J. Psychiatr. Res. 2014, 59, 220–228. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dionne, O.; Corbin, F. An “Omic” Overview of Fragile X Syndrome. Biology 2021, 10, 433. https://doi.org/10.3390/biology10050433
Dionne O, Corbin F. An “Omic” Overview of Fragile X Syndrome. Biology. 2021; 10(5):433. https://doi.org/10.3390/biology10050433
Chicago/Turabian StyleDionne, Olivier, and François Corbin. 2021. "An “Omic” Overview of Fragile X Syndrome" Biology 10, no. 5: 433. https://doi.org/10.3390/biology10050433
APA StyleDionne, O., & Corbin, F. (2021). An “Omic” Overview of Fragile X Syndrome. Biology, 10(5), 433. https://doi.org/10.3390/biology10050433