Efficacy of Immunohistochemistry for SDHB in the Screening of Hereditary Pheochromocytoma–Paraganglioma


 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Microarray (TMA) Construction
2.2. IHC of SDHB
2.3. IHC Interpretation
2.4. Genetic Analysis
2.4.1. Sanger Sequencing
2.4.2. Next Generation Sequencing (NGS)
2.5. Statistical Anlysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jochmanová, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef]
- Dahia, P.L.M. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Woodward, E.R.; Maher, E.R. Von Hippel-Lindau disease and endocrine tumour susceptibility. Endocr. Relat. Cancer 2006, 13, 415–425. [Google Scholar] [CrossRef]
- Raue, F.; Frank-Raue, K. Update multiple endocrine neoplasia type 2. Fam. Cancer 2010, 9, 449–457. [Google Scholar] [CrossRef]
- Bausch, B.; Borozdin, W.; Neumann, H.P. Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma. N. Engl. J. Med. 2006, 354, 2729–2731. [Google Scholar] [CrossRef]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef] [PubMed]
- van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.C.A.; Sleddens, H.F.B.M.; Derkx, P.; Rivière, J.; Dannenberg, H.; et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine, S. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, L.A.; Xie, Q.; Pang, J.; Wang, L.; Yi, Y.; Zhang, J.; Zhang, Y.; Chen, R.; Lan, W.; et al. Germline SDHB and SDHD mutations in pheochromocytoma and paraganglioma patients. Endocr. Connect. 2018, 7, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, G.; Klink, B.; Richter, S.; Lenders, J.W.; Robledo, M. Metabologenomics of Phaeochromocytoma and Paraganglioma: An Integrated Approach for Personalised Biochemical and Genetic Testing. Clin. Biochem. Rev. 2017, 38, 69–100. [Google Scholar]
- Young, A.L.; Baysal, B.E.; Deb, A.; Young, W.F., Jr. Familial malignant catecholamine-secreting paraganglioma with prolonged survival associated with mutation in the succinate dehydrogenase B gene. J. Clin. Endocrinol. Metab. 2002, 87, 4101–4105. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Dahia, P.L. Next-generation sequencing for the diagnosis of hereditary pheochromocytoma and paraganglioma syndromes. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef]
- Gill, A.J.; Benn, D.E.; Chou, A.; Clarkson, A.; Muljono, A.; Meyer-Rochow, G.Y.; Richardson, A.L.; Sidhu, S.B.; Robinson, B.G.; Clifton-Bligh, R.J. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2010, 41, 805–814. [Google Scholar] [CrossRef]
- Pai, R.; Manipadam, M.T.; Singh, P.; Ebenazer, A.; Samuel, P.; Rajaratnam, S. Usefulness of Succinate dehydrogenase B (SDHB) immunohistochemistry in guiding mutational screening among patients with pheochromocytoma-paraganglioma syndromes. APMIS Acta Pathol. Microbiol. Et Immunol. Scand. 2014, 122, 1130–1135. [Google Scholar] [CrossRef]
- Menara, M.; Oudijk, L.; Badoual, C.; Bertherat, J.; Lepoutre-Lussey, C.; Amar, L.; Iturrioz, X.; Sibony, M.; Zinzindohoué, F.; de Krijger, R.; et al. SDHD immunohistochemistry: A new tool to validate SDHx mutations in pheochromocytoma/paraganglioma. J. Clin. Endocrinol. Metab. 2015, 100, E287–E291. [Google Scholar] [CrossRef] [Green Version]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.K. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelblanco, E.; Santacana, M.; Valls, J.; de Cubas, A.; Cascón, A.; Robledo, M.; Matias-Guiu, X. Usefulness of negative and weak-diffuse pattern of SDHB immunostaining in assessment of SDH mutations in paragangliomas and pheochromocytomas. Endocr. Pathol. 2013, 24, 199–205. [Google Scholar] [CrossRef]
- Blank, A.; Schmitt, A.M.; Korpershoek, E.; van Nederveen, F.; Rudolph, T.; Weber, N.; Strebel, R.T.; de Krijger, R.; Komminoth, P.; Perren, A. SDHB loss predicts malignancy in pheochromocytomas/sympathethic paragangliomas, but not through hypoxia signalling. Endocr. Relat. Cancer 2010, 17, 919–928. [Google Scholar] [CrossRef]
- Neumann, H.P.; Erlic, Z.; Boedeker, C.C.; Rybicki, L.A.; Robledo, M.; Hermsen, M.; Schiavi, F.; Falcioni, M.; Kwok, P.; Bauters, C.; et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: Cost reduction strategy in genetic diagnostic process as fall-out. Cancer Res. 2009, 69, 3650–3656. [Google Scholar] [CrossRef] [Green Version]
- Pillai, S.; Gopalan, V.; Lam, A.K. Review of sequencing platforms and their applications in phaeochromocytoma and paragangliomas. Crit. Rev. Oncol. Hematol. 2017, 116, 58–67. [Google Scholar] [CrossRef]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Douwes Dekker, P.B.; Hogendoorn, P.C.; Kuipers-Dijkshoorn, N.; Prins, F.A.; van Duinen, S.G.; Taschner, P.E.; van der Mey, A.G.; Cornelisse, C.J. SDHD mutations in head and neck paragangliomas result in destabilization of complex II in the mitochondrial respiratory chain with loss of enzymatic activity and abnormal mitochondrial morphology. J. Pathol. 2003, 201, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Burnichon, N.; Cascon, A.; Benn, D.E.; Bayley, J.P.; Welander, J.; Tops, C.M.; Firth, H.; Dwight, T.; Ercolino, T.; et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef] [PubMed]


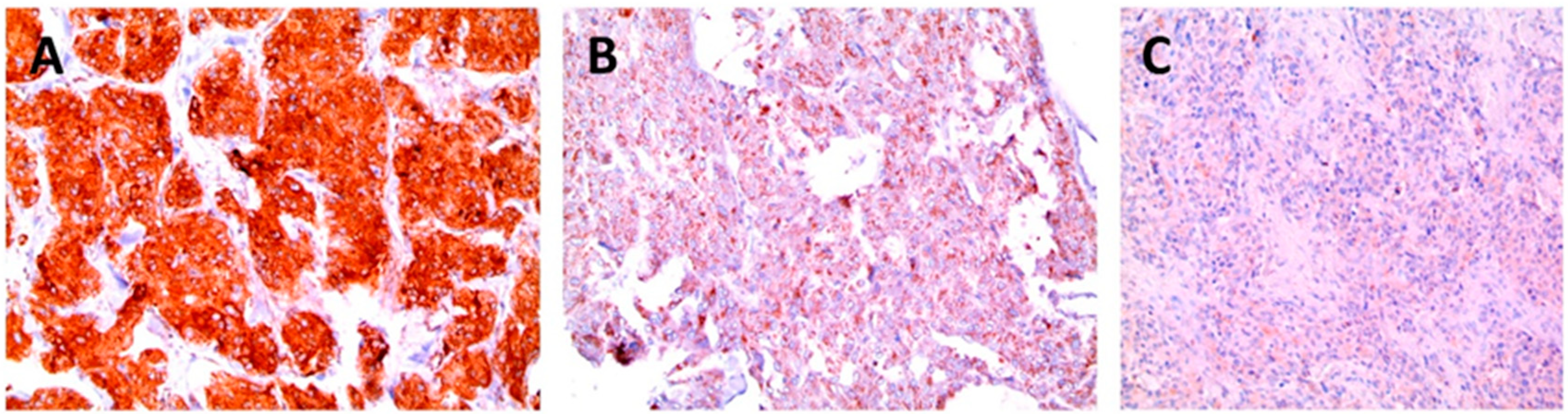{kind=link}
| Gene Mutated | (n) | M/F | Age (y, Mean) | PCC (PASS ≥ 4) | PGL (PASS ≥ 4) | SDHB IHC Neg/Weak/Pos |
|---|---|---|---|---|---|---|
| SDHB | 9 | 5/4 | 16–67 (37) | 5 (4) | 4 (2) | 8/1/0 |
| SDHD | 2 | 0/2 | 24–56 (40) | 1 (0) | 1 (0) | 2/0/0 |
| RET | 9 | 4/5 | 13–62 (48) | 9 (0) | 0 | 0/2/7 |
| VHL | 4 | 3/1 | 30–55 (48) | 3 (0) | 1 (0) | 0/1/3 |
| NF1 | 2 | 1/1 | 36–55 (52) | 2 (0) | 0 | 0/0/2 |
| Unknown | 26 | 10/16 | 19–68 (49) | 25 (1) | 1 (0) | 0/3/23 |
| Total | 52 | 23/29 | 13–68 (49) | 45 (5) | 7 (2) | 10/7/35 |
| SDHB IHC | PASS | (n) | Gene Mutation |
|---|---|---|---|
| SDHB neg | PASS < 4 | 5 | SDHB (3), SDHD (2) |
| PASS ≥ 4 | 5 | SDHB (5) | |
| SDHB weak | PASS < 4 | 12 | RET (2), VHL (1), unknown (9) |
| PASS ≥ 4 | 1 | SDHB (1) | |
| SDHB pos | PASS < 4 | 34 | RET (7), VHL (3), NF1 (2), unknown (22) |
| PASS ≥ 4 | 1 | Unknown (1) | |
| Total | 58 | SDHB (9), SDHD (2), RET (9), VHL (4), NF1 (2), unknown (32) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.-R.; Koo, J.-S.; Lee, C.-R.; Lee, J.-D.; Kang, S.-W.; Jo, Y.-S.; Chung, W.-Y. Efficacy of Immunohistochemistry for SDHB in the Screening of Hereditary Pheochromocytoma–Paraganglioma. Biology 2021, 10, 677. https://doi.org/10.3390/biology10070677
Choi H-R, Koo J-S, Lee C-R, Lee J-D, Kang S-W, Jo Y-S, Chung W-Y. Efficacy of Immunohistochemistry for SDHB in the Screening of Hereditary Pheochromocytoma–Paraganglioma. Biology. 2021; 10(7):677. https://doi.org/10.3390/biology10070677
Chicago/Turabian StyleChoi, Hye-Ryeon, Ja-Seung Koo, Cho-Rok Lee, Jan-Dee Lee, Sang-Wook Kang, Young-Seok Jo, and Woong-Youn Chung. 2021. "Efficacy of Immunohistochemistry for SDHB in the Screening of Hereditary Pheochromocytoma–Paraganglioma" Biology 10, no. 7: 677. https://doi.org/10.3390/biology10070677
APA StyleChoi, H.-R., Koo, J.-S., Lee, C.-R., Lee, J.-D., Kang, S.-W., Jo, Y.-S., & Chung, W.-Y. (2021). Efficacy of Immunohistochemistry for SDHB in the Screening of Hereditary Pheochromocytoma–Paraganglioma. Biology, 10(7), 677. https://doi.org/10.3390/biology10070677