
Examination of the Transcriptional Response to LaMIR166a Overexpression in Larix kaempferi (Lamb.) Carr

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Preparation and Detection
2.3. Library Preparation, RNA Sequencing, and Data Quality Control
2.4. De Novo Assembly and Gene Functional Annotation
2.5. Differentially Expressed Genes and Pathway Enrichment Analysis
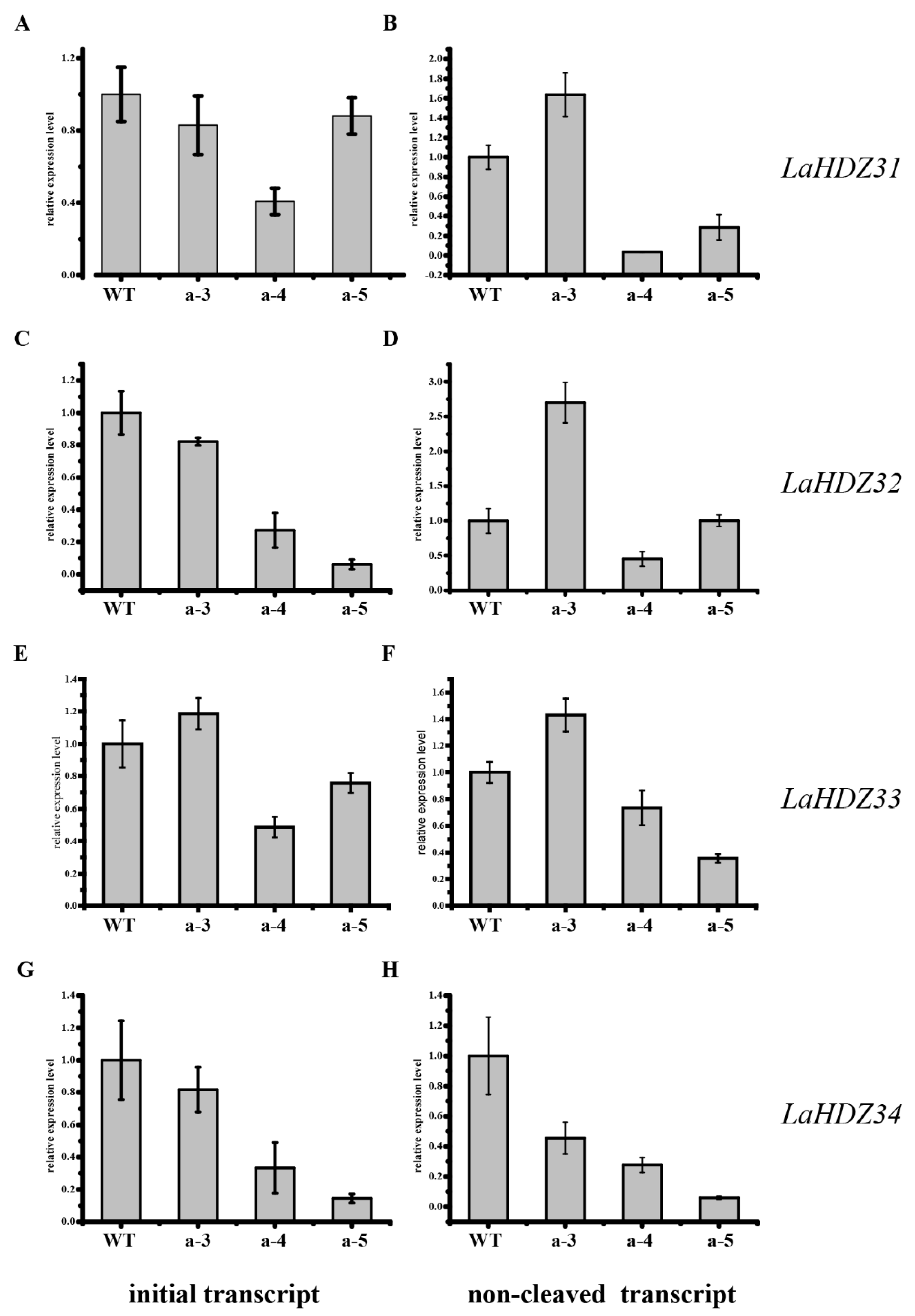
2.6. LaHDZ31–34 Expression Patterns Detected by Quantitative RT-PCR
2.7. Discovery of the LaHDZ31–34 Candidate Genes in Response to LaMIR166a Overexpression in L. kaempferi
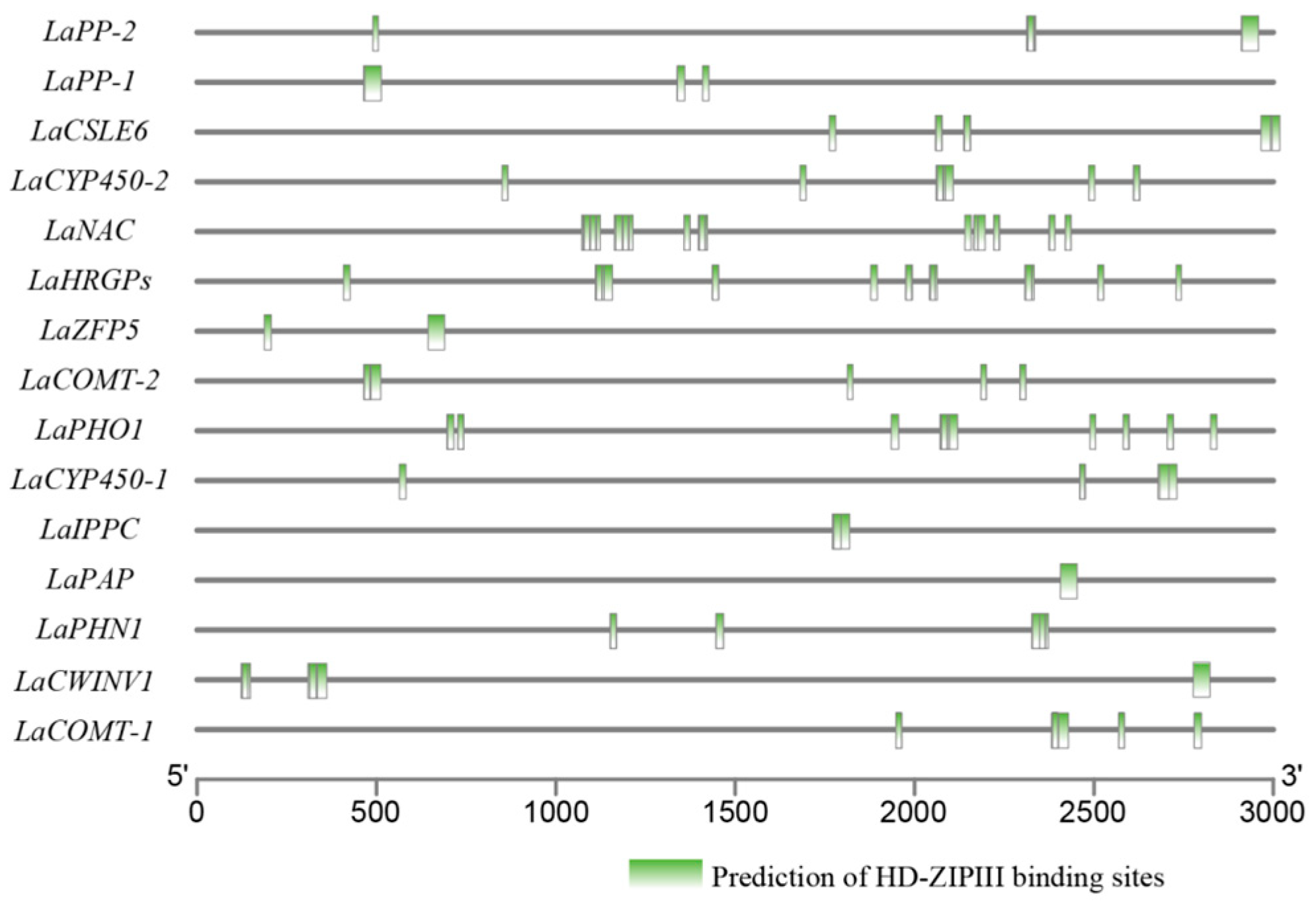
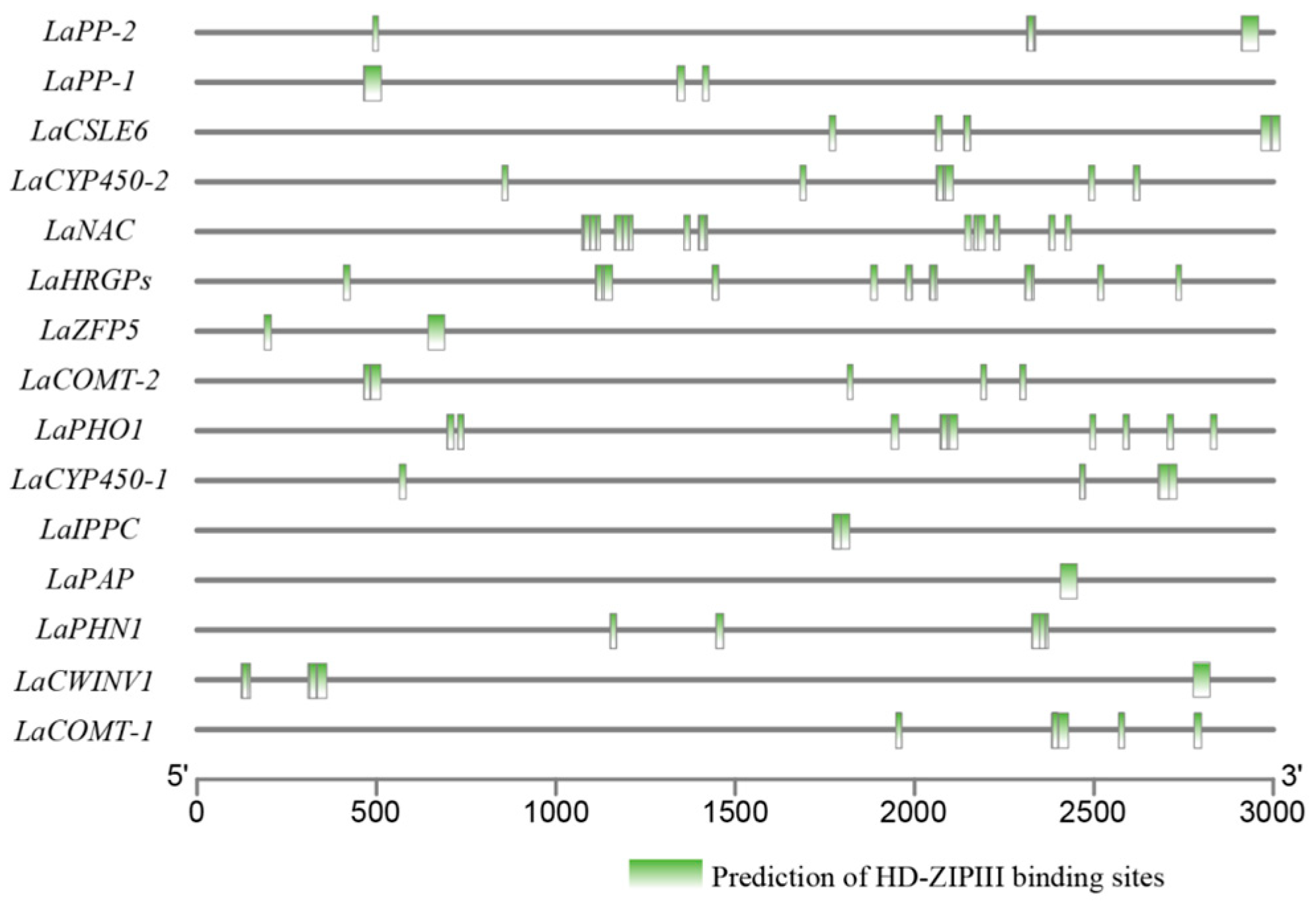
2.8. Cis-Elements in Promoter Prediction
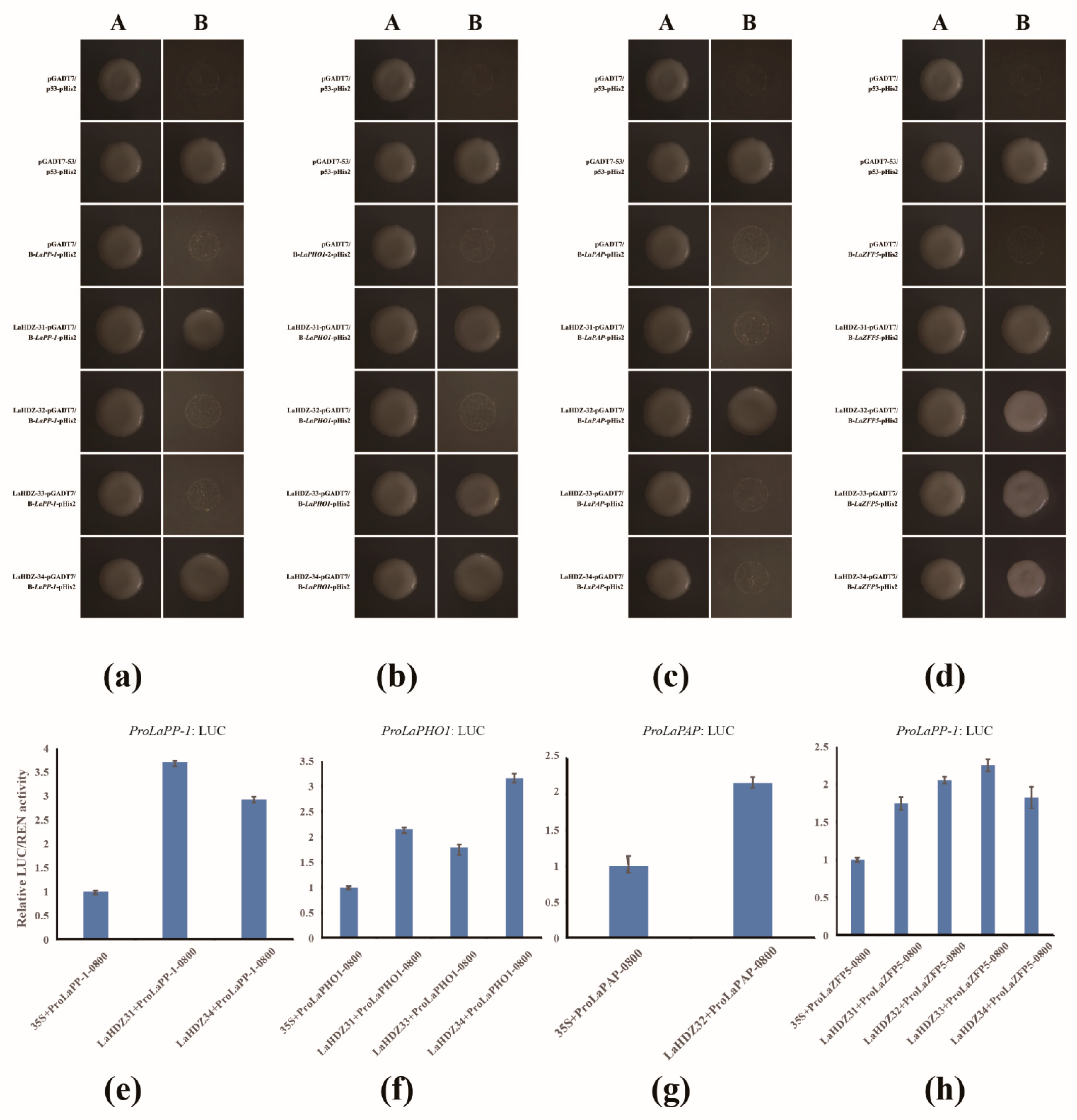
2.9. Cloning of Related Gene Promoters and Y1H, Dual-Luciferase Report Assay
3. Results
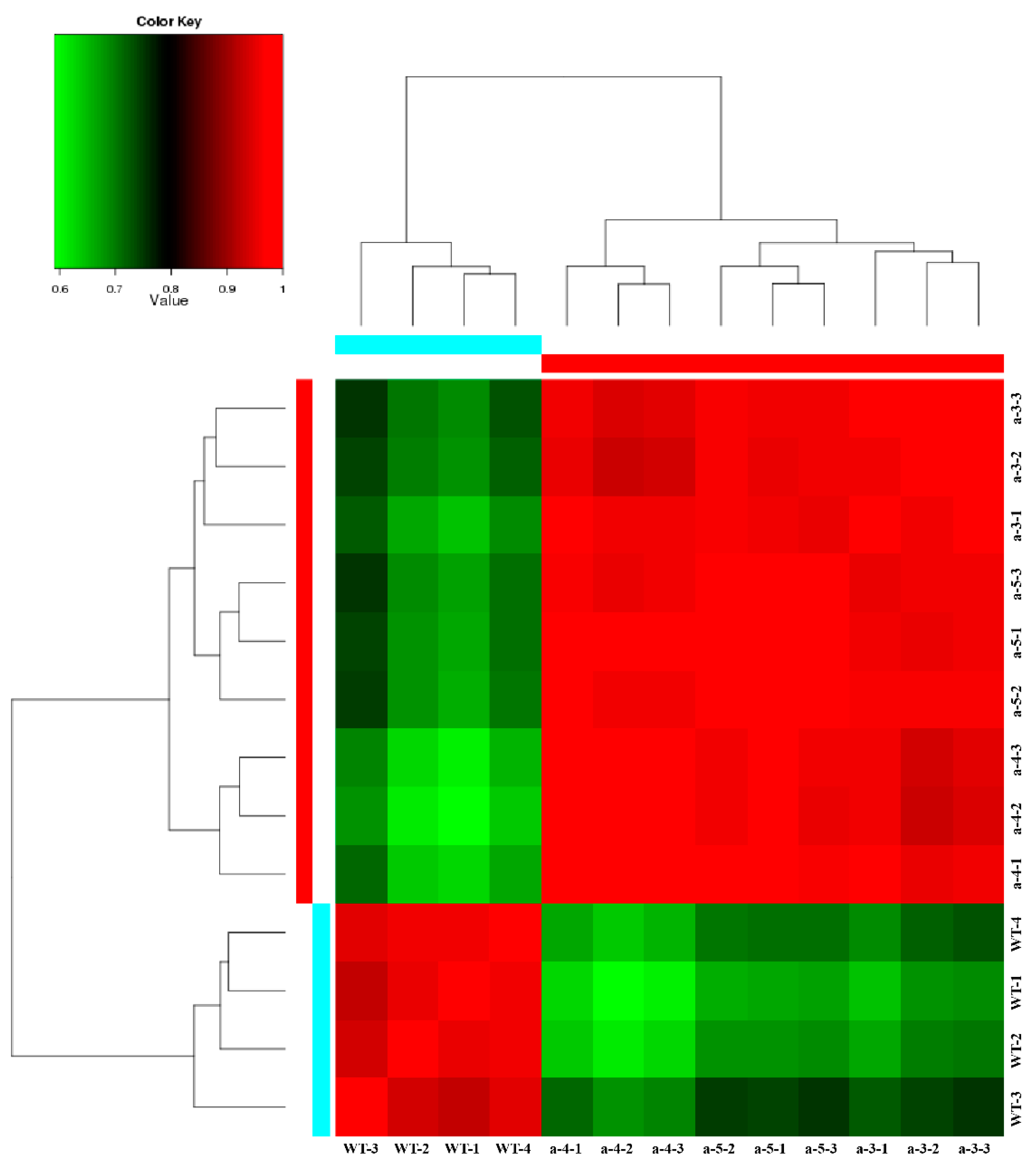
3.1. De Novo Transcriptome Assembly and Gene Expression Profiles
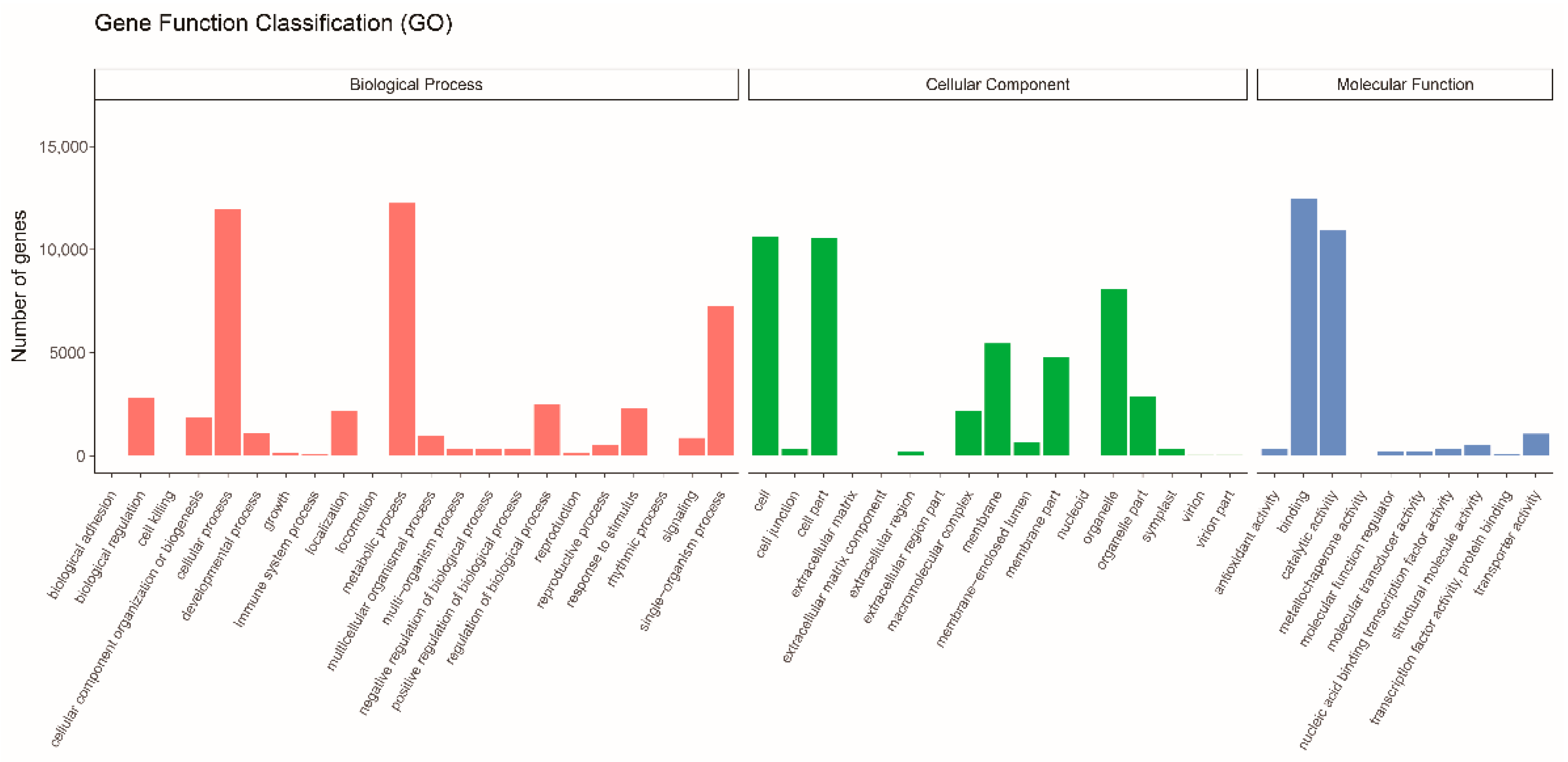
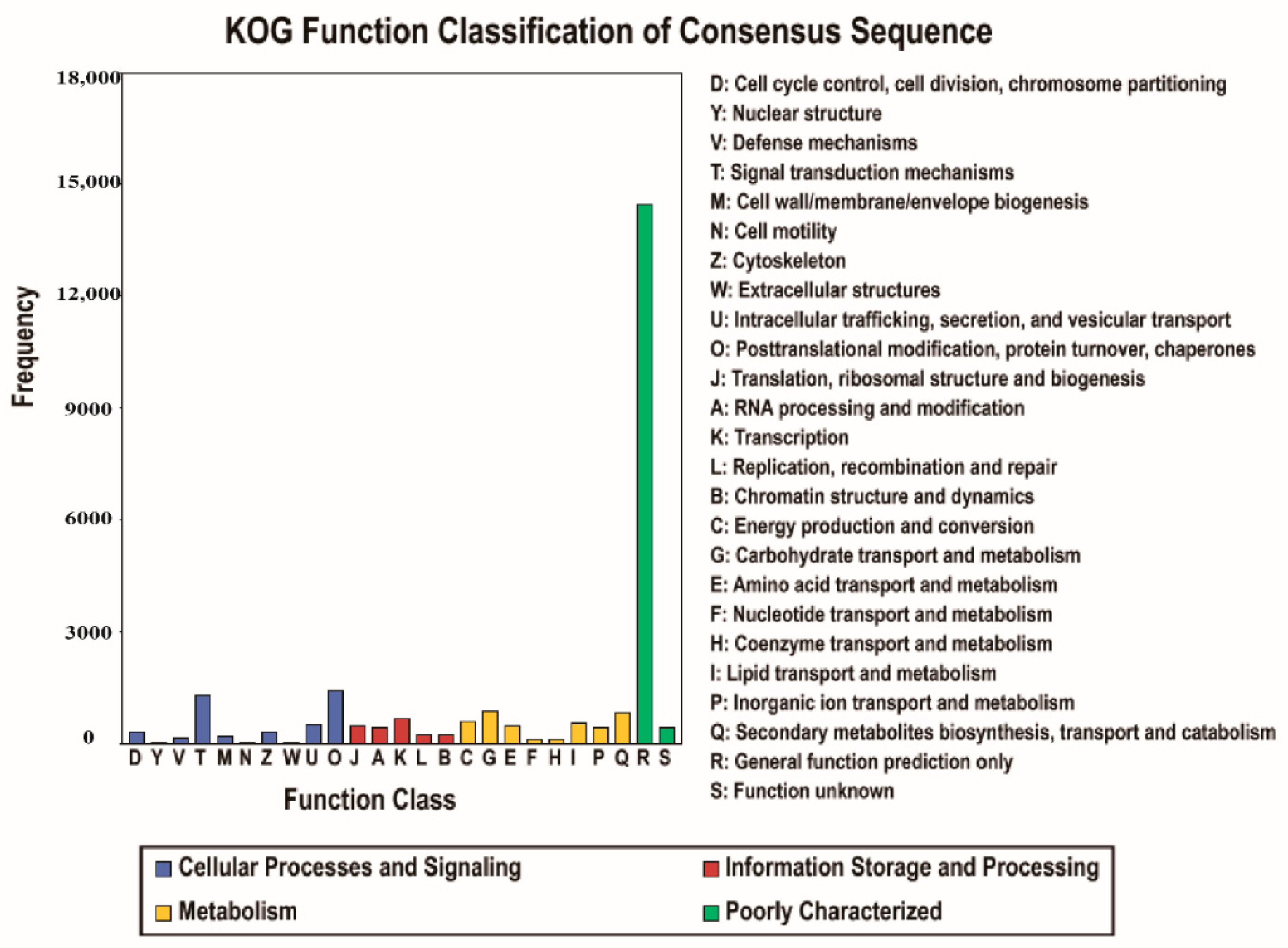
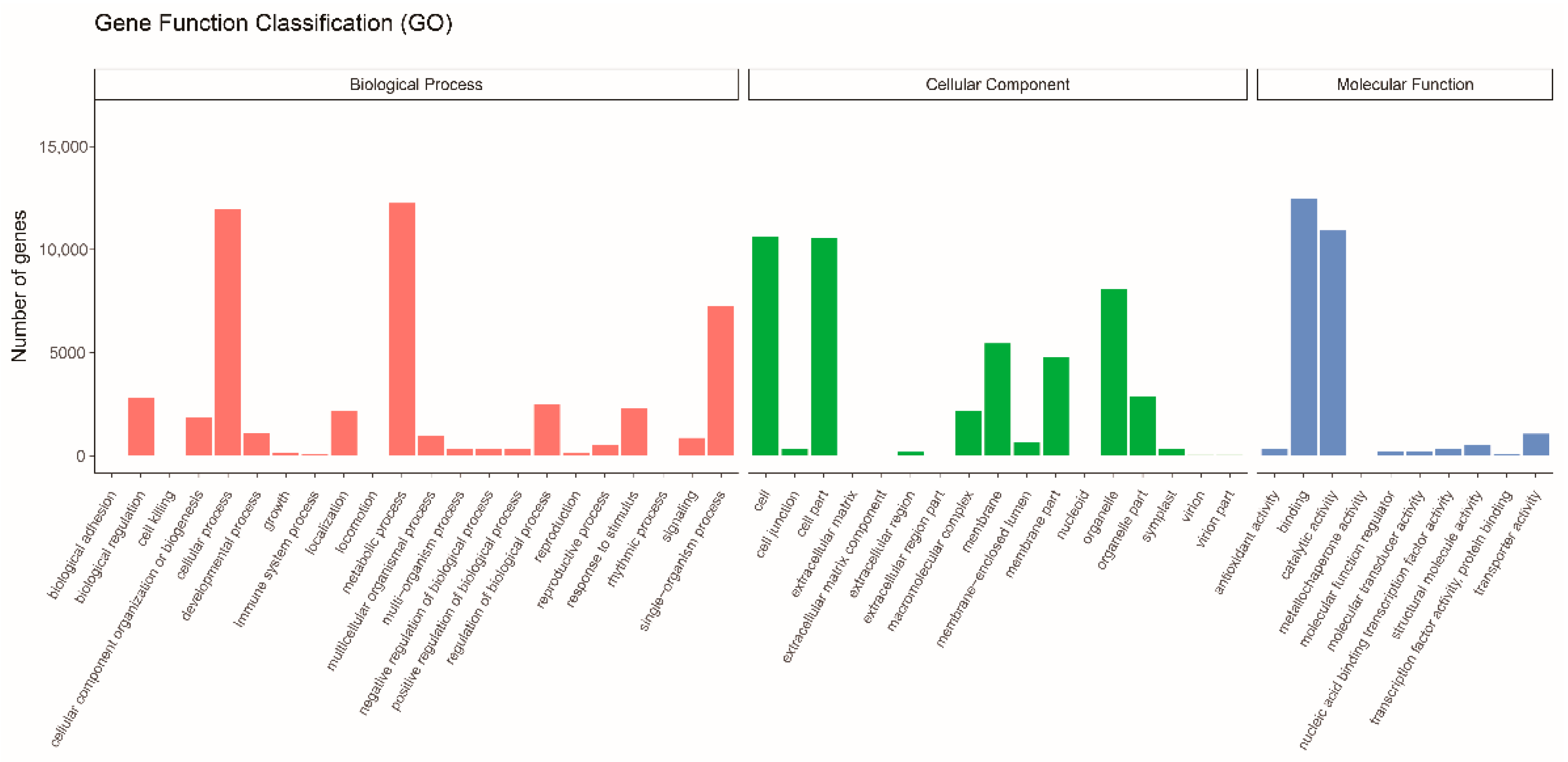
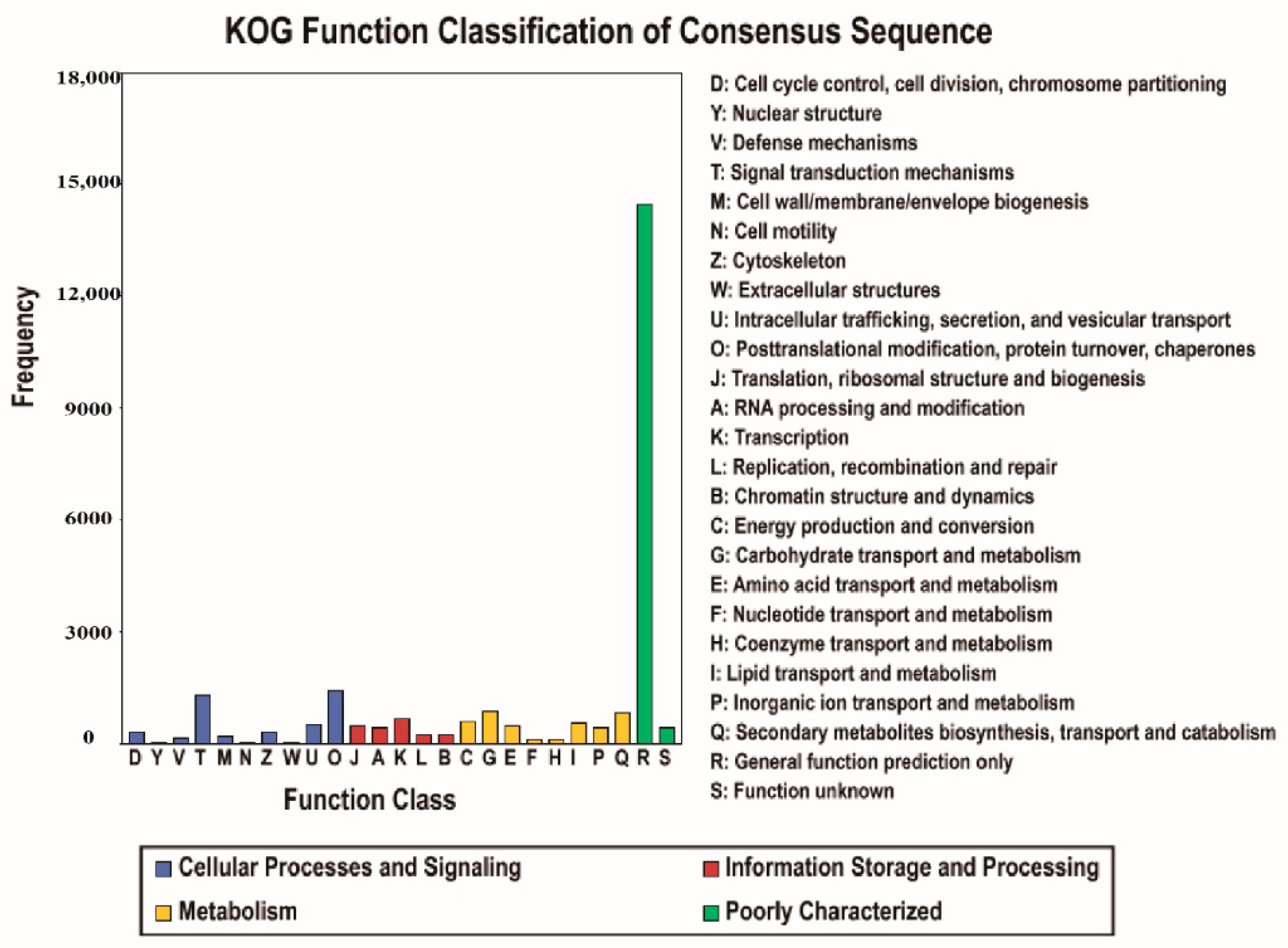
3.2. Gene Function Annotation
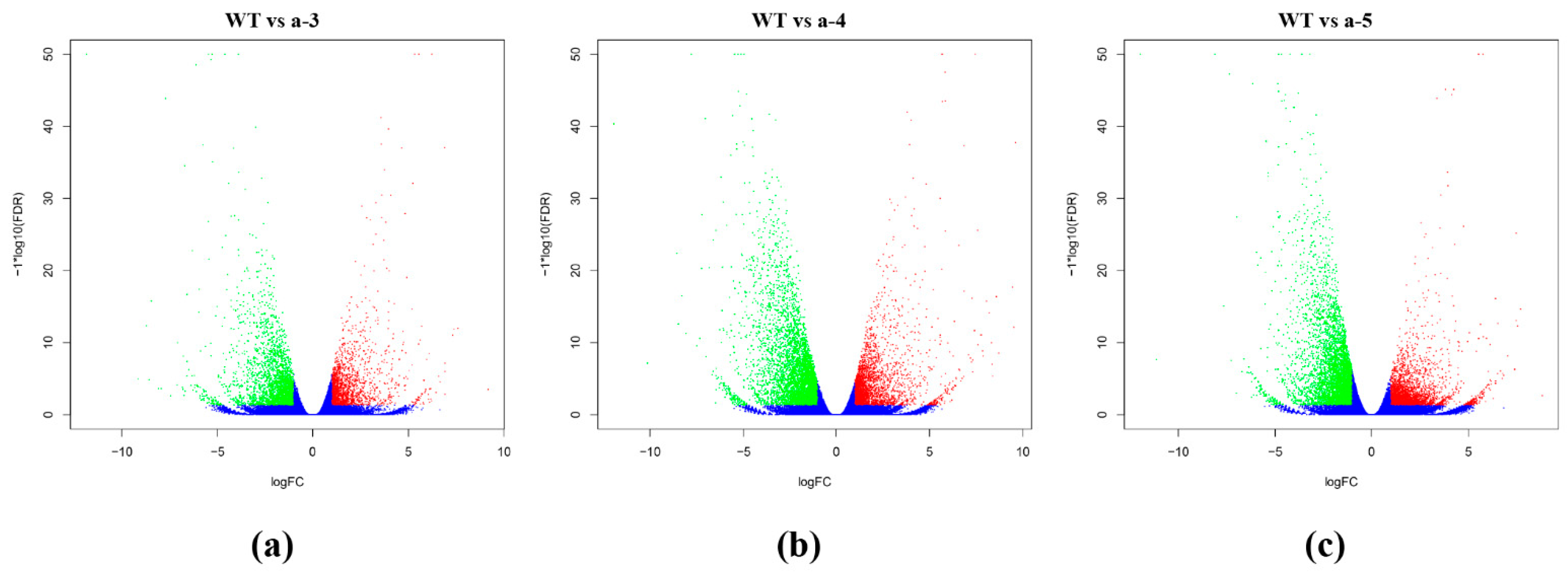
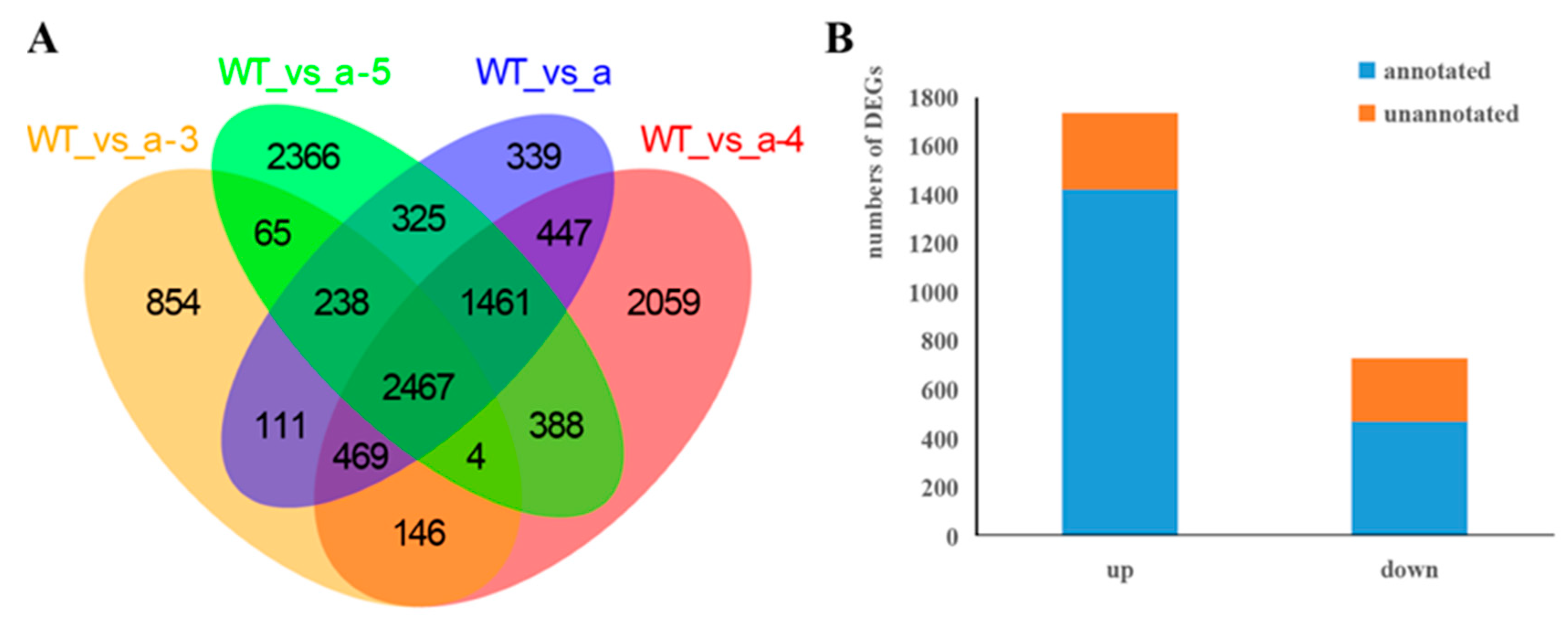
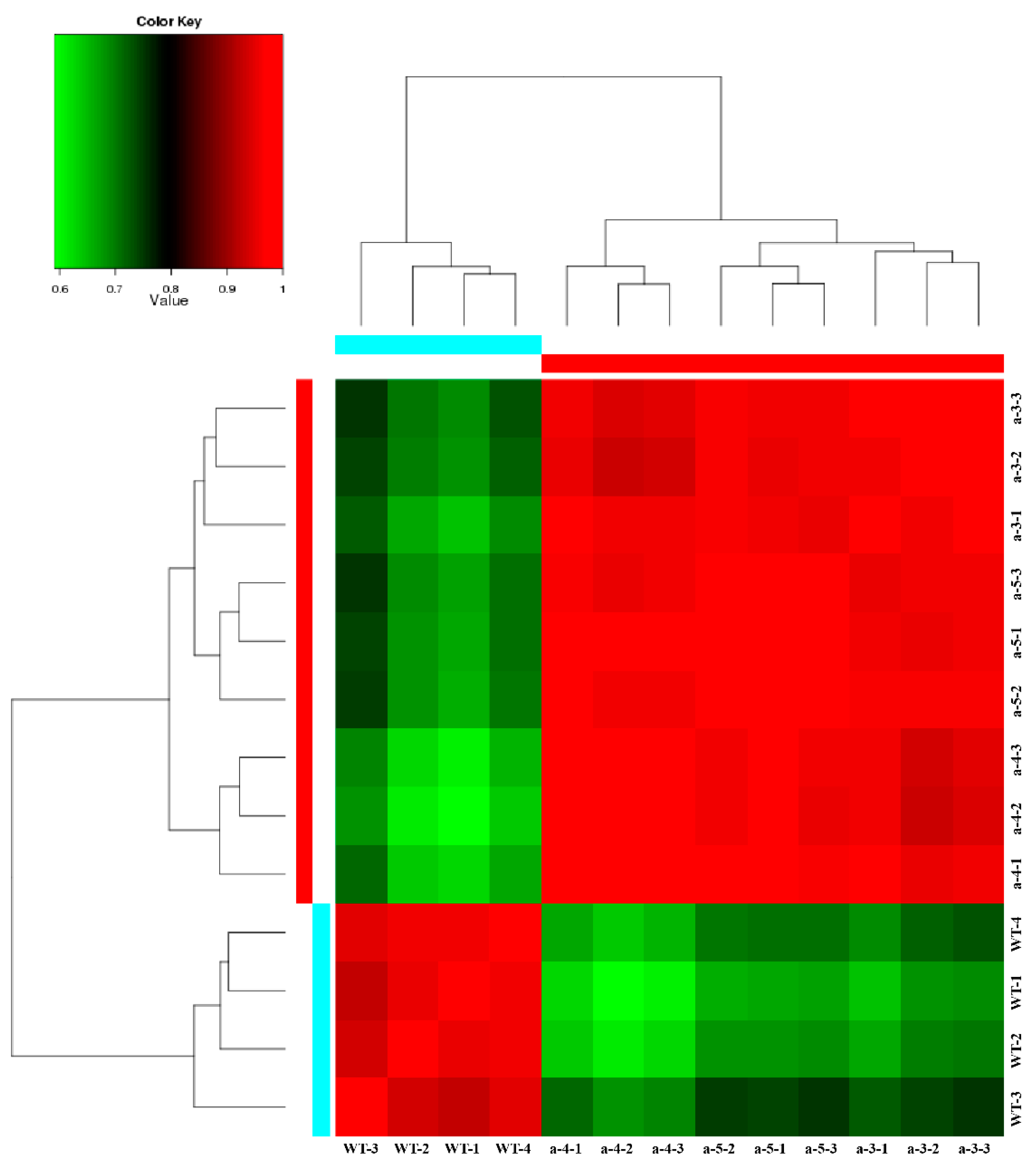
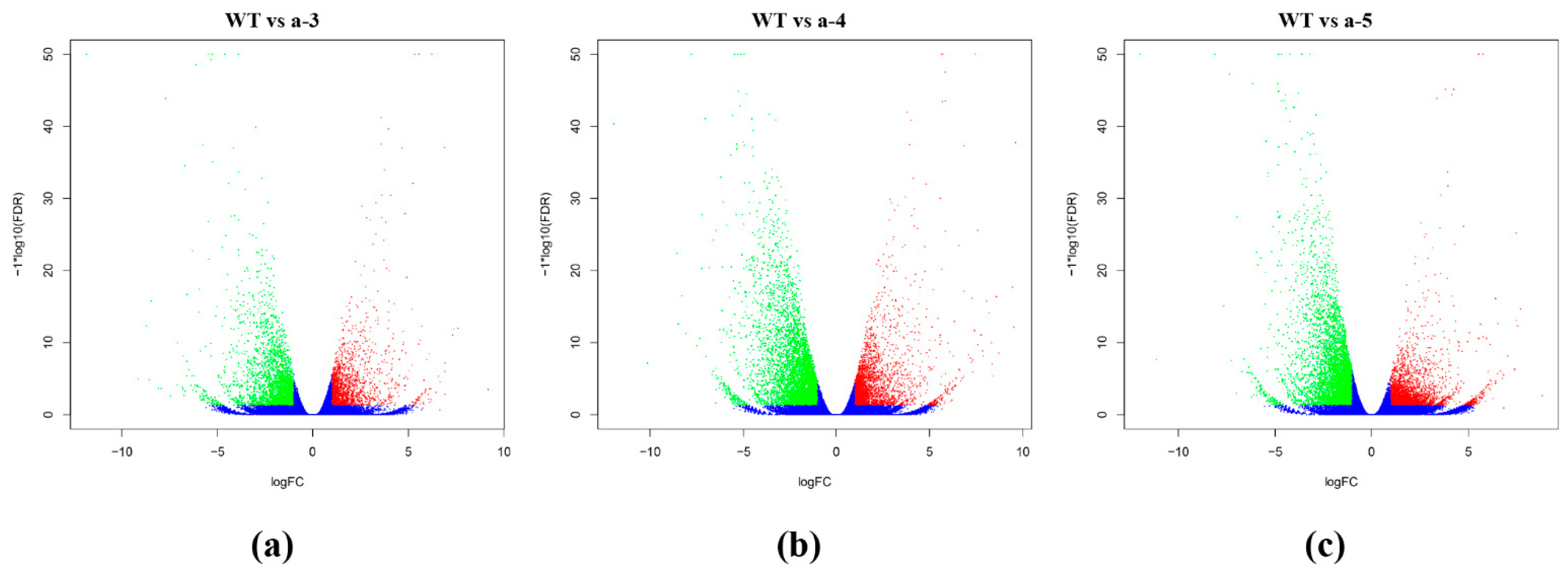
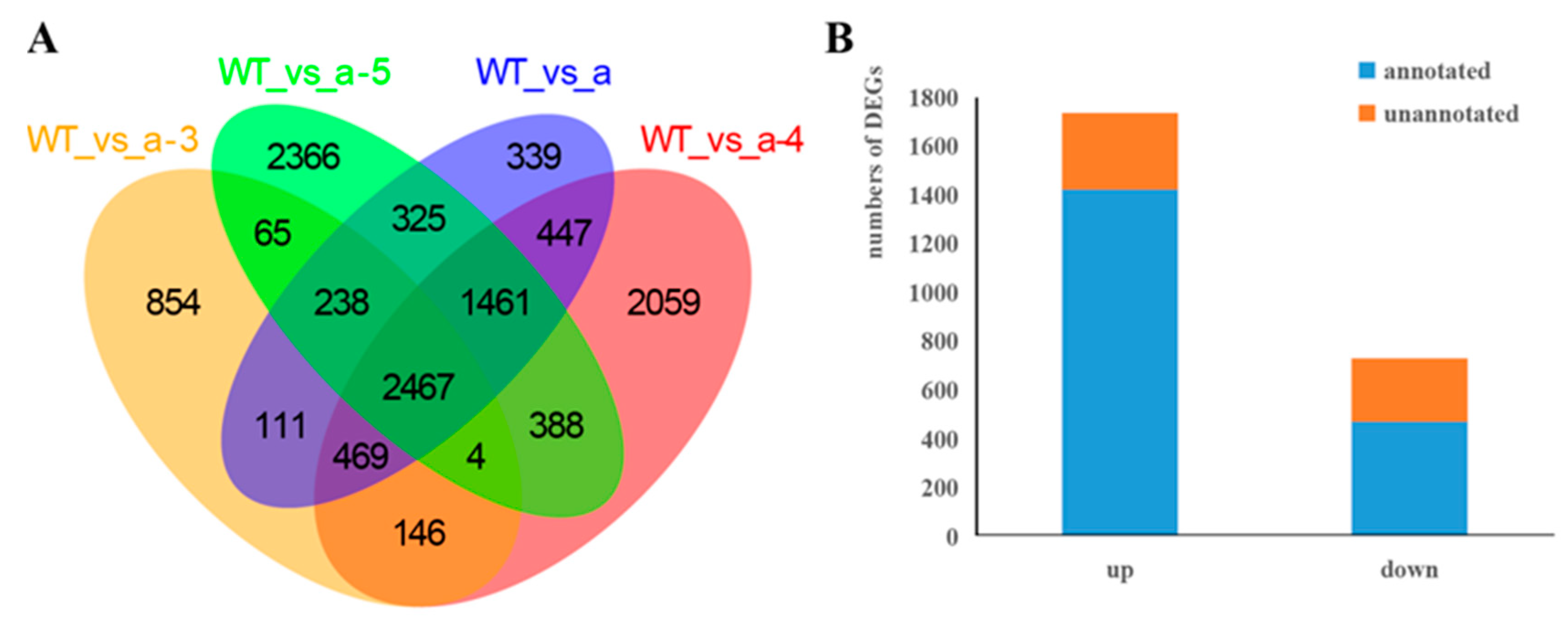
3.3. Screening DEGs between WT and Transgenic Lines
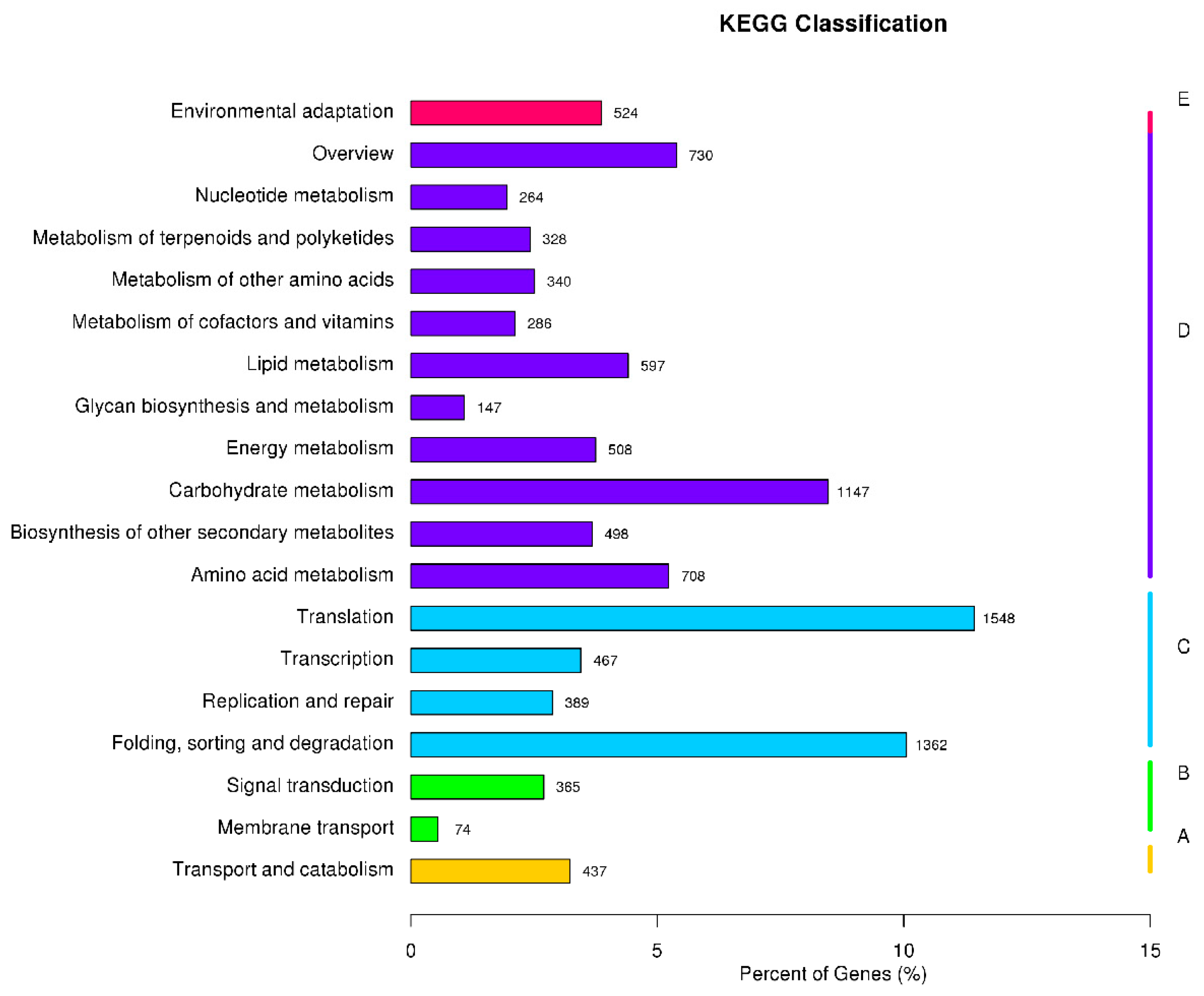
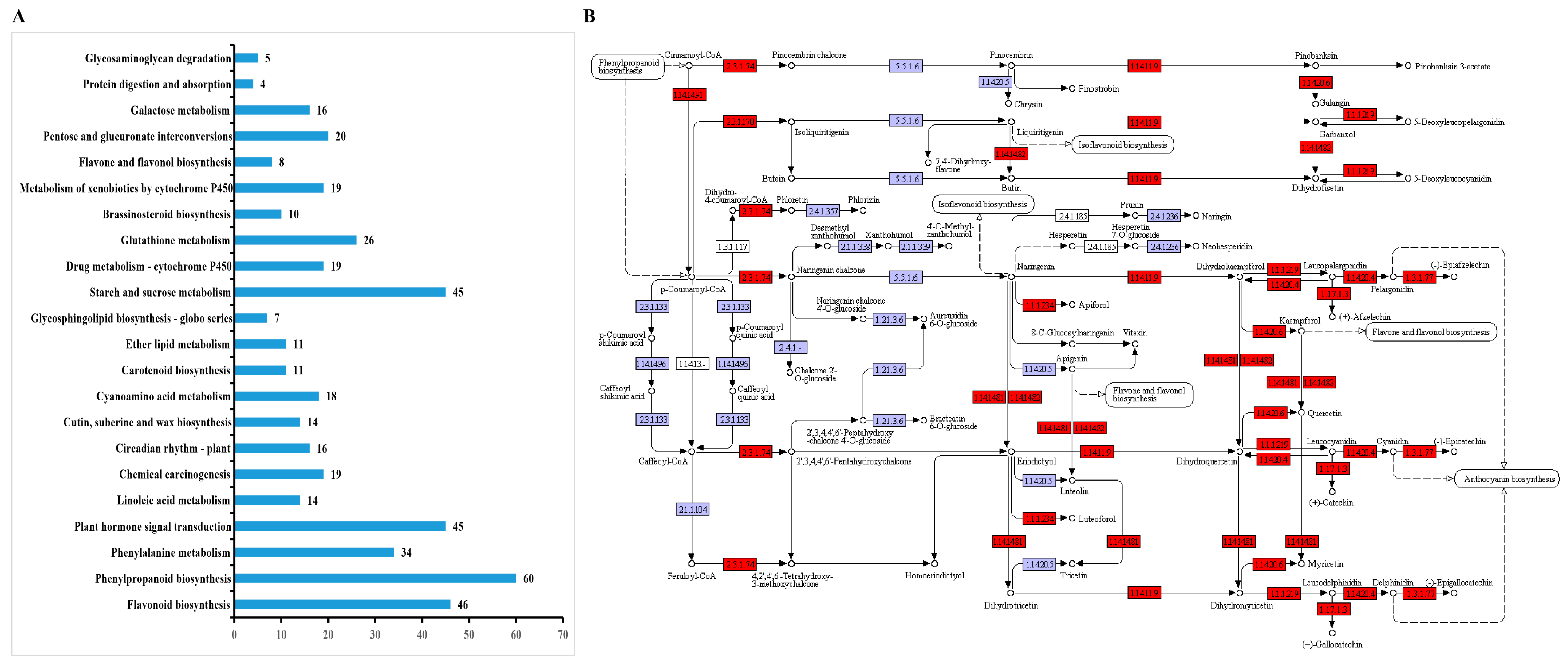
3.4. GO Enrichment and KEGG Pathway Analysis of DEGs
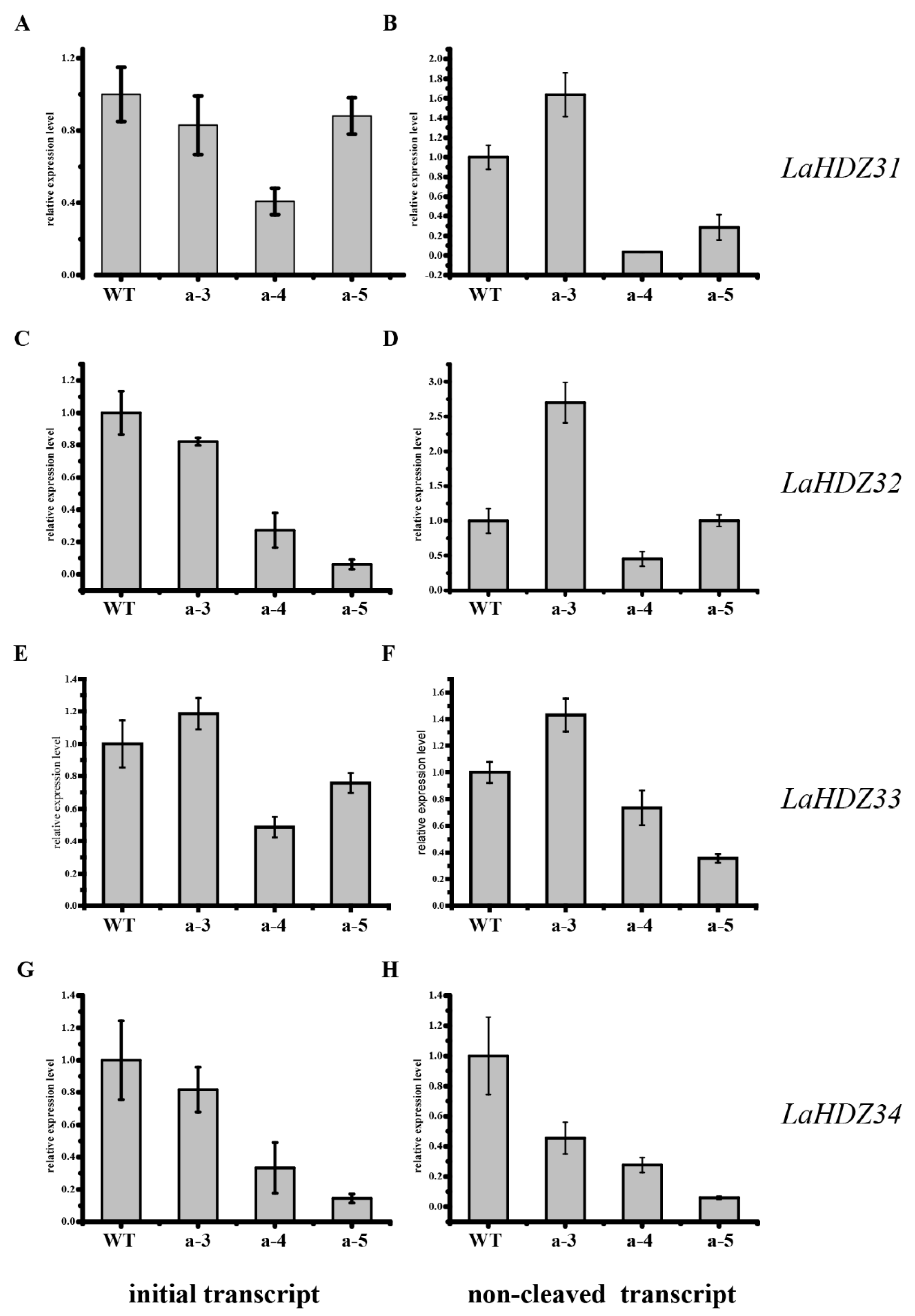
3.5. LaHDZ31–34 Expression Patterns
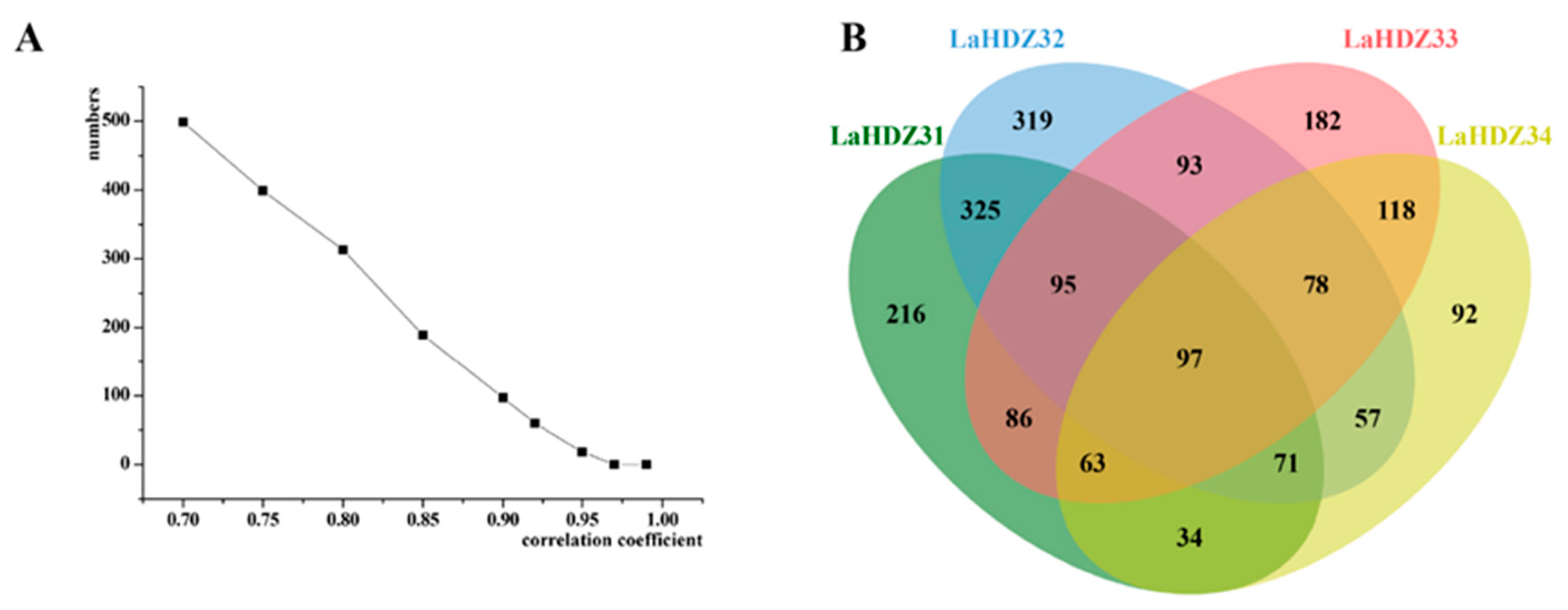
3.6. Pearson’s Correlation Analysis of DEGs Related to the Expression of LaHDZ31–34 in Response to LaMIR166a Overexpression
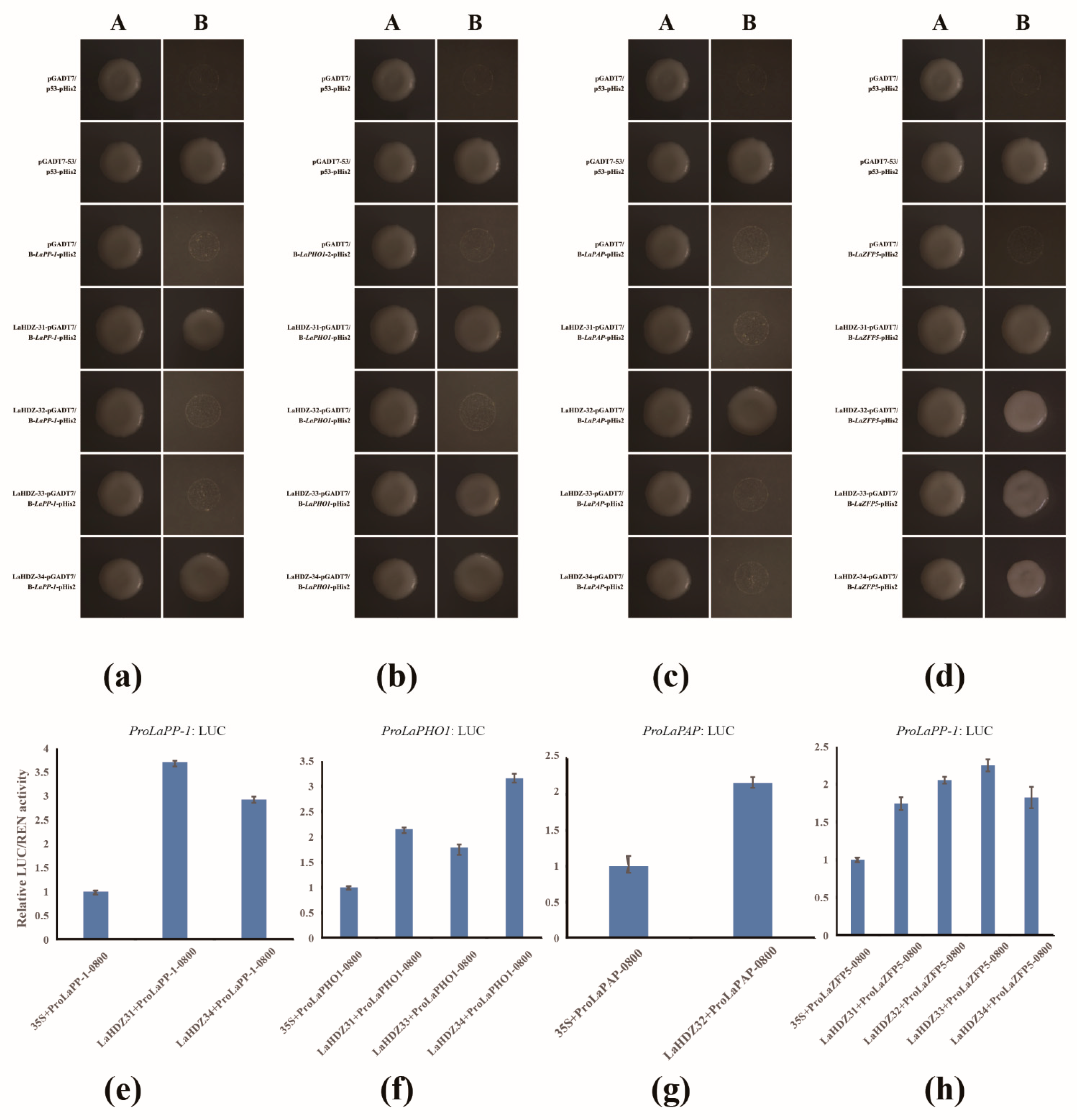
3.7. LaHDZ31–34 Bind to the Promoters of LaPAP, LaPP1, LaZFP5, and LaPHO1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhang, S.; Zhou, J.; Han, S.; Yang, W.; Li, W.; Wei, H.; Li, X.; Qi, L. Four abiotic stress-induced miRNA families differentially regulated in the embryogenic and non-embryogenic callus tissues of Larix leptolepis. Biochem. Biophys. Res. Commun. 2010, 398, 355–360. [Google Scholar] [CrossRef]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of Plant MicroRNA Targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.K.; Bowman, J.L. Ancient microRNA target sequences in plants. Nat. Cell Biol. 2004, 428, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Merelo, P.; Ram, H.; Caggiano, M.P.; Ohno, C.; Ott, F.; Straub, D.; Graeff, M.; Cho, S.K.; Yang, S.W.; Wenkel, S.; et al. Regulation of MIR165/166 by class II and class III homeodomain leucine zipper proteins establishes leaf polarity. Proc. Natl. Acad. Sci. USA 2016, 113, 11973–11978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, M.E. Shoot Meristem Function and Leaf Polarity: The Role of Class III HD–ZIP Genes. PLoS Genet. 2006, 2, e89. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turchi, L.; Carabelli, M.; Ruzza, V.; Possenti, M.; Sassi, M.; Peñalosa, A.; Sessa, G.; Salvi, S.; Forte, V.; Morelli, G.; et al. Arabidopsis HD-Zip II transcription factors control apical embryo development and meristem function. Development 2013, 140, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Hawker, N.P.; Bowman, J.L. Roles for Class III HD-Zip and KANADI Genes in Arabidopsis Root Development. Plant Physiol. 2004, 135, 2261–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhang, B.; Ma, R.; Yu, M.; Guo, S.; Guo, L. Isolation and expression analysis of four HD-ZIP III family genes targeted by microRNA166 in peach. Genet. Mol. Res. 2015, 14, 14151–14161. [Google Scholar] [CrossRef]
- Huang, T.; Harrar, Y.; Lin, C.; Reinhart, B.; Newell, N.R.; Talavera-Rauh, F.; Hokin, S.A.; Barton, M.K.; Kerstetter, R.A. Arabidopsis KANADI1 Acts as a Transcriptional Repressor by Interacting with a Specific Cis-Element and Regulates Auxin Biosynthesis, Transport, and Signaling in Opposition to HD-ZIPIII Factors. Plant Cell 2014, 26, 246–262. [Google Scholar] [CrossRef] [Green Version]
- Turchi, L.; Baima, S.; Morelli, G.; Ruberti, I. Interplay of HD-Zip II and III transcription factors in auxin-regulated plant development. J. Exp. Bot. 2015, 66, 5043–5053. [Google Scholar] [CrossRef]
- Izhaki, A.; Bowman, J.L. KANADI and Class III HD-Zip Gene Families Regulate Embryo Patterning and Modulate Auxin Flow during Embryogenesis in Arabidopsis. Plant Cell 2007, 19, 495–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, J.F.; Floyd, S.K.; Alvarez, J.; Eshed, Y.; Hawker, N.P.; Izhaki, A.; Baum, S.F.; Bowman, J.L. Radial Patterning of Arabidopsis Shoots by Class III HD-ZIP and KANADI Genes. Curr. Biol. 2003, 13, 1768–1774. [Google Scholar] [CrossRef] [Green Version]
- McHale, N.A.; Koning, R.E. MicroRNA-Directed Cleavage of Nicotiana sylvestris PHAVOLUTA mRNA Regulates the Vascular Cambium and Structure of Apical Meristems. Plant Cell 2004, 16, 1730–1740. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, J.-H.; Reyes, J.L.; Kim, Y.-S.; Kim, S.-Y.; Chung, K.-S.; Kim, J.A.; Lee, M.; Lee, Y.; Kim, V.N.; et al. microRNA-directed cleavage of ATHB15 mRNA regulates vascular development in Arabidopsis inflorescence stems. Plant J. 2005, 42, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.; Grigg, S.P.; Xie, M.; Christensen, S.; Fletcher, J.C. Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 2005, 132, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Grigg, S.P.; Canales, C.; Hay, A.; Tsiantis, M. SERRATE coordinates shoot meristem function and leaf axial patterning in Arabidopsis. Nat. Cell Biol. 2005, 437, 1022–1026. [Google Scholar] [CrossRef]
- Li, Z.-X.; Li, S.-G.; Zhang, L.-F.; Han, S.-Y.; Li, W.-F.; Xu, H.-Y.; Yang, W.-H.; Liu, Y.-L.; Fan, Y.-R.; Qi, L.-W. Over-expression of miR166a inhibits cotyledon formation in somatic embryos and promotes lateral root development in seedlings of Larix leptolepis. Plant Cell Tissue Organ Cult. 2016, 127, 461–473. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.-F.; Zhang, S.-G.; Han, S.-Y.; Wu, T.; Zhang, J.-H.; Qi, L.-W. The post-transcriptional regulation of LaSCL6 by miR171 during maintenance of embryogenic potential in Larix kaempferi (Lamb.) Carr. Tree Genet. Genomes 2014, 10, 223–229. [Google Scholar] [CrossRef]
- Yang, Z.; Li, C.; Wang, Y.; Zhang, C.; Wu, Z.; Zhang, X.; Liu, C.; Li, F. GhAGL15s, preferentially expressed during somatic embryogenesis, promote embryogenic callus formation in cotton (Gossypium hirsutum L.). Mol. Genet. Genom. 2014, 289, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.W.-K.; Zhang, C.; Miller, M.; Palmer, G.; Whiteley, M.; Tholl, D.; Chen, Z.J. cis- and trans-Regulation of miR163 and Target Genes Confers Natural Variation of Secondary Metabolites in Two Arabidopsis Species and Their Allopolyploids. Plant Cell 2011, 23, 1729–1740. [Google Scholar] [CrossRef] [Green Version]
- Gou, J.-Y.; de Felippes, F.F.; Liu, C.-J.; Weigel, D.; Wang, J.-W. Negative Regulation of Anthocyanin Biosynthesis in Arabidopsis by a miR156-Targeted SPL Transcription Factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [Green Version]
- Davuluri, G.R.; Van Tuinen, A.; Fraser, P.D.; Manfredonia, A.; Newman, R.; Burgess, D.; Brummell, D.; King, S.R.; Palys, J.; Uhlig, J.; et al. Fruit-specific RNAi-mediated suppression of DET1 enhances carotenoid and flavonoid content in tomatoes. Nat. Biotechnol. 2005, 23, 890–895. [Google Scholar] [CrossRef]
- Green, K.A.; Prigge, M.J.; Katzman, R.B.; Clark, S.E. CORONA, a Member of the Class III Homeodomain Leucine Zipper Gene Family in Arabidopsis, Regulates Stem Cell Specification and Organogenesis. Plant Cell 2005, 17, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Du, H.; Wuyun, T.-N. Genome-Wide Identification of MicroRNAs and Their Targets in the Leaves and Fruits of Eucommia ulmoides Using High-Throughput Sequencing. Front. Plant Sci. 2016, 7, 1632. [Google Scholar] [CrossRef] [Green Version]
- Mao, R.; Li, W.; He, Z.; Bai, Z.; Xia, P.; Liang, Z.; Liu, Y. Physiological, transcriptional, and metabolic alterations in spaceflight-subjected Senna obtusifolia. Plant Physiol. Biochem. 2019, 139, 33–43. [Google Scholar] [CrossRef]
- Huang, D.; Dai, W. Molecular characterization of the basic helix-loop-helix (bHLH) genes that are differentially expressed and induced by iron deficiency in Populus. Plant Cell Rep. 2015, 34, 1211–1224. [Google Scholar] [CrossRef]
- Wang, L.; Liu, D. Functions and regulation of phosphate starvation-induced secreted acid phosphatases in higher plants. Plant Sci. 2018, 271, 108–116. [Google Scholar] [CrossRef]
- Pomeranz, M.C.; Hah, C.; Lin, P.-C.; Kang, S.G.; Finer, J.J.; Blackshear, P.J.; Jang, J.-C. The Arabidopsis Tandem Zinc Finger Protein AtTZF1 Traffics between the Nucleus and Cytoplasmic Foci and Binds Both DNA and RNA. Plant Physiol. 2009, 152, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.C.; Lee, S.H.; Cheong, Y.H.; Yoo, C.; Lee, S.I.; Chun, H.J.; Yun, D.; Hong, J.C.; Lim, C.O.; Cho, M.J. A novel cold-inducible zinc finger protein from soybean, SCOF-1, enhances cold tolerance in transgenic plants. Plant J. 2001, 25, 247–259. [Google Scholar] [CrossRef]
- An, L.; Zhou, Z.; Sun, L.; Yan, A.; Xi, W.; Yu, N.; Cai, W.; Chen, X.; Yu, H.; Schiefelbein, J.; et al. A zinc finger protein gene ZFP5 integrates phytohormone signaling to control root hair development in Arabidopsis. Plant J. 2012, 72, 474–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Jiang, Q.; Wu, J.; An, L.; Zhou, Z.; Wong, C.; Wu, M.; Yu, H.; Gan, Y. Zinc finger protein 5 (ZFP5) associates with ethylene signaling to regulate the phosphate and potassium deficiency-induced root hair development in Arabidopsis. Plant Mol. Biol. 2020, 102, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Hyoung, S.; So, W.M.; Shin, J.S. The novel transcription factor TRP interacts with ZFP5, a trichome initiation-related transcription factor, and negatively regulates trichome initiation through gibberellic acid signaling. Plant Mol. Biol. 2018, 96, 315–326. [Google Scholar] [CrossRef]
- Zhou, Z.; An, L.; Sun, L.; Zhu, S.; Xi, W.; Broun, P.; Yu, H.; Gan, Y. Zinc Finger Protein5 Is Required for the Control of Trichome Initiation by Acting Upstream of Zinc Finger Protein8 in Arabidopsis. Plant Physiol. 2011, 157, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads (bp) | Clean Reads (bp) | Clean Bases (bp) | GC Percentage (%) | Combined Non-Redundant Unigene (bp) | Total Length (bp) | Mean Length (bp) | N50 (bp) |
|---|---|---|---|---|---|---|---|---|
| WT-1 | 16,885,888 | 16,764,729 | 5,065,766,400 | 46.75 | 203,256 | 160,145,128 | 787.90 | 996 |
| WT-2 | 48,658,992 | 48,169,554 | 14,597,697,600 | 46.04 | ||||
| WT-3 | 48,260,203 | 45,731,695 | 13,719508500 | 44.83 | ||||
| WT-4 | 50,838,449 | 48,265,826 | 14,479747800 | 46.04 | ||||
| a-3-1 | 39,548,734 | 39,163,538 | 11,864620200 | 44.85 | ||||
| a-3-2 | 23,648,726 | 23,439,052 | 7,094,617,800 | 44.7 | ||||
| a-3-3 | 46,286,892 | 43,875,416 | 13,162,624,800 | 45.07 | ||||
| a-4-1 | 42,151,448 | 39,915,863 | 11,974,758,900 | 45.69 | ||||
| a-4-2 | 36,001,158 | 34,168,865 | 10,250,659,500 | 45.21 | ||||
| a-4-3 | 35,848,191 | 33,974,592 | 10,192,377,600 | 45.68 | ||||
| a-5-1 | 46,286892 | 40,263,192 | 12,078,957,600 | 46.42 | ||||
| a-5-2 | 45,867,514 | 43,432,516 | 13,029,800,000 | 46.19 | ||||
| a-5-3 | 52,606,986 | 49,961,824 | 14,988,547,200 | 45.6 |
| Number of Annotated Unigenes | 0 ≤ Length < 1000 | 1000 ≤ Length < 2000 | 2000 ≤ Length < 3000 | 3000 ≤ Length < 6000 | Length ≥ 6000 | |
|---|---|---|---|---|---|---|
| GO_Annotation | 26,234 | 13,063 | 6461 | 3730 | 2744 | 236 |
| KEGG_Annotation | 13,546 | 5245 | 3767 | 2417 | 1916 | 201 |
| KOG_Annotation | 23,331 | 13,101 | 5091 | 2714 | 2198 | 227 |
| NR_Annotation | 51,321 | 28,042 | 11,901 | 6133 | 4776 | 469 |
| NT_Annotation | 32,963 | 14,512 | 8830 | 5118 | 4111 | 392 |
| Swissprot_Annotation | 25,452 | 11,086 | 6618 | 4038 | 3370 | 340 |
| All_Annotated | 58,487 | 33,324 | 13,206 | 6502 | 4977 | 478 |
| Sample | Number | Downregulated | Upregulated |
|---|---|---|---|
| WT vs. a-3 | 4354 | 1786 | 2568 |
| WT vs. a-4 | 7441 | 2724 | 4717 |
| WT vs. a-5 | 7314 | 2958 | 4356 |
| WT vs. a | 5857 | 2227 | 3630 |
| DEGs | 2467 | 731 | 1736 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Li, W.; Li, Z.; Dang, S.; Han, S.; Zhang, L.; Qi, L. Examination of the Transcriptional Response to LaMIR166a Overexpression in Larix kaempferi (Lamb.) Carr. Biology 2021, 10, 576. https://doi.org/10.3390/biology10070576
Fan Y, Li W, Li Z, Dang S, Han S, Zhang L, Qi L. Examination of the Transcriptional Response to LaMIR166a Overexpression in Larix kaempferi (Lamb.) Carr. Biology. 2021; 10(7):576. https://doi.org/10.3390/biology10070576
Chicago/Turabian StyleFan, Yanru, Wanfeng Li, Zhexin Li, Shaofei Dang, Suying Han, Lifeng Zhang, and Liwang Qi. 2021. "Examination of the Transcriptional Response to LaMIR166a Overexpression in Larix kaempferi (Lamb.) Carr" Biology 10, no. 7: 576. https://doi.org/10.3390/biology10070576
APA StyleFan, Y., Li, W., Li, Z., Dang, S., Han, S., Zhang, L., & Qi, L. (2021). Examination of the Transcriptional Response to LaMIR166a Overexpression in Larix kaempferi (Lamb.) Carr. Biology, 10(7), 576. https://doi.org/10.3390/biology10070576