1. Introduction
The gastrointestinal tract is a dynamic and varied ecosystem made up of trillions of bacteria, viruses, protozoa, fungi and archaea that co-evolved with the host [
1]. As a result, mutually beneficial interactions have developed over a prolonged period of time. Recently, there has been an explosion of studies focusing on the microbiome and its role in host health and disease. Nonetheless, such studies are largely anthropocentric and focus mainly on bacterial microbiota [
2,
3,
4,
5,
6]. Microbiome-based research of non-human vertebrates mainly encompasses livestock [
7,
8,
9], companion animals [
10,
11] and other notable species, such as those at risk of extinction [
12,
13]. A common objective of animal studies has been to explore the extent of bacterial community perturbations in the gut caused by anthropogenic intervention and investigate resulting ramifications on animal fitness and longevity [
13,
14,
15,
16,
17,
18]. Recent investigations have focused on comparing gut microbiota of captive animals and their wild counterparts in order to assess links to the captive lifestyle [
14,
19,
20,
21]. Roles of microbiota on host survival upon release have also been examined. Collectively, these studies support monitoring of the microbiota of animals involved in re-introduction and/or translocation projects. Regrettably, the majority of studies fail to include intestinal protozoa, even though emerging evidence suggests that some species persist as asymptomatic colonisers of the intestinal tract [
13,
22,
23,
24,
25,
26,
27,
28]. Recent studies have showcased that water voles constitute an attractive model for examining these questions [
22,
23].
The European water vole (
Arvicola amphibius) is a semi-aquatic rodent that was widespread across Britain in the early 1900s. However, in the past few decades its population has decreased drastically, disappearing from over 89% of previously occupied sites. This has been attributed mainly to habitat destruction and an invasive alien species (IAS), the American mink (
Neovison vison) [
29]. In the UK, attempts are being made to repair fractured populations and re-introduce this mammal into protected wetlands [
30,
31]. An emerging factor in achieving both of these objectives is the gut microbiota, whose composition in water voles has not been explored. Despite this, previous studies have demonstrated high prevalence and co-occurrence of several eukaryotic microbes in the stool of water voles, hinting at potentially important roles as well [
23]. Specifically, the prevalence of protozoan parasites in captive and wild water voles and their associated gut flora remain little explored. The few studies that have examined gut protists in these animals have shown that the most common organism is
Blastocystis, a microbial eukaryote of questionable pathogenicity.
Herein we investigate the gut microbiota of captive water voles, some of which are involved in re-introductory and breeding schemes. Our annotation and characterisation of the gut microbiota encompassed not only the bacterial component, but also included the protozoan parasites Cryptosporidium, Giardia, and the questionable pathogen Blastocystis. Information on the prokaryotic and eukaryotic components was collectively considered in order to explore their associations in the gut of voles. This study provides the first investigation on association of Blastocystis with bacterial communities in the gut of captive water voles. This type of information can assist in re-shaping the strategies for re-introduction programmes of voles into the wild.
2. Materials and Methods
2.1. Study Site, Animals and Sample Preparation
A conservation park situated in the Southeast, United Kingdom was the subject of this study; Wildwood Trust, Herne Bay, Kent, United Kingdom (51°19′54.1″ N 1°07′10.1″ E) is a wildlife park that houses native British wildlife, housing vertebrate and invertebrate species, that also includes a selection of non-native species that assist with the organisation’s education programme. The park aims to educate the general public on the ecology and status of resident animals in addition to participating in several conservation programmes aiming to ‘re-wild’ Britain. The park is actively involved in breeding, re-introduction and mitigation services for the European water vole (Arvicola amphibius) and in recent years has been involved in the release of several individuals. There are over 60 of these rodents currently housed within the park, and their health and breeding status are closely monitored by a licenced veterinarian and keepers. The animals are also monitored for presence of infectious disease agents. During one of these health screenings, Giardia was detected in a large number of water voles. Thus, the animals were categorised as either infected or non-infected with Giardia and housed accordingly; non-infected and suspected infected individuals were housed in separate enclosures. To minimise risk of transmission, enclosures housing non-infected individuals are cleaned first using separate housekeeping equipment. Following the screening, definitive diagnosis for giardiasis was made by the park veterinarian as follows: faecal samples were collected from three adjacent cages at a time and collated faecal samples were examined for Giardia by direct smear. The veterinarian would group collated faecal from three adjacent cages at a time and screen for Giardia spp. If they tested positive, all individuals were placed on anti-protozoal treatment with either metronidazole or fenbendazole. Metronidazole treatment lasted 5 days; the dosage was 0.8 mL of a 5 mg/mL injectable solution (approximately 4 mg/vole). Fenbendazole treatments varied in duration; dosage was typically 0.25 mL of a 20% dilution (200 mg/mL original concentration) per 200 g vole. All drug treatments had ended between 10 and 14 days prior to sample collection.
2.2. Sample Collection
In this study, a total of 16 water voles housed within the park were sampled between 18 January 2019 and 20 February 2019. A total of 29 faecal samples were collected. Sampling successfully occurred across two collection dates for 12 voles, and a single sample collection took place for four of the voles (R4, Q49, R95 and R34); this was due to no suitable faecal sample for R4 (first collection), Q49 and R95 (second collection), and the death of R34 before the second collection. R13 also died on the morning of the second collection; however, a faecal sample for this vole was obtained before the enclosure was cleaned. Therefore, a total of 12 voles had two successful collections. Samples were collected shortly after enclosures had been cleaned and with the guidance of the park keepers. Only fresh faecal samples were collected. Upon collection, samples were placed in sterile tubes and stored at 4 °C within one hour of collection. At the time of study all voles were considered healthy as stated by a licenced veterinarian and lacked symptoms of gastrointestinal disease.
2.3. DNA Extraction, Polymerase Chain Reaction, Cloning
Genomic DNA was extracted directly from approximately 250 mg of the fresh faecal sample using the Microbiome DNA Purification Kit, Purelink (Fisher, California, USA) according to the manufacturer’s instructions. DNA was eluted in 100 μL elution buffer and the working stock stored at −20 °C. Genomic DNA was used for polymerase chain reaction (PCR) with primers targeting gene regions of
Blastocystis,
Cryptosporidium and
Giardia according to previously described protocols [
23] (
Appendix A Table A1). The purified gel extracts were eluted, of which 1.5 μL was used for cloning with the pGEM-T easy vector system I (Promega, Madison, WI, USA). Between five and ten colonies per transformation were inoculated and grown overnight in 5 mL LB media. The plasmid DNA was extracted using the GeneJet Plasmid Miniprep Kit and clones were confirmed as positive using
EcoRI (Promega) restriction digestion. Positive clones were sent for sequencing using the T7 and/or SP6 universal primers (Eurofins, Ebersberg Germany).
2.4. Amplicon Sequencing of 16S rRNA
Twenty-eight genomic DNA samples were used for microbiome profiling analysis using the Illumina platform (paired-end sequencing). Bacterial taxonomic profiling was carried out by targeting the V1–V3 hypervariable regions of the 16S rRNA gene using the following primers: forward sequence fD2: AGAGTTTGATCATGGCTCAG [
32] and reverse sequence S-D-Bact-0008-a-S-20, S-*-Univ-0519-a-A-18: GTATTACCGCGGCTGCTG [
33]. All data have been submitted to GenBank under Bioproject number SUB9442672.
2.5. Microbiome and Statistical Analysis
Initial bioinformatics analysis was performed using the INVIEW Microbiome Profiling 3.0. Reads with ambiguous bases were removed and chimeric sequences were detected and removed based on the algorithm of UCHIME via the VSEARCH package [
34,
35]. Where necessary, reads were merged using FLASH software (Baltimore, Maryland) (V2.2.00
http://ccb.jhu.edu/software/FLASH/—access date: February 2020) [
36]. Primer and adaptor sequences were removed using Cutadapt [
37]. High-quality reads were processed using Minimum Entropy Decomposition (MED) [
38,
39]. Taxonomy assignment of OTUs was carried using the NCBI sequence database (version 10 October 2019). The most specific taxonomic assignment for each OTU was then transferred from the best-matching reference sequence set and a sequence identity of 70% across a minimum of 80% of representative sequences was the threshold for consideration of a reference sequence (representing a 97% threshold). Further processing of OTUs and taxonomy assignments as part of the INVIEW Microbiome Profiling pipeline was performed using QIIME (Arizona, USA) (version 1.9.1
http://qiime.org/, accessed on 1 February 2020) and OTU abundance normalisation was employed using CopyRighter [
40].
All subsequent analyses, including microbial diversity analysis, were performed using the MicrobiomeAnalyst pipeline [
41]. Data filtering of OTU data consisted of low count filtering to remove reads with low counts across few samples this was set to a minimum count of four with a prevalence of 20% in samples. Data variance was measured using the inter-quartile range (IQR) and low-variance filtering was implemented to remove features that were close to constant throughout the dataset. Data normalisation was used to facilitate data comparison and to account for unevenness in sampling sparsity and depth; normalisation approaches considered herein included rarefaction to even sequencing depth, data scaling and data transformation.
Microbial diversity was analysed at different taxonomic levels between infected and uninfected voles taking into consideration collection date and protist. Specifically, microbial communities of voles infected with only Blastocystis, Cryptosporidium, or Giardia were compared against uninfected control voles. Moreover, voles infected with more than one protist were also compared against the controls.
Diversity analysis included alpha (within sample) and beta (between samples) measures. Alpha diversity was measured using the Observed species (OS), Chao1 and Shannon indices accounting for OTU richness and evenness. Corresponding statistical significance was determined using the Mann-Whitney U test. The results were displayed using boxplots. Beta diversity was measured using Bray-Curtis Index distance. Corresponding statistical significance was assessed via Permutational Multivariate Analysis of Variance Using Distance Matrices (PERMANOVA). The results were presented as principal coordinates analysis plots (PCoA) to illustrate relationships between the vole microbiomes based on infection category.
To assess the microbial community abundances between different experimental variables, we employed two analyses to identify differentially abundant communities. Classical univariate analysis was used to identify differentially abundant community profiles using Mann-Whitney U (two variables) and Kruskal-Wallis (three variables) analysis based on a single grouping experimental variable. Bioconductor MetagenomeSeq analysis [
42], accessed via the Microbiomeanalyst platform, was also used to account for the effects of normalisation and under-sampling of microbial communities. This method was implemented in addition to classical univariate analysis as it is specifically designed to address normalisation and biases in measurements across taxonomic features by way of a zero-inflated Gaussian distribution model to account for the variety in sequencing depth. This method is beneficial, as it aids in the detection of differentially abundant rare taxa.
LEfSE (Linear discriminant analysis Effect Size) was used to perform non-parametric factorial Kruskal-Wallis sum-rank test to identify which community abundance features were significantly different with regard to the experimental factor and most likely to explain differences between experimental variables.
4. Discussion
This pilot study provides the first investigation exploring the association of Blastocystis with bacterial communities in the gut of captive water voles (Arvicola amphibius) presenting no gastrointestinal symptoms at the time of collection. Twenty-nine samples were collected over two time points and the microbiome of twenty-eight of these was characterised. Of these, 44% were Blastocystis positive and were subsequently subtyped, and associations with bacterial microbiome were examined for the first time in this rodent.
Several studies on other rodents do exist, including those of the subfamily
Arvicolinae [
43,
44,
45]. Generally, the core
Arvicolinae microbiota was made up predominantly of
Firmicutes and
Bacteroidetes, and this was the case here, with the majority of the water voles reporting
Bacteroidetes and
Firmicutes as the two dominant phyla, reflecting results across other mammalian studies [
46]. Specifically, the abundance of
Bacteroidetes was higher (65%) in comparison to that of
Firmicutes. Previous vole-based studies of captive and wild-captured cohorts have shown similar abundances [
43]. In general, studies in humans and rodents have shown that
Bacteroidetes-driven microbiota may be the result of low fat/high fibre diet [
47,
48,
49,
50]. Nonetheless, rats showed a less than 10% abundance of
Bacteroidetes, possibly reflecting their omnivorous nature [
51]. The water voles in this study were fed a diet rich in fibrous material including fruits, legumes, willow leaves and bark, which likely accounts towards the high abundance of
Bacteroidetes [
45]. Besides the two dominant phyla, the bacterial communities of the water voles diversified further at the lower taxonomic levels, in accordance with numerous microbiome-based studies [
52,
53]. More research investigating composition of the bacterial community and abundance of individual taxa in a wide range of vole species will help establish the gut microbiome makeup of these small rodents.
An exception to the above was noted in one water vole, which displayed a markedly different abundance profile from the rest during the second collection. Specifically, this water vole (R13) had a drastic change in the relative abundance of
Proteobacteria between collections, with the value increasing from <1% in the first collection to over 30% in just one month. This alteration was also accompanied by a decrease in the relative abundance of
Bacteroidetes from 85% to 49% between collections. The water vole that died from unknown causes on the morning of the second collection had a high abundance of
Proteobacteria, which is often associated with dysbiosis in animal studies including humans, where it has been linked to both intestinal related diseases, such as Crohn’s disease, as well as extra-intestinal disease possibly indicating a disruption of enteric homeostasis [
54,
55,
56,
57,
58]. This implies a use for
Proteobacteria as a biomarker for intestinal dysbiosis in captive water voles.
In recent years, hypotheses regarding the association of opportunistic protists with distinct microbial profiles have been brought forth [
59,
60,
61,
62,
63]. Among those, the most-studied protist is
Blastocystis, which has been suggested to be an “ecosystem engineer” [
64]. Presence of
Blastocystis has been associated with an increase in overall bacterial diversity and richness. A negative association with
Bacteroides and presence of
Blastocystis has been consistently found across human studies [
64,
65,
66,
67]. Positive associations with
Roseburia and
Faecalibacterium, which are often associated with eubiosis, have also been noted [
68,
69]. Presence of this protist has also been linked to a decrease in
Hymenolepis nana, which has been associated with alterations in the microbiota [
70,
71], these results led to the hypothesis that
Blastocystis is part of the healthy intestinal microbiome in humans. Contrary to this, a single study focusing on chimpanzees demonstrated that bearing
Blastocystis was associated with decreased microbial richness and decline in the ‘protective’ species
Faecalibacterium prausnitzii and increase in
Enterobacteriaceae, a marker of poor intestinal health in humans [
62]. Herein, contrary to the human studies, we found no significant differences between the microbial profiles of water voles with and without
Blastocystis. A possible explanation could be that the presence of
Blastocystis in water voles might not have the same associations as those observed in humans. Other possible hypotheses could be the homogenisation of bacterial taxa due to captivity and/or disturbances due to drug administration. Due to the small size (16 individuals, <30 samples), we cannot at this point draw a definitive conclusion.
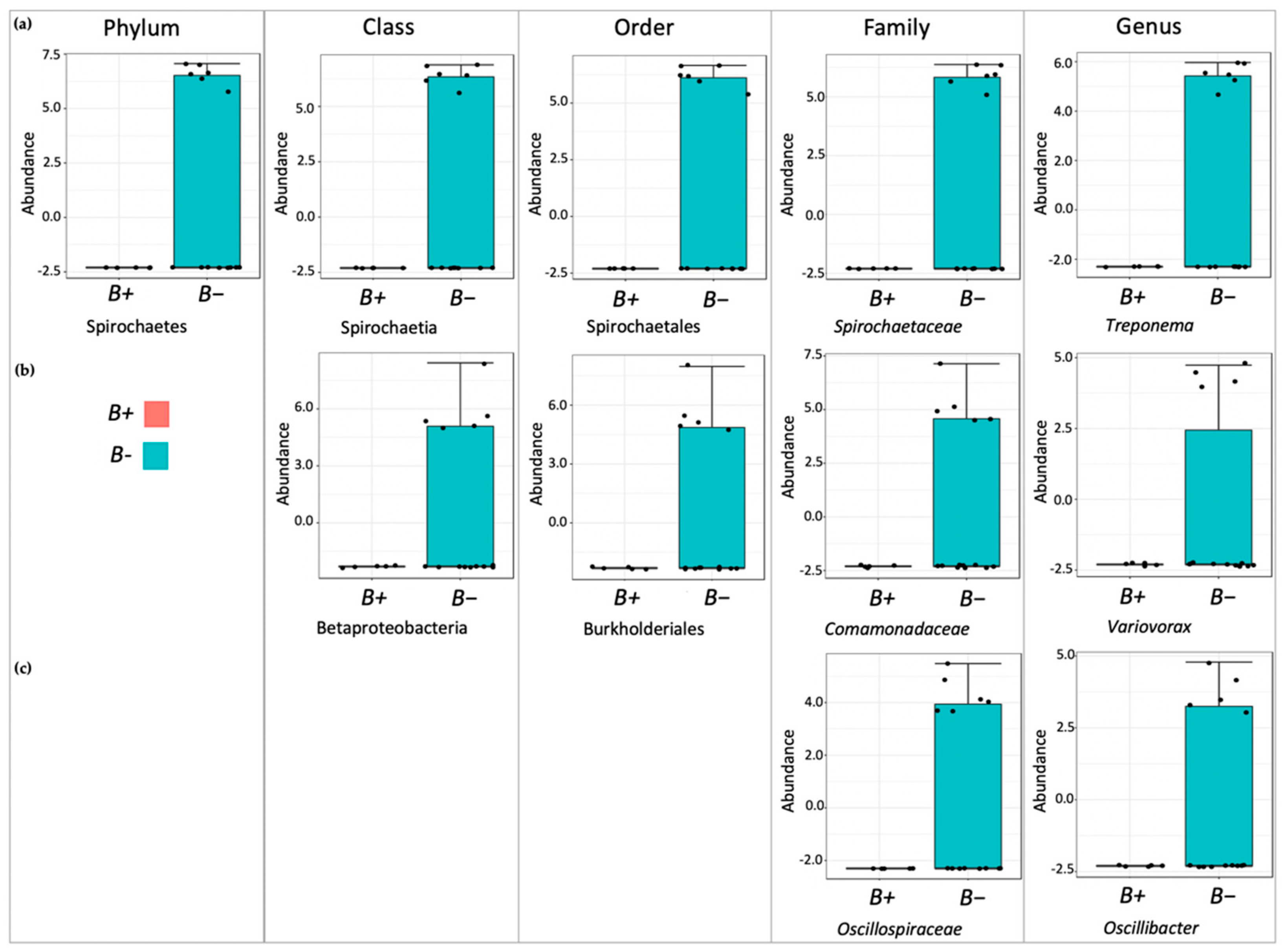
Although the overall microbial community richness was similar in water voles with and without
Blastocystis, closer inspection of community comparisons indicated that reductions in
Treponema and
Kineothrix were strongly associated with
Blastocystis presence.
Treponema has been associated with degradation of plant materials in the rumen [
72]. In support of this,
Treponema has been found in significantly higher abundances in the gut of humans living in rural areas and eating fibre-rich diets, while it is typically absent in urbanites consuming fibre-poor diets [
73,
74]. The bacterium has also been associated with the vole microbiota, where it likely has similar roles [
44]. Nonetheless, it is worth noting that other common degraders, such as
Prevotella,
Ruminococcus and
Oscillospira, were not significantly decreased in
Blastocystis carriers herein. This suggests that the observed decline in
Treponema likely does not impair the ability of the water vole to degrade plant materials.
Kineothrix was also significantly decreased in positive voles, this bacterium produces butyrate, a metabolite that serves as energy source of enterocytes and has notable anti-inflammatory and immunomodulatory properties [
75]. Furthermore, the
Firmicutes genera
Thermoclostridium, Anaeromassilibacillus and
Anaerotignum were also decreased in positive water voles; however, most of these have uncharacterised roles in the murine microbiota. Nonetheless,
Firmicutes are generally associated with fermentation of dietary fibre and production of short-chain fatty acids [
76]. Overall, the observed significant reductions of specific bacterial taxa may represent the beginnings of a disturbed gut, which is a hallmark of the transition from free-living to a more confined lifestyle. Nonetheless, definitive conclusions cannot be drawn, as there was redundancy of function in the gut, which may be enhanced by the presence of
Blastocystis, as the organism has been associated with increased species richness in the gut [
67,
77,
78,
79]. Alternatively, the observed shifts might be associated with an as-yet-unidentified factor other than
Blastocystis.
Several of the water voles were also colonised with either Giardia or Cryptosporidium and in several cases with more than just one of these microbial eukaryotes. Notably, the water voles also received the antiprotozoal drugs fenbendazole and metronidazole against Giardia and treatments had ended by the time of sampling. Despite this, there were no differences in either alpha or beta diversities in any of these groups. This, along with the absence of diarrhoea or other GI symptoms, suggests that the water voles might be carriers of these parasites.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





