
Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2



{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
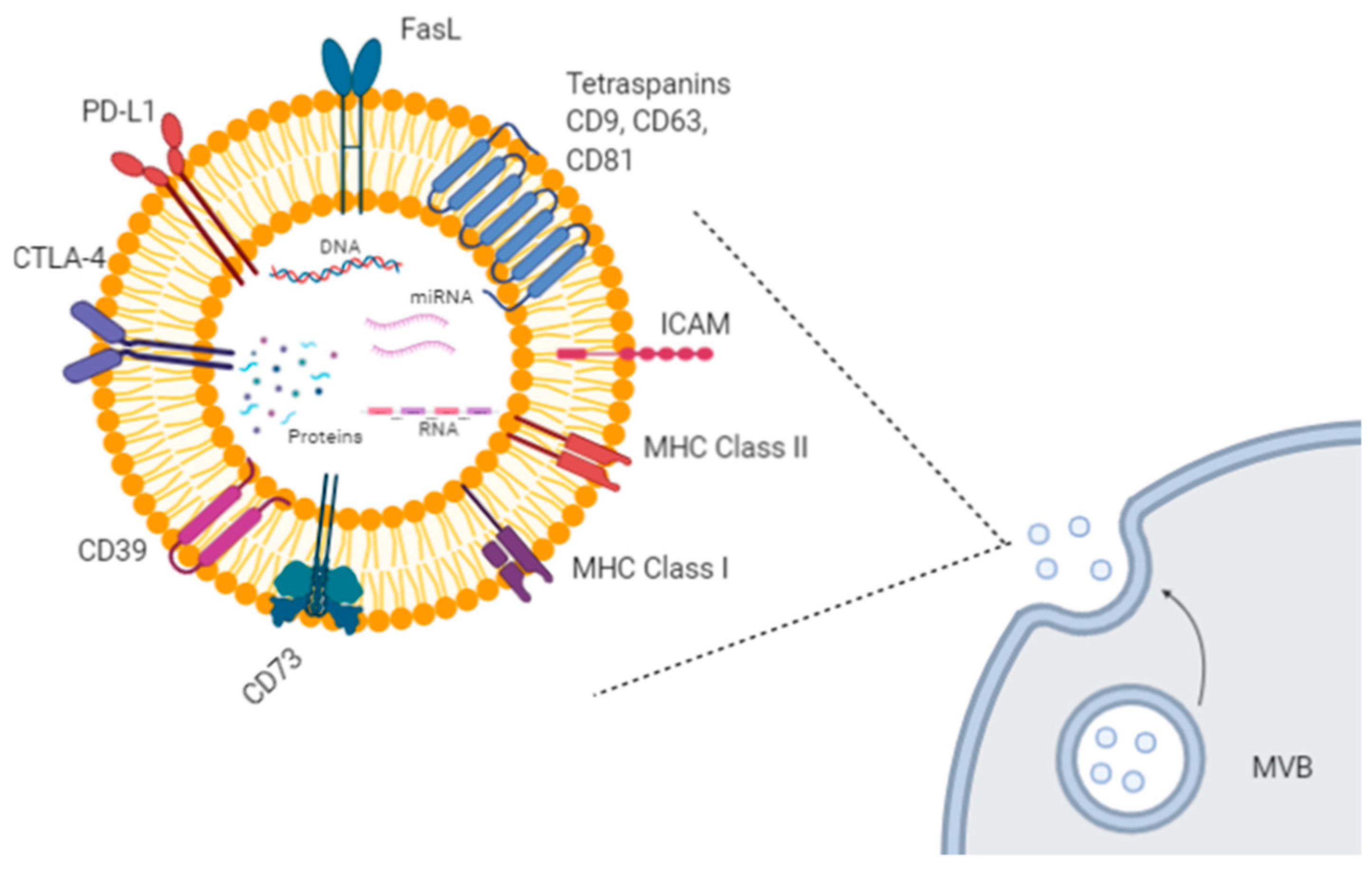
1. Introduction
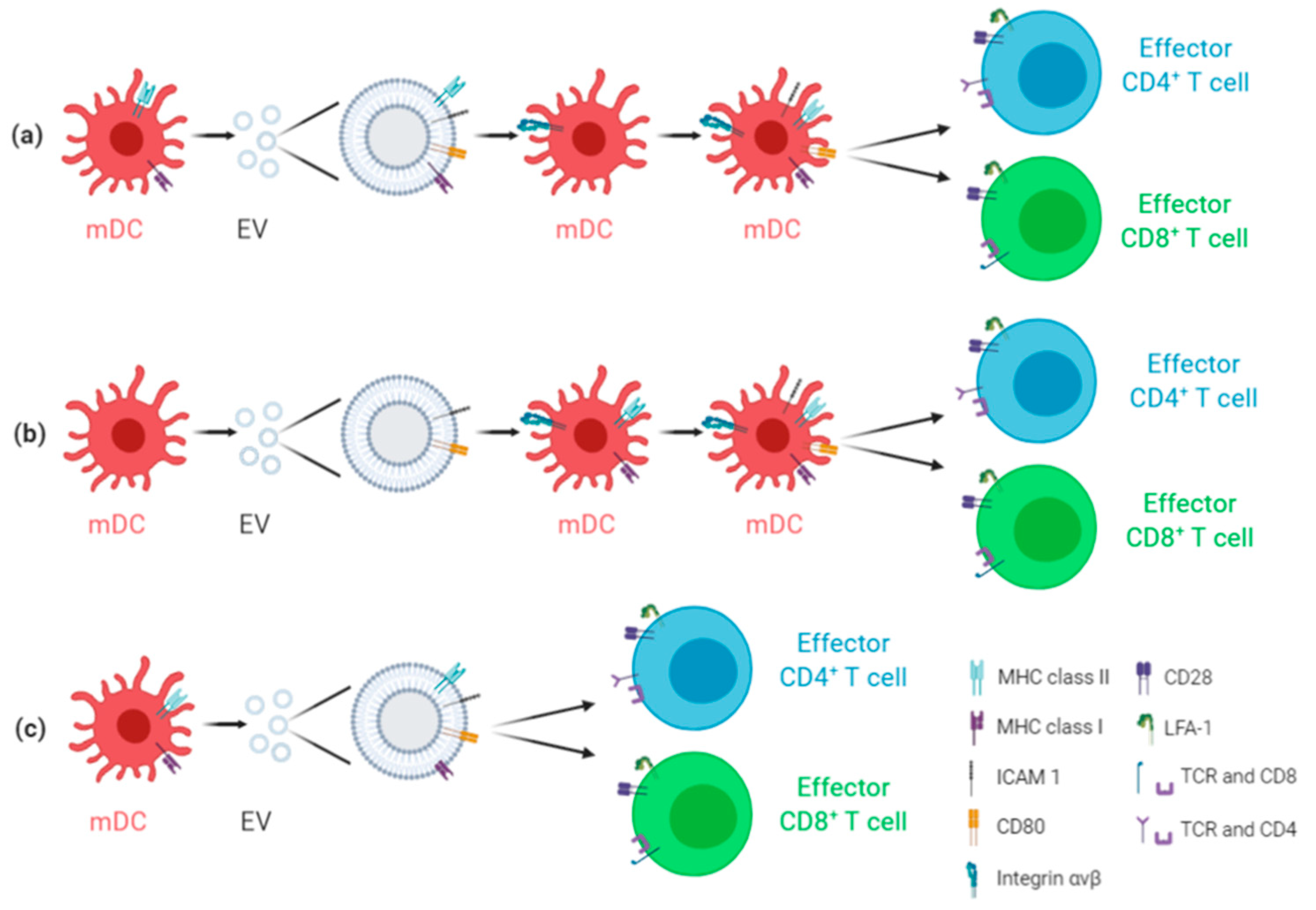
2. Antiviral EV-Based Vaccines: General Overview
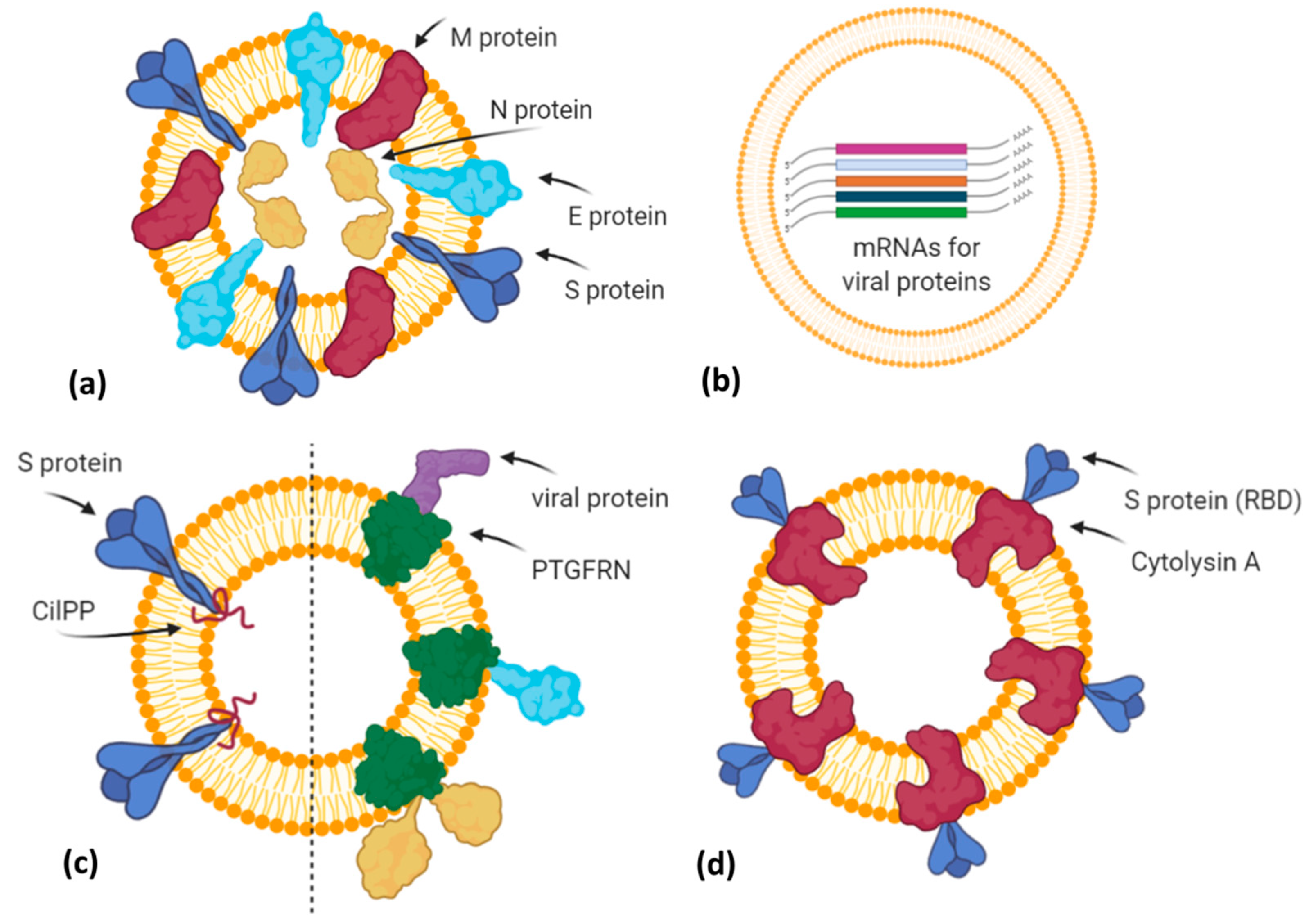
3. EV-Based Vaccines: Focus on COVID-19
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N.; Dittmer, D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Microbiol. 2017, 15, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Lorizate, M.; Krausslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef]
- Lorizate, M.; Sachsenheimer, T.; Glass, B.; Habermann, A.; Gerl, M.J.; Krausslich, H.G.; Brugger, B. Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell Microbiol. 2013, 15, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef]
- Tamai, K.; Shiina, M.; Tanaka, N.; Nakano, T.; Yamamoto, A.; Kondo, Y.; Kakazu, E.; Inoue, J.; Fukushima, K.; Sano, K.; et al. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 2012, 422, 377–385. [Google Scholar] [CrossRef]
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.L.; Mutsafi, Y.; De Jesus-Diaz, D.A.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I.; et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-organismal Viral Transmission. Cell Host Microbe 2018, 24, 208–220. [Google Scholar] [CrossRef]
- Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. Cloaked Viruses and Viral Factors in Cutting Edge Exosome-Based Therapies. Front. Cell Dev. Biol. 2020, 8, 376. [Google Scholar] [CrossRef]
- Zicari, S.; Arakelyan, A.; Palomino, R.A.N.; Fitzgerald, W.; Vanpouille, C.; Lebedeva, A.; Schmitt, A.; Bomsel, M.; Britt, W.; Margolis, L. Human cytomegalovirus-infected cells release extracellular vesicles that carry viral surface proteins. Virology 2018, 524, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Pleet, M.L.; Mathiesen, A.; DeMarino, C.; Akpamagbo, Y.A.; Barclay, R.A.; Schwab, A.; Iordanskiy, S.; Sampey, G.C.; Lepene, B.; Nekhai, S.; et al. Ebola VP40 in Exosomes Can Cause Immune Cell Dysfunction. Front. Microbiol 2016, 7, 1765. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Du, T.; Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996. [Google Scholar] [CrossRef] [PubMed]
- Chahar, H.S.; Corsello, T.; Kudlicki, A.S.; Komaravelli, N.; Casola, A. Respiratory Syncytial Virus Infection Changes Cargo Composition of Exosome Released from Airway Epithelial Cells. Sci. Rep. 2018, 8, 387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, D.; Ma, X.; Wang, J.; Hou, W.; Zhang, W. Exosomes as drug carriers for cancer therapy and challenges regarding exosome uptake. Biomed. Pharmacother. 2020, 128, 110237. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Zou, X.; Yuan, M.; Zhang, T.; Wei, H.; Xu, S.; Jiang, N.; Zheng, N.; Wu, Z. Extracellular vesicles expressing a single-chain variable fragment of an HIV-1 specific antibody selectively target Env(+) tissues. Theranostics 2019, 9, 5657–5671. [Google Scholar] [CrossRef]
- Walker, S.; Busatto, S.; Pham, A.; Tian, M.; Suh, A.; Carson, K.; Quintero, A.; Lafrence, M.; Malik, H.; Santana, M.X.; et al. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics 2019, 9, 8001–8017. [Google Scholar] [CrossRef]
- Imai, T.; Takahashi, Y.; Nishikawa, M.; Kato, K.; Morishita, M.; Yamashita, T.; Matsumoto, A.; Charoenviriyakul, C.; Takakura, Y. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J. Extracel. Vesicles 2015, 4, 26238. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nishikawa, M.; Shinotsuka, H.; Matsui, Y.; Ohara, S.; Imai, T.; Takakura, Y. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 in mice after intravenous injection. J. Biotechnol. 2013, 165, 77–84. [Google Scholar] [CrossRef]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed]
- Giulietti, M.; Bastianoni, M.; Cecati, M.; Ruzzo, A.; Bracci, M.; Malavolta, M.; Piacenza, F.; Giacconi, R.; Piva, F. MetaTropismDB: A database of organ-specific metastasis induced by human cancer cell lines in mouse models. Database 2020. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.; Yan, I.K.; Parasramka, M.; Mohankumar, S.; Matsuda, A.; Patel, T. In vitro toxicology studies of extracellular vesicles. J. Appl. Toxicol. 2017, 37, 310–318. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerakar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef]
- Saleh, A.F.; Lazaro-Ibanez, E.; Forsgard, M.A.; Shatnyeva, O.; Osteikoetxea, X.; Karlsson, F.; Heath, N.; Ingelsten, M.; Rose, J.; Harris, J.; et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale 2019, 11, 6990–7001. [Google Scholar] [CrossRef]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; La Perle, K.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef]
- Lewis, N.D.; Sia, C.L.; Kirwin, K.; Haupt, S.; Mahimkar, G.; Zi, T.; Xu, K.; Dooley, K.; Jang, S.C.; Choi, B.; et al. Exosome surface display of IL-12 results in tumor-retained pharmacology with superior potency and limited systemic exposure compared to recombinant IL-12. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef]
- Yi, Y.W.; Lee, J.H.; Kim, S.Y.; Pack, C.G.; Ha, D.H.; Park, S.R.; Youn, J.; Cho, B.S. Advances in Analysis of Biodistribution of Exosomes by Molecular Imaging. Int. J. Mol. Sci. 2020, 21, 665. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Yi, J.; Liu, Y.; Li, D.; Wang, J.; Hou, D.; Jiang, X.; Zhang, J.; Wang, J.; et al. Identification and Characterization of 293T Cell-Derived Exosomes by Profiling the Protein, mRNA and MicroRNA Components. PLoS ONE 2016, 11, e0163043. [Google Scholar] [CrossRef] [PubMed]
- Admyre, C.; Johansson, S.M.; Paulie, S.; Gabrielsson, S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur. J. Immunol. 2006, 36, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Montaner-Tarbes, S.; Borras, F.E.; Montoya, M.; Fraile, L.; Del Portillo, H.A. Serum-derived exosomes from non-viremic animals previously exposed to the porcine respiratory and reproductive virus contain antigenic viral proteins. Vet. Res. 2016, 47, 59. [Google Scholar] [CrossRef] [PubMed]
- Montaner-Tarbes, S.; Novell, E.; Tarancon, V.; Borras, F.E.; Montoya, M.; Fraile, L.; Del Portillo, H.A. Targeted-pig trial on safety and immunogenicity of serum-derived extracellular vesicles enriched fractions obtained from Porcine Respiratory and Reproductive virus infections. Sci. Rep. 2018, 8, 17487. [Google Scholar] [CrossRef] [PubMed]
- Jafari, D.; Shajari, S.; Jafari, R.; Mardi, N.; Gomari, H.; Ganji, F.; Forouzandeh Moghadam, M.; Samadikuchaksaraei, A. Designer Exosomes: A New Platform for Biotechnology Therapeutics. BioDrugs 2020, 34, 567–586. [Google Scholar] [CrossRef]
- Kanuma, T.; Yamamoto, T.; Kobiyama, K.; Moriishi, E.; Masuta, Y.; Kusakabe, T.; Ozasa, K.; Kuroda, E.; Jounai, N.; Ishii, K.J. CD63-Mediated Antigen Delivery into Extracellular Vesicles via DNA Vaccination Results in Robust CD8(+) T Cell Responses. J. Immunol. 2017, 198, 4707–4715. [Google Scholar] [CrossRef]
- Anticoli, S.; Manfredi, F.; Chiozzini, C.; Arenaccio, C.; Olivetta, E.; Ferrantelli, F.; Capocefalo, A.; Falcone, E.; Ruggieri, A.; Federico, M. An Exosome-Based Vaccine Platform Imparts Cytotoxic T Lymphocyte Immunity Against Viral Antigens. Biotechnol. J. 2018, 13, e1700443. [Google Scholar] [CrossRef]
- System Biosciences, LLC. Exosome Engineering. Available online: https://systembio.com/products/exosome-research/exosome-engineering/ (accessed on 26 January 2021).
- Delcayre, A.; Estelles, A.; Sperinde, J.; Roulon, T.; Paz, P.; Aguilar, B.; Villanueva, J.; Khine, S.; Le Pecq, J.B. Exosome Display technology: Applications to the development of new diagnostics and therapeutics. Blood Cells Mol. Dis. 2005, 35, 158–168. [Google Scholar] [CrossRef]
- Hong, S.; Ruan, S.; Kubota, P.G.; He, M.; McGill, J.L. Immunogenic potency of engineered exosomes for prevention of respiratory syncytial virus. J. Immunol. 2020, 204, 245.22. [Google Scholar]
- Afrough, B.; Dowall, S.; Hewson, R. Emerging viruses and current strategies for vaccine intervention. Clin. Exp. Immunol. 2019, 196, 157–166. [Google Scholar] [CrossRef]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [PubMed]
- Qazi, K.R.; Gehrmann, U.; Domange Jordo, E.; Karlsson, M.C.; Gabrielsson, S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood 2009, 113, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Jesus, S.; Soares, E.; Cruz, M.T.; Borges, O. Exosomes as adjuvants for the recombinant hepatitis B antigen: First report. Eur. J. Pharm. Biopharm. 2018, 133, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rappazzo, C.G.; Watkins, H.C.; Guarino, C.M.; Chau, A.; Lopez, J.L.; DeLisa, M.P.; Leifer, C.A.; Whittaker, G.R.; Putnam, D. Recombinant M2e outer membrane vesicle vaccines protect against lethal influenza A challenge in BALB/c mice. Vaccine 2016, 34, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.C.; Rappazzo, C.G.; Higgins, J.S.; Sun, X.; Brock, N.; Chau, A.; Misra, A.; Cannizzo, J.P.B.; King, M.R.; Maines, T.R.; et al. Safe Recombinant Outer Membrane Vesicles that Display M2e Elicit Heterologous Influenza Protection. Mol. Ther. 2017, 25, 989–1002. [Google Scholar] [CrossRef]
- Ciloa. Adjuvant and Virus-Free Vaccines. Available online: https://www.ciloa.fr/vaccines/ (accessed on 26 January 2021).
- Yang, Y.; Peng, F.; Wang, R.; Yange, M.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun 2020, 109, 102434. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Piva, F.; Sabanovic, B.; Cecati, M.; Giulietti, M. Expression and co-expression analyses of TMPRSS2, a key element in COVID-19. Eur. J. Clin. Microbiol. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Kuate, S.; Cinatl, J.; Doerr, H.W.; Uberla, K. Exosomal vaccines containing the S protein of the SARS coronavirus induce high levels of neutralizing antibodies. Virology 2007, 362, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Ghaebi, M.; Osali, A.; Valizadeh, H.; Roshangar, L.; Ahmadi, M. Vaccine development and therapeutic design for 2019-nCoV/SARS-CoV-2: Challenges and chances. J. Cell Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Omolo, C.A.; Soni, N.; Fasiku, V.O.; Mackraj, I.; Govender, T. Update on therapeutic approaches and emerging therapies for SARS-CoV-2 virus. Eur. J. Pharmacol. 2020, 173348. [Google Scholar] [CrossRef]
- Capricor Therapeutics. Available online: https://capricor.com/covid-19/ (accessed on 26 January 2021).
- Morel, P.A.; Falkner, D.; Plowey, J.; Larregina, A.T.; Falo, L.D. DNA immunisation: Altering the cellular localisation of expressed protein and the immunisation route allows manipulation of the immune response. Vaccine 2004, 22, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.; Guo, C.; Atai, N.A.; Gould, S.J. Exosome-Mediated mRNA Delivery For SARS-CoV-2 Vaccination. bioRxiv 2020. [Google Scholar] [CrossRef]
- Polak, K.; Greze, N.; Lachat, M.; Merle, D.; Chiumento, S.; Bertrand-Gaday, C.; Trentin, B.; Mamoun, R.Z. Extracellular vesicle-based vaccine platform displaying native viral envelope proteins elicits a robust anti-SARS-CoV-2 response in mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Codiak BioSciences. The exoVACC Vaccine Platform for SARS-CoV-2. Available online: https://ir.codiakbio.com/news-releases/news-release-details/codiak-biosciences-collaborates-ragon-institute-evaluate (accessed on 26 January 2021).
- Dooley, K.; McConnell, R.E.; Xu, K.; Lewis, N.D.; Haupt, S.; Youniss, M.R.; Martin, S.; Sia, C.L.; McCoy, C.; Moniz, R.J.; et al. A Versatile Platform for Generating Engineered Extracellular Vesicles with Defined Therapeutic Properties. Mol. Therapy 2020. [Google Scholar] [CrossRef]
- Allele Biotechnology and Pharmaceuticals Inc. COVID-19. Available online: https://www.allelebiotech.com/covid19 (accessed on 26 January 2021).
- Versatope Therapeutics, Inc. Available online: https://www.versatope.com/ (accessed on 26 January 2021).
- Li, M.; Zhou, H.; Yang, C.; Wu, Y.; Zhou, X.; Liu, H.; Wang, Y. Bacterial outer membrane vesicles as a platform for biomedical applications: An update. J. Control. Release 2020, 323, 253–268. [Google Scholar] [CrossRef]
- Shehata, M.M.; Mostafa, A.; Teubner, L.; Mahmoud, S.H.; Kandeil, A.; Elshesheny, R.; Frantz, R.; La Pietra, L.; Pleschka, S.; Osman, A.; et al. Bacterial Outer Membrane Vesicles (OMVs)-based Dual Vaccine for Influenza A H1N1 Virus and MERS-CoV. Vaccines 2019, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Schetters, S.T.T.; Jong, W.S.P.; Horrevorts, S.K.; Kruijssen, L.J.W.; Engels, S.; Stolk, D.; Daleke-Schermerhorn, M.H.; Garcia-Vallejo, J.; Houben, D.; Unger, W.W.J.; et al. Outer membrane vesicles engineered to express membrane-bound antigen program dendritic cells for cross-presentation to CD8(+) T cells. Acta Biomater. 2019, 91, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Rossi, O.; Citiulo, F.; Mancini, F. Outer membrane vesicles: Moving within the intricate labyrinth of assays that can predict risks of reactogenicity in humans. Hum. Vaccin Immunother. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. A prefusion SARS-CoV-2 spike RNA vaccine is highly immunogenic and prevents lung infection in non-human primates. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombácz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Karikó, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther. Nucleic Acids 2019, 15, 36–47. [Google Scholar] [CrossRef]
- Sengupta, V.; Sengupta, S.; Lazo, A.; Woods, P.; Nolan, A.; Bremer, N. Exosomes Derived from Bone Marrow Mesenchymal Stem Cells as Treatment for Severe COVID-19. Stem Cells Dev. 2020, 29, 747–754. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabanovic, B.; Piva, F.; Cecati, M.; Giulietti, M. Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2. Biology 2021, 10, 94. https://doi.org/10.3390/biology10020094
Sabanovic B, Piva F, Cecati M, Giulietti M. Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2. Biology. 2021; 10(2):94. https://doi.org/10.3390/biology10020094
Chicago/Turabian StyleSabanovic, Berina, Francesco Piva, Monia Cecati, and Matteo Giulietti. 2021. "Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2" Biology 10, no. 2: 94. https://doi.org/10.3390/biology10020094
APA StyleSabanovic, B., Piva, F., Cecati, M., & Giulietti, M. (2021). Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2. Biology, 10(2), 94. https://doi.org/10.3390/biology10020094