Long-Term Aspartame Administration Leads to Fibrosis, Inflammasome Activation, and Gluconeogenesis Impairment in the Liver of Mice


 ,
,
Abstract
Simple Summary
Abstract
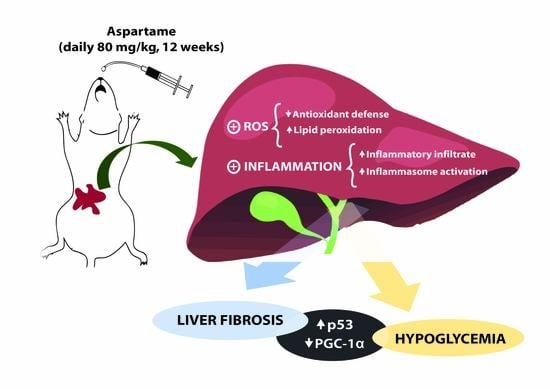
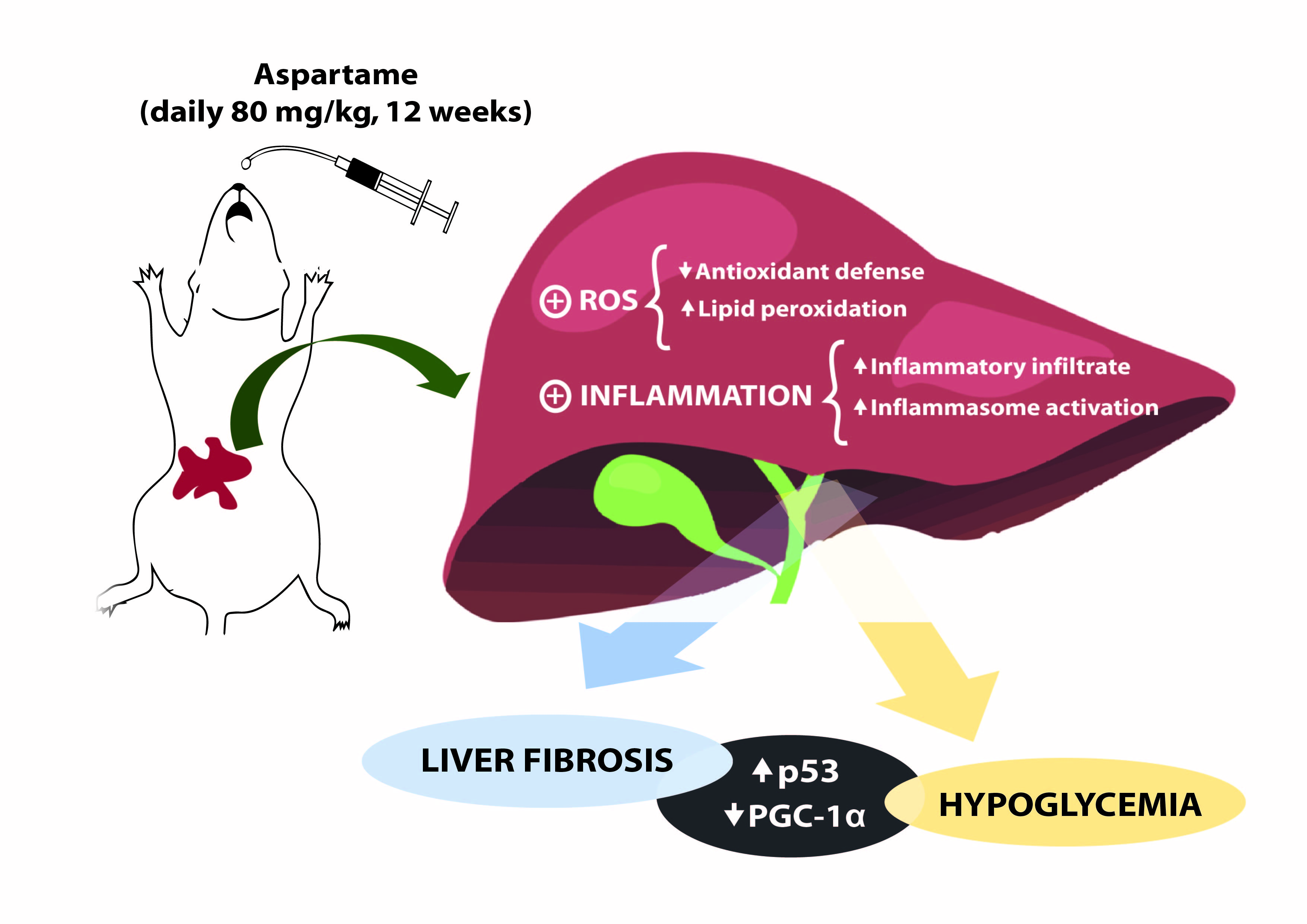
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Protocol
2.3. Assays
2.3.1. Transaminases, Glucose, and Lipid Profile
2.3.2. Histology
2.3.3. RT-qPCR
2.3.4. Western Blotting
2.3.5. Malondialdehyde
2.3.6. Antioxidant Enzymes
2.3.7. Myeloperoxidase
2.3.8. Protein
2.4. Statistical Analysis
3. Results
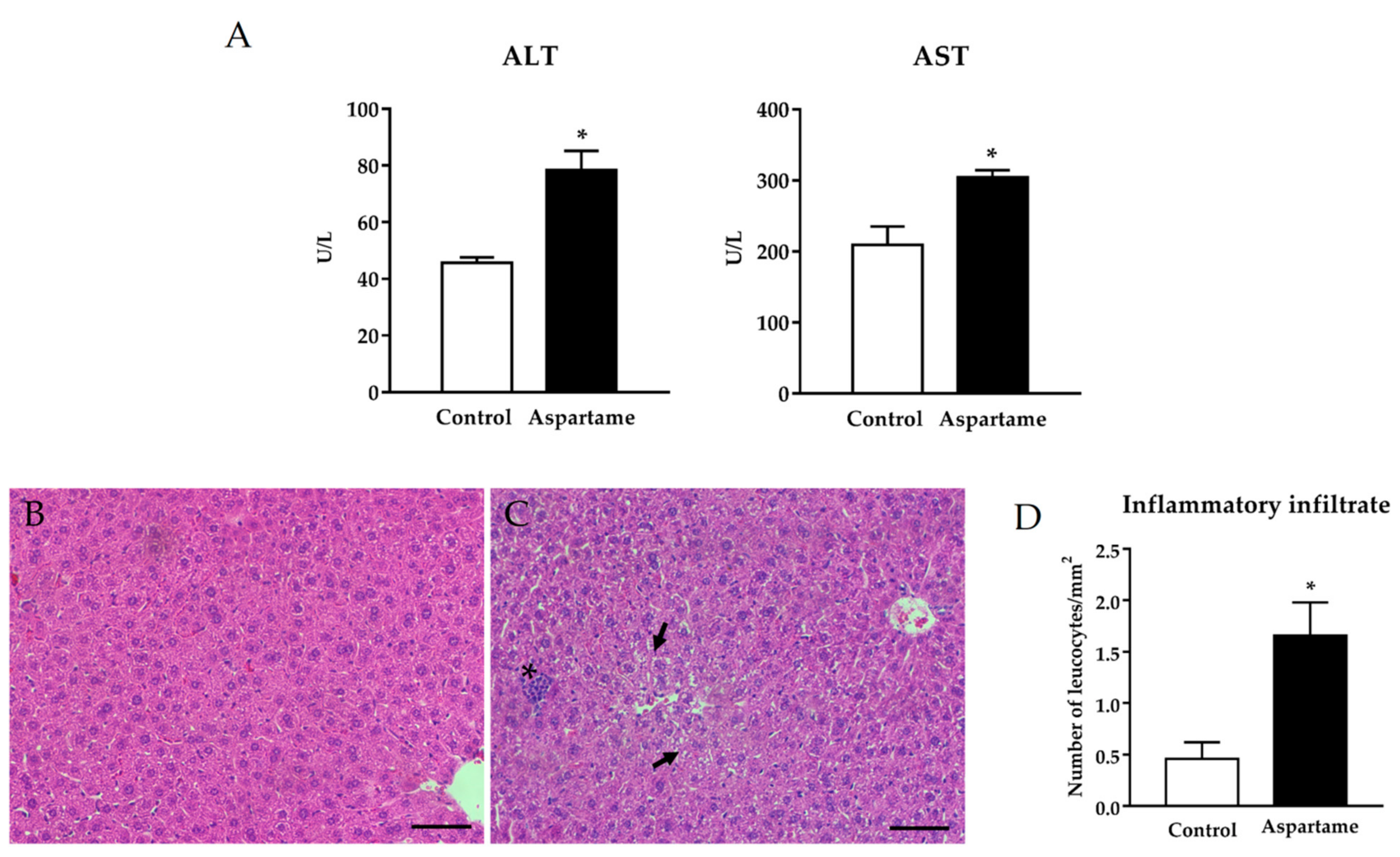
3.1. Aspartame Leads to Liver Injury
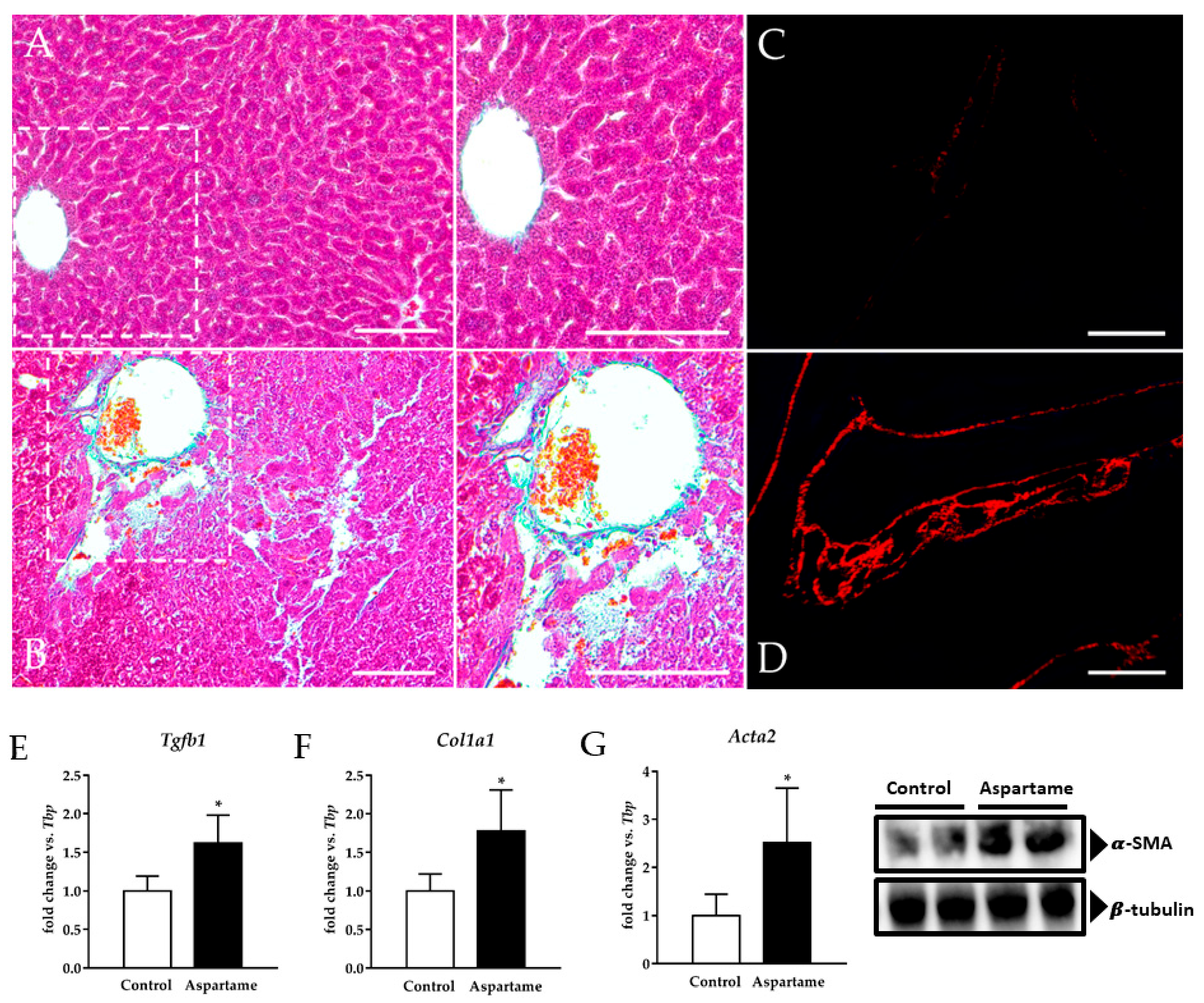
3.2. Aspartame Causes Liver Fibrosis
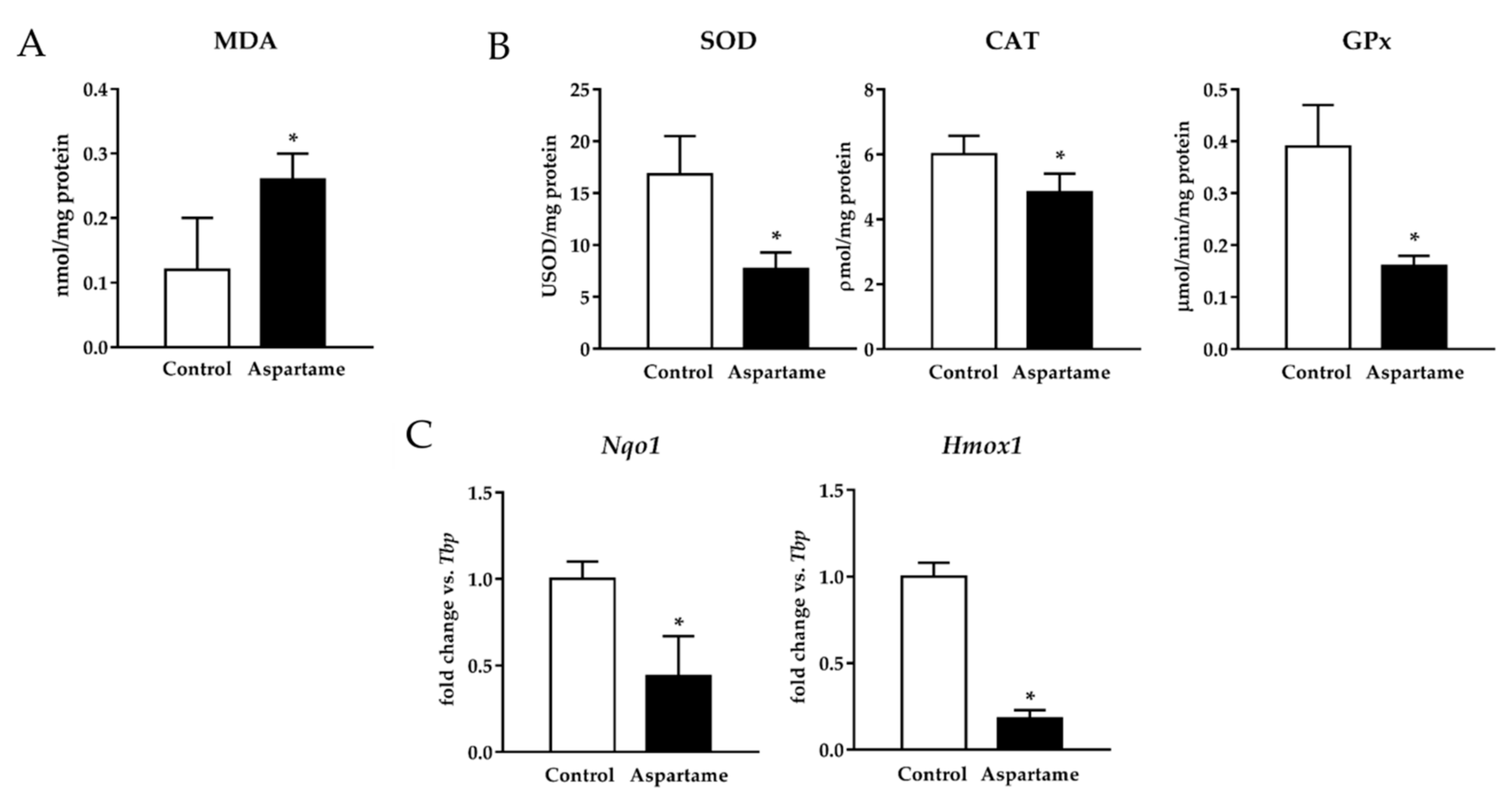
3.3. Aspartame Triggers Lipid Peroxidation and Decreases the Activity of Antioxidant Enzymes
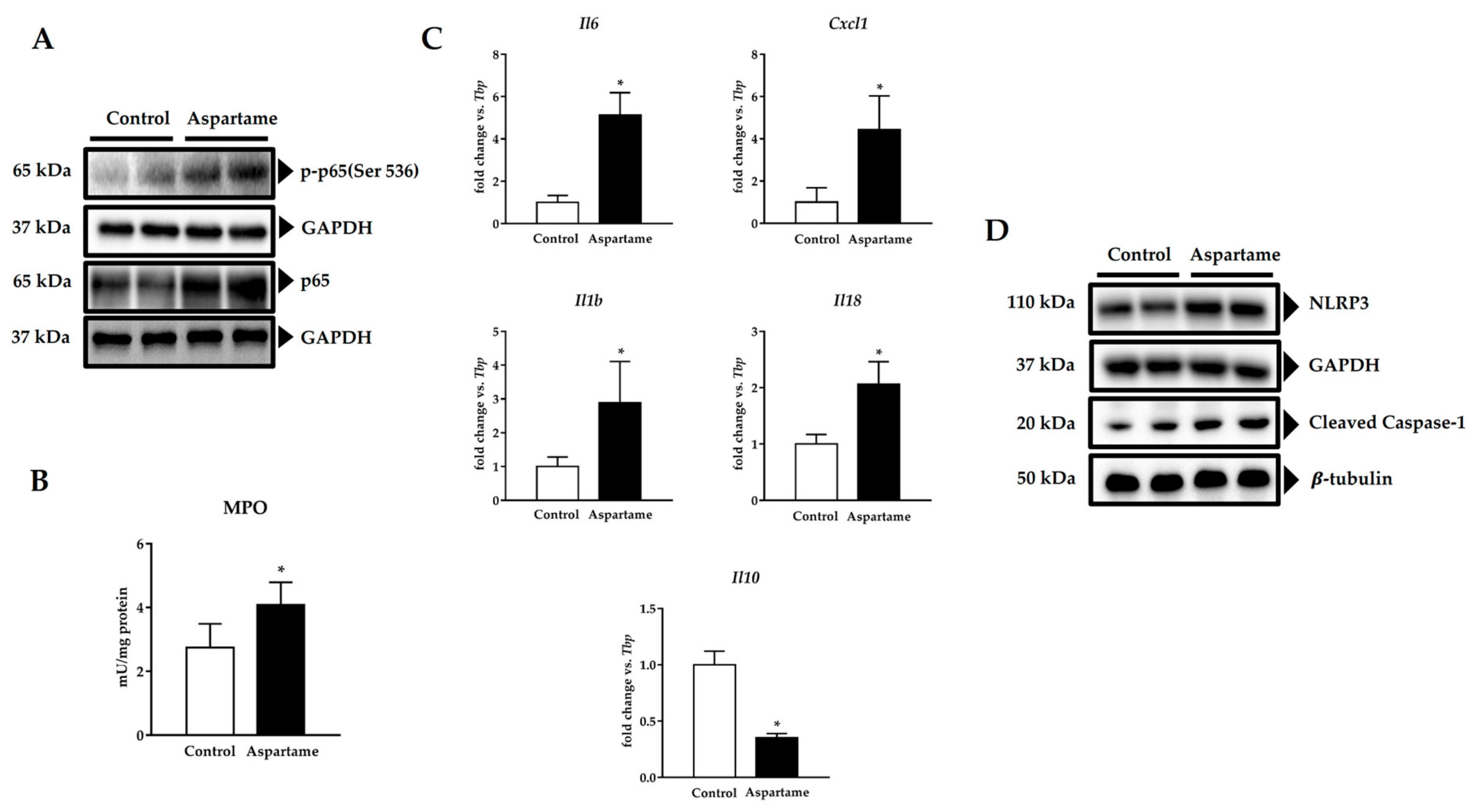
3.4. Aspartame Promotes Phosphorylation of p65 Subunit of NF-κB and NLRP3 Inflammasome Activation
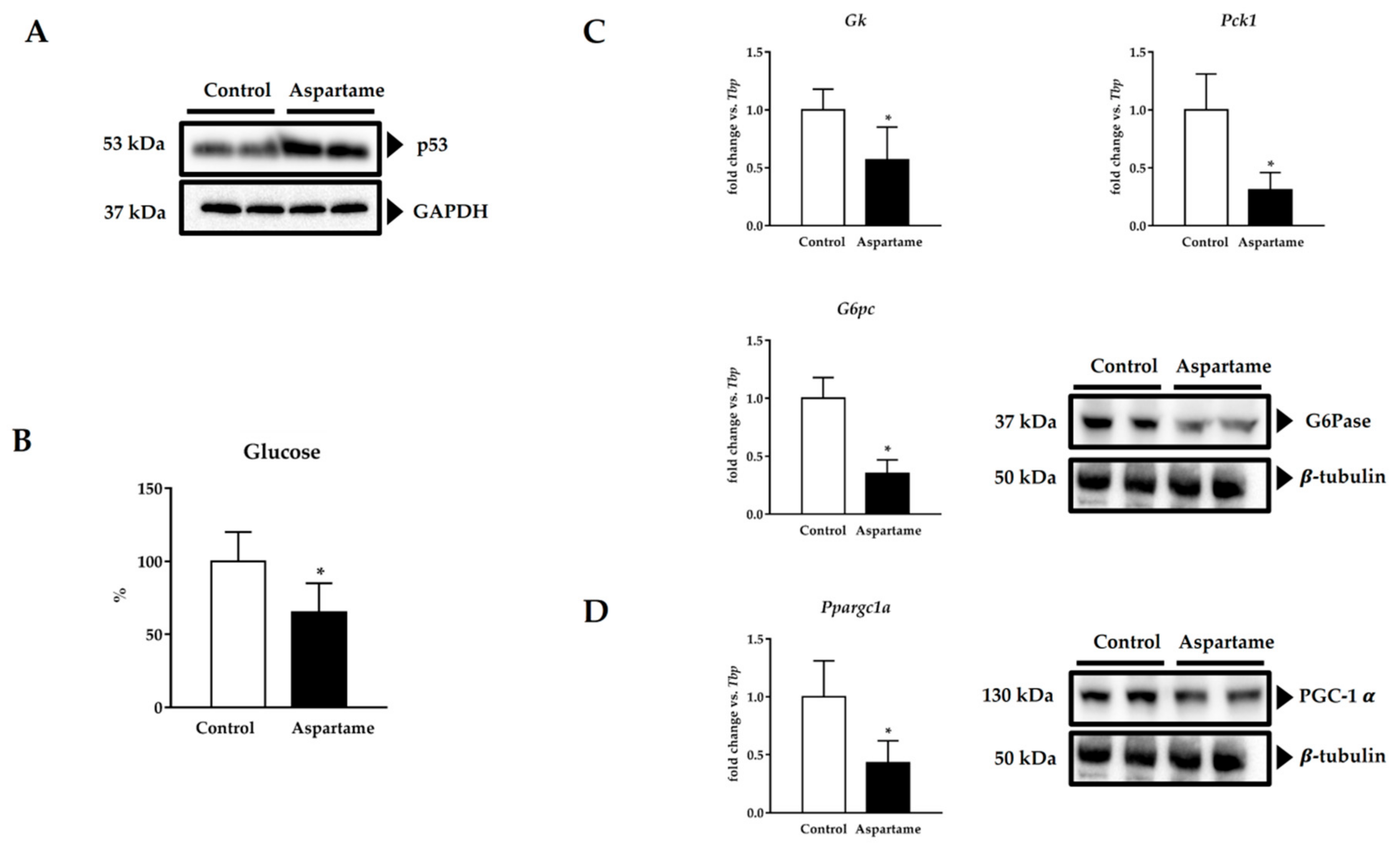
3.5. Aspartame Induces p53
3.6. Aspartame Reduces Fasting Glucose Levels and Impairs Gluconeogenesis via PGC-1α
3.7. Aspartame Affects the Lipid Profile and Generates the Accumulation of Total Lipids in Liver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Butchko, H.H.; Stargel, W.W.; Comer, C.P.; Mayhew, D.A.; Benninger, C.; Blackburn, G.L.; de Sonneville, L.M.J.; Geha, R.S.; Hertelendy, Z.; Koestner, A.; et al. Aspartame: Review of Safety. Regul. Toxicol. Pharmacol. 2002, 35, S1–S93. [Google Scholar] [CrossRef] [PubMed]
- Ranney, R.E.; Oppermann, J.A.; Muldoon, E.; McMahon, F.G. Comparative Metabolism of Aspartame in Experimental Animals and Humans. J. Toxicol. Environ. Health 1976, 2, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Lebda, M.A.; Tohamy, H.G.; El-Sayed, Y.S. Long-Term Soft Drink and Aspartame Intake Induces Hepatic Damage via Dysregulation of Adipocytokines and Alteration of the Lipid Profile and Antioxidant Status. Nutr. Res. 2017, 41, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Abhilash, M.; Paul, M.V.S.; Varghese, M.V.; Nair, R.H. Effect of Long Term Intake of Aspartame on Antioxidant Defense Status in Liver. Food Chem. Toxicol. 2011, 49, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Ashok, I.; Wankhar, D.; Sheeladevi, R.; Wankhar, W. Long-Term Effect of Aspartame on the Liver Antioxidant Status and Histopathology in Wistar Albino Rats. Biomed. Prev. Nutr. 2014, 4, 299–305. [Google Scholar] [CrossRef]
- Finamor, I.; Pérez, S.; Bressan, C.A.; Brenner, C.E.; Rius-Pérez, S.; Brittes, P.C.; Cheiran, G.; Rocha, M.I.; da Veiga, M.; Sastre, J.; et al. Chronic Aspartame Intake Causes Changes in the Trans-Sulphuration Pathway, Glutathione Depletion and Liver Damage in Mice. Redox Biol. 2017, 11, 701–707. [Google Scholar] [CrossRef]
- Hamza, R.Z.; Al-Eisa, R.A.; Mehana, A.E.; El-Shenawy, N.S. Effect of L-Carnitine on Aspartame-Induced Oxidative Stress, Histopathological Changes, and Genotoxicity in Liver of Male Rats. J. Basic Clin. Physiol. Pharmacol. 2019, 30, 219–232. [Google Scholar] [CrossRef]
- Alkafafy, M.E.-S.; Ibrahim, Z.S.; Ahmed, M.M.; El-Shazly, S.A. Impact of Aspartame and Saccharin on the Rat Liver: Biochemical, Molecular, and Histological Approach. Int. J. Immunopathol. Pharmacol. 2015, 28, 247–255. [Google Scholar] [CrossRef]
- Khidr, B.M.; El-Sokkary, G.H.; Saleh, S.M.M. Study on Morphological Changes Induced by Aspartame on Liver of Normal and Diabetic Male Albino Rats. J. Histol. Histopathol. 2017, 4, 1. [Google Scholar] [CrossRef]
- Byass, P. The Global Burden of Liver Disease: A Challenge for Methods and for Public Health. BMC Med. 2014, 12, 159. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of Liver Fibrosis: A Translational Success Story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Robino, G. Oxidative Stress-Related Molecules and Liver Fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Reyes-Gordillo, K.; Shah, R.; Muriel, P. Oxidative Stress and Inflammation in Hepatic Diseases: Current and Future Therapy. Oxid. Med. Cell. Longev. 2017, 2017, 3140673. [Google Scholar] [CrossRef] [PubMed]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired Endothelial Autophagy Promotes Liver Fibrosis by Aggravating the Oxidative Stress Response during Acute Liver Injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 Inflammasome Activation Results in Hepatocyte Pyroptosis, Liver Inflammation, and Fibrosis in Mice. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Ju, D. Inflammasome: A Double-Edged Sword in Liver Diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2014, 22, 1111–1129. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 Inflammasome Blockade Reduces Liver Inflammation and Fibrosis in Experimental NASH in Mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the Concentration of Low-Density Lipoprotein Cholesterol in Plasma, without Use of the Preparative Ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Da Rosa, V.M.; Ariotti, K.; Bressan, C.A.; da Silva, E.G.; Dallaporta, M.; Júnior, G.B.; da Costa, S.T.; de Vargas, A.C.; Baldisserotto, B.; Finamor, I.A.; et al. Dietary Addition of Rutin Impairs Inflammatory Response and Protects Muscle of Silver Catfish (Rhamdia Quelen) from Apoptosis and Oxidative Stress in Aeromonas Hydrophila-Induced Infection. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2019, 226, 108611. [Google Scholar] [CrossRef]
- Misra, H.P.; Fridovich, I. The Role of Superoxide Anion in the Autoxidation of Epinephrine and a Simple Assay for Superoxide Dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Aebi, H. Catalase In Vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar]
- Flohé, L.; Günzler, W.A. Assays of Glutathione Peroxidase. Methods Enzymol. 1984, 105, 114–121. [Google Scholar] [PubMed]
- Lopez-Font, I.; Gea-Sorlí, S.; de-Madaria, E.; Gutiérrez, L.M.; Pérez-Mateo, M.; Closa, D. Pancreatic and Pulmonary Mast Cells Activation during Experimental Acute Pancreatitis. World J. Gastroenterol. 2010, 16, 3411–3417. [Google Scholar] [CrossRef] [PubMed]
- Pulli, B.; Ali, M.; Forghani, R.; Schob, S.; Hsieh, K.L.C.; Wojtkiewicz, G.; Linnoila, J.J.; Chen, J.W. Measuring Myeloperoxidase Activity in Biological Samples. PLoS ONE 2013, 8, e67976. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Uchida, D.; Takaki, A.; Oyama, A.; Adachi, T.; Wada, N.; Onishi, H.; Okada, H. Oxidative Stress Management in Chronic Liver Diseases and Hepatocellular Carcinoma. Nutrients 2020, 12, 1576. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-KappaB and IkappaB Proteins: Implications in Cancer and Inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Boro, M.; Balaji, K.N. CXCL1 and CXCL2 Regulate NLRP3 Inflammasome Activation via G-Protein-Coupled Receptor CXCR2. J. Immunol. 2017, 199, 1660–1671. [Google Scholar] [CrossRef]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in P53 Expression Induce CTGF Synthesis by Mouse and Human Hepatocytes and Result in Liver Fibrosis in Mice. J. Clin. Investig. 2011, 121, 3343–3356. [Google Scholar] [CrossRef]
- Tian, X.-F.; Ji, F.-J.; Zang, H.-L.; Cao, H. Activation of the MiR-34a/SIRT1/P53 Signaling Pathway Contributes to the Progress of Liver Fibrosis via Inducing Apoptosis in Hepatocytes but Not in HSCs. PLoS ONE 2016, 11, e0158657. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB Regulates Hepatic Gluconeogenesis through the Coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of Hepatic Gluconeogenesis through the Transcriptional Coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Koo, S.-H.; Satoh, H.; Herzig, S.; Lee, C.-H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 Promotes Insulin Resistance in Liver through PPAR-Alpha-Dependent Induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef]
- Estall, J.L.; Kahn, M.; Cooper, M.P.; Fisher, F.M.; Wu, M.K.; Laznik, D.; Qu, L.; Cohen, D.E.; Shulman, G.I.; Spiegelman, B.M. Sensitivity of Lipid Metabolism and Insulin Signaling to Genetic Alterations in Hepatic Peroxisome Proliferator-Activated Receptor-Gamma Coactivator-1alpha Expression. Diabetes 2009, 58, 1499–1508. [Google Scholar] [CrossRef]
- Morris, E.M.; Meers, G.M.E.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1α Overexpression Results in Increased Hepatic Fatty Acid Oxidation with Reduced Triacylglycerol Accumulation and Secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, J.W.; Kim, J.H.; Lim, C.W.; Kim, B. Differential Roles of Angiogenesis in the Induction of Fibrogenesis and the Resolution of Fibrosis in Liver. Biol. Pharm. Bull. 2015, 38, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Alleva, R.; Borghi, B.; Santarelli, L.; Strafella, E.; Carbonari, D.; Bracci, M.; Tomasetti, M. In Vitro Effect of Aspartame in Angiogenesis Induction. Toxicol. Vitr. 2011, 25, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of Inflammatory Response in Liver Diseases: Therapeutic Strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Okada, K.; Shoda, J.; Warabi, E.; Ishige, K.; Ueda, T.; Taguchi, K.; Yanagawa, T.; Nakahara, A.; Hyodo, I.; et al. Deletion of Nuclear Factor-E2-Related Factor-2 Leads to Rapid Onset and Progression of Nutritional Steatohepatitis in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G283–G294. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Caldas, C.; Santos, A.G.; Silva-Gomes, S.; Santos-Gonçalves, A.; Martins, M.J.; Porto, G.; Lopes, J.M. Genetic Disruption of NRF2 Promotes the Development of Necroinflammation and Liver Fibrosis in a Mouse Model of HFE-Hereditary Hemochromatosis. Redox Biol. 2017, 11, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.J.; Kim, J.-Y.; Min, A.-K.; Park, K.-G.; Harris, R.A.; Kim, H.-J.; Lee, I.-K. Sulforaphane Attenuates Hepatic Fibrosis via NF-E2-Related Factor 2-Mediated Inhibition of Transforming Growth Factor-β/Smad Signaling. Free Radic. Biol. Med. 2012, 52, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Alwaleedi, S. Alterations in Antioxidant Defense System in Hepatic and Renal Tissues of Rats Following Aspartame Intake. J. Appl. Biol. Biotechnol. 2016, 4, 046–052. [Google Scholar]
- Adaramoye, O.A.; Akanni, O.O. Effects of Long-Term Administration of Aspartame on Biochemical Indices, Lipid Profile and Redox Status of Cellular System of Male Rats. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 29–37. [Google Scholar] [CrossRef]
- MacDonald, G.A.; Bridle, K.R.; Ward, P.J.; Walker, N.I.; Houglum, K.; George, D.K.; Smith, J.L.; Powell, L.W.; Crawford, D.H.; Ramm, G.A. Lipid Peroxidation in Hepatic Steatosis in Humans Is Associated with Hepatic Fibrosis and Occurs Predominately in Acinar Zone 3. J. Gastroenterol. Hepatol. 2001, 16, 599–606. [Google Scholar] [CrossRef]
- Tanwar, S.; Rhodes, F.; Srivastava, A.; Trembling, P.M.; Rosenberg, W.M. Inflammation and Fibrosis in Chronic Liver Diseases Including Non-Alcoholic Fatty Liver Disease and Hepatitis C. World J. Gastroenterol. 2020, 26, 109–133. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate Tracing Reveals Hepatic Stellate Cells as Dominant Contributors to Liver Fibrosis Independent of Its Aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef]
- Lee, K.S.; Buck, M.; Houglum, K.; Chojkier, M. Activation of Hepatic Stellate Cells by TGF Alpha and Collagen Type I Is Mediated by Oxidative Stress through C-Myb Expression. J. Clin. Investig. 1995, 96, 2461–2468. [Google Scholar] [CrossRef]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic Recruitment of the Inflammatory Gr1+ Monocyte Subset upon Liver Injury Promotes Hepatic Fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Pulli, B.; Ali, M.; Iwamoto, Y.; Zeller, M.W.G.; Schob, S.; Linnoila, J.J.; Chen, J.W. Myeloperoxidase-Hepatocyte-Stellate Cell Cross Talk Promotes Hepatocyte Injury and Fibrosis in Experimental Nonalcoholic Steatohepatitis. Antioxid. Redox Signal. 2015, 23, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Dorokhov, Y.L.; Shindyapina, A.V.; Sheshukova, E.V.; Komarova, T.V. Metabolic Methanol: Molecular Pathways and Physiological Roles. Physiol. Rev. 2015, 95, 603–644. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef]
- Alegre, F.; Pelegrin, P.; Feldstein, A.E. Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 2017, 37, 119–127. [Google Scholar] [CrossRef]
- Licandro, G.; Ling Khor, H.; Beretta, O.; Lai, J.; Derks, H.; Laudisi, F.; Conforti-Andreoni, C.; Liang Qian, H.; Teng, G.G.; Ricciardi-Castagnoli, P.; et al. The NLRP3 Inflammasome Affects DNA Damage Responses after Oxidative and Genotoxic Stress in Dendritic Cells. Eur. J. Immunol. 2013, 43, 2126–2137. [Google Scholar] [CrossRef]
- Collison, K.S.; Makhoul, N.J.; Zaidi, M.Z.; Al-Rabiah, R.; Inglis, A.; Andres, B.L.; Ubungen, R.; Shoukri, M.; Al-Mohanna, F.A. Interactive Effects of Neonatal Exposure to Monosodium Glutamate and Aspartame on Glucose Homeostasis. Nutr. Metab. (Lond) 2012, 9, 58. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere Dysfunction Induces Metabolic and Mitochondrial Compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef]
- Palmnäs, M.S.A.; Cowan, T.E.; Bomhof, M.R.; Su, J.; Reimer, R.A.; Vogel, H.J.; Hittel, D.S.; Shearer, J. Low-Dose Aspartame Consumption Differentially Affects Gut Microbiota-Host Metabolic Interactions in the Diet-Induced Obese Rat. PLoS ONE 2014, 9, e109841. [Google Scholar]
- Prokic, M.D.; Paunovic, M.G.; Matic, M.M.; Djordjevic, N.Z.; Ognjanovic, B.I.; Stajn, A.S.; Saicic, Z.S. Prooxidative Effects of Aspartame on Antioxidant Defense Status in Erythrocytes of Rats. J. Biosci. 2014, 39, 859–866. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene (mm) | Direct/Reverse Oligonucleotide |
|---|---|
| Acta2 (Gene ID: 11475) | F→ TTCATTGGGATGGAGTCAGC R→ CTTCTGCATCCTGTCAGCAA |
| Col1a1 (Gene ID: 12842) | F→ GCCAAGAAGACATCCCTGAA R→ GGTTGGGACAGTCCAGTTCT |
| G6pc (Gene ID: 14377) | F→ CAACGTATGGATTCCGGTGT R→ GAAAGTTTCAGCCACAGCAA |
| Gk (Gene ID: 14933) | F→ GTTGGAAGGTTCCGTGGCTA R→ AACCCTGAAAATGCTGGAAC |
| Hmox1 (Gene ID: 15368) | F→ AAGAGGCTAAGACCGCCTTC R→ GCCACATTGGACAGAGTTCA |
| Nqo1 (Gene ID: 18104) | F→ TTCTCTGGCCGATTCAGAGT R→ GGCTGCTTGGAGCAAAATA |
| Pck1 (Gene ID: 18534) | F→ CCAAGGCAACTTAAGGGCTA R→ TAAACACCCCCATCGCTAGT |
| Ppargc1a (Gene ID: 19017) | F→ TTAAAGTTCATGGGGCAAGC R→ TAGGAATGGCTGAAGGGATG |
| Tbp (Gene ID: 21374) | F→CAGCCTTCCACCTTATGCTC R→ CCGTAAGGCATCATTGGACT |
| Tgfb1 (Gene ID: 21803) | F→ TTGCTTCAGCTCCACAGAGA R→ TGGTTGTAGAGGGCAAGGAC |
| Target Gene (mm) | TaqMan Probe |
|---|---|
| Cxcl1 (Gene ID: 14825) | Mm04207460_m1 |
| Il1b (Gene ID: 16176) | Mm00434228_m1 |
| Il6 (Gene ID: 16193) | Mm00446190_m1 |
| Il10 (Gene ID: 16153) | Mm01288386_m1 |
| Il18 (Gene ID: 16173) | Mm00434226_m1 |
| Tbp (Gene ID: 21374) | Mm01277042_m1 |
| Control | Aspartame | |
|---|---|---|
| Total Cholesterol (mg/dL) | 117.0 ± 16.4 | 153.0 ± 22.8 * |
| HDL cholesterol (mg/dL) | 37.0 ± 11.8 | 25.5 ± 12.1 * |
| LDL cholesterol (mg/dL) | 47.6 ± 22.9 | 84.2 ± 21.5 * |
| Triglycerides (mg/dL) | 120.0 ± 29.0 | 191.5 ± 27.0 * |
| Total lipids (mg/g protein) | 8.55 ± 3.59 | 15.02 ± 2.22 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finamor, I.A.; Bressan, C.A.; Torres-Cuevas, I.; Rius-Pérez, S.; da Veiga, M.; Rocha, M.I.; Pavanato, M.A.; Pérez, S. Long-Term Aspartame Administration Leads to Fibrosis, Inflammasome Activation, and Gluconeogenesis Impairment in the Liver of Mice. Biology 2021, 10, 82. https://doi.org/10.3390/biology10020082
Finamor IA, Bressan CA, Torres-Cuevas I, Rius-Pérez S, da Veiga M, Rocha MI, Pavanato MA, Pérez S. Long-Term Aspartame Administration Leads to Fibrosis, Inflammasome Activation, and Gluconeogenesis Impairment in the Liver of Mice. Biology. 2021; 10(2):82. https://doi.org/10.3390/biology10020082
Chicago/Turabian StyleFinamor, Isabela A., Caroline A. Bressan, Isabel Torres-Cuevas, Sergio Rius-Pérez, Marcelo da Veiga, Maria I. Rocha, Maria A. Pavanato, and Salvador Pérez. 2021. "Long-Term Aspartame Administration Leads to Fibrosis, Inflammasome Activation, and Gluconeogenesis Impairment in the Liver of Mice" Biology 10, no. 2: 82. https://doi.org/10.3390/biology10020082
APA StyleFinamor, I. A., Bressan, C. A., Torres-Cuevas, I., Rius-Pérez, S., da Veiga, M., Rocha, M. I., Pavanato, M. A., & Pérez, S. (2021). Long-Term Aspartame Administration Leads to Fibrosis, Inflammasome Activation, and Gluconeogenesis Impairment in the Liver of Mice. Biology, 10(2), 82. https://doi.org/10.3390/biology10020082