Formyl Peptide Receptor 1 Signaling in Acute Inflammation and Neural Differentiation Induced by Traumatic Brain Injury

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  , , ,
, , ,  ,
,  ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
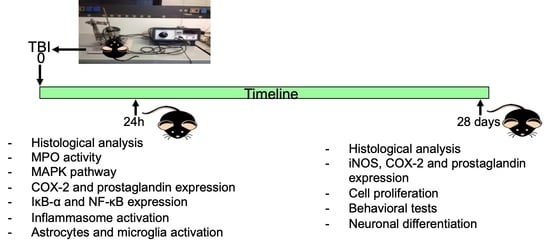
2.2. Induction of Experimental Traumatic Brain Injury TBI
2.3. Experimental Groups
- TBI WT group: mice were subjected to CCI as described above.
- TBI Fpr1 KO group: Fpr1 KO mice were subjected to CCI as well as the WT group.
- Sham WT group: Mice were subjected to the surgical procedures as per the above group (anesthesia and craniotomy) except that the impact tip was not applied.
- Sham Fpr1 KO group: Mice were subjected to the surgical procedures as per the above group (anesthesia and craniotomy) except that the impact tip was not applied.
- Exp 1—to investigate the early stage of acute inflammation, animals were sacrificed at 24 h after TBI.
- Exp 2—to investigate the neurogenesis, animals were sacrificed four weeks after the injury.
2.4. Open Field
2.5. Social Interaction Test
2.6. Novel Object Recognition Test
2.7. Morris Water Maze Test
2.8. Histological Examination
2.9. Assessment of Lesion Volume
2.10. Myeloperoxidase Activity
2.11. Western Blot Analysis
2.12. Bromodeoxyuridine (BrdU) Treatment
2.13. Immunohistochemical Analysis
2.14. ELISA Analysis of IL-1 β and IL-18
2.15. Materials
2.16. Statistical Evaluation
3. Results
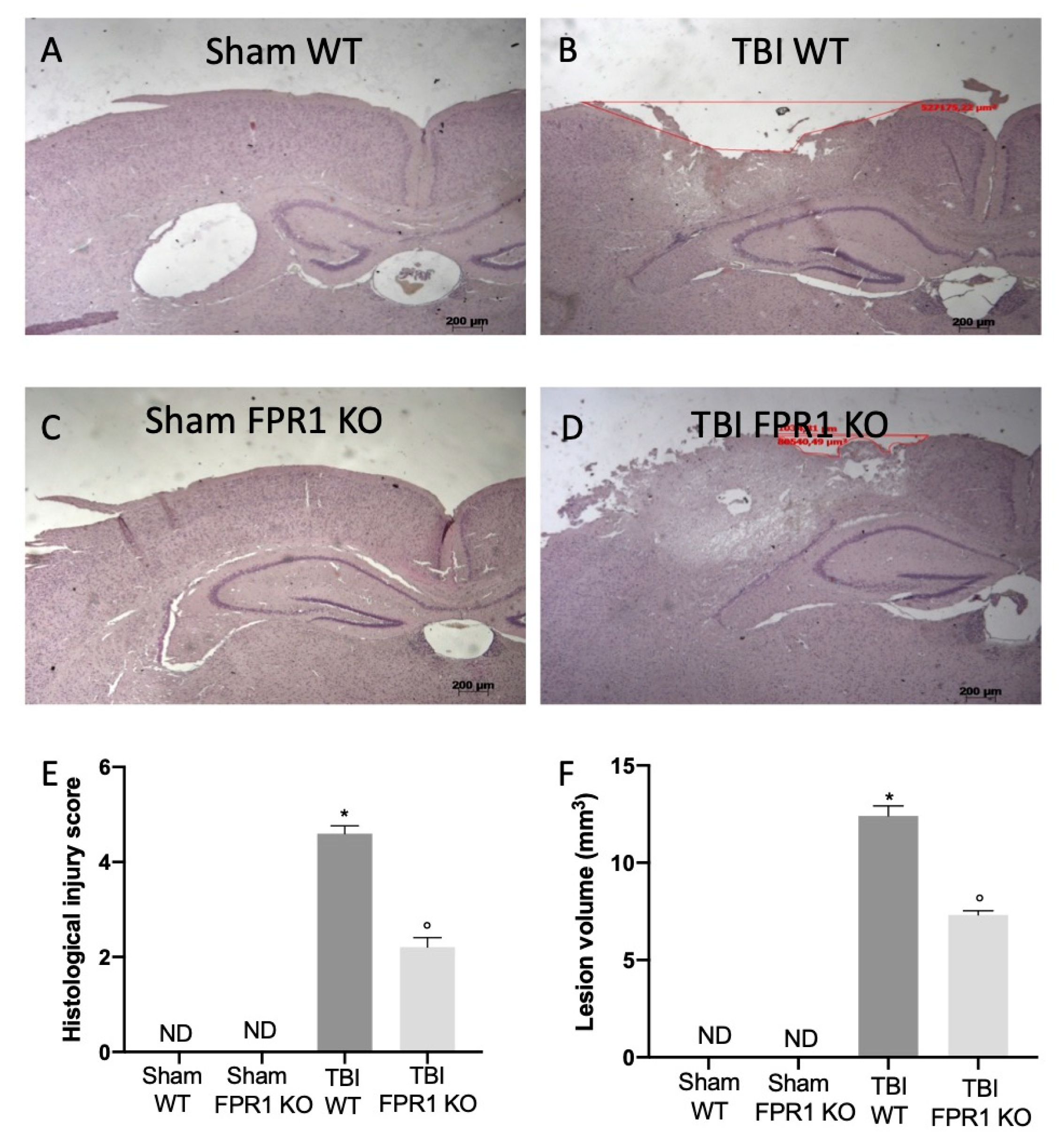
3.1. Effect of Absence of Fpr1 on Severity of Tissue Damage 24 h Following Traumatic Brain Injury
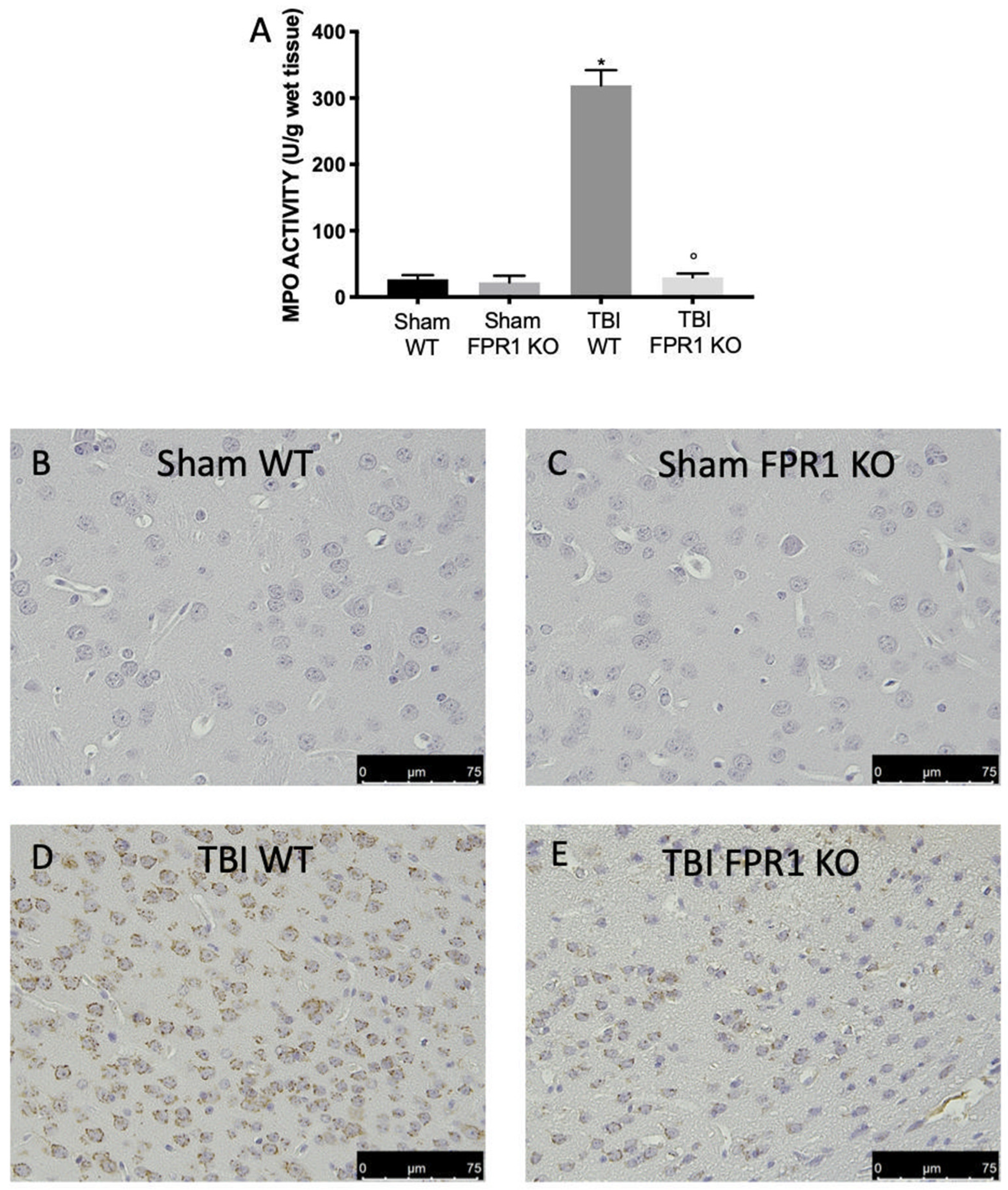
3.2. Effect of Absence of Fpr1 on MPO Activity 24 h Following Traumatic Brain Injury
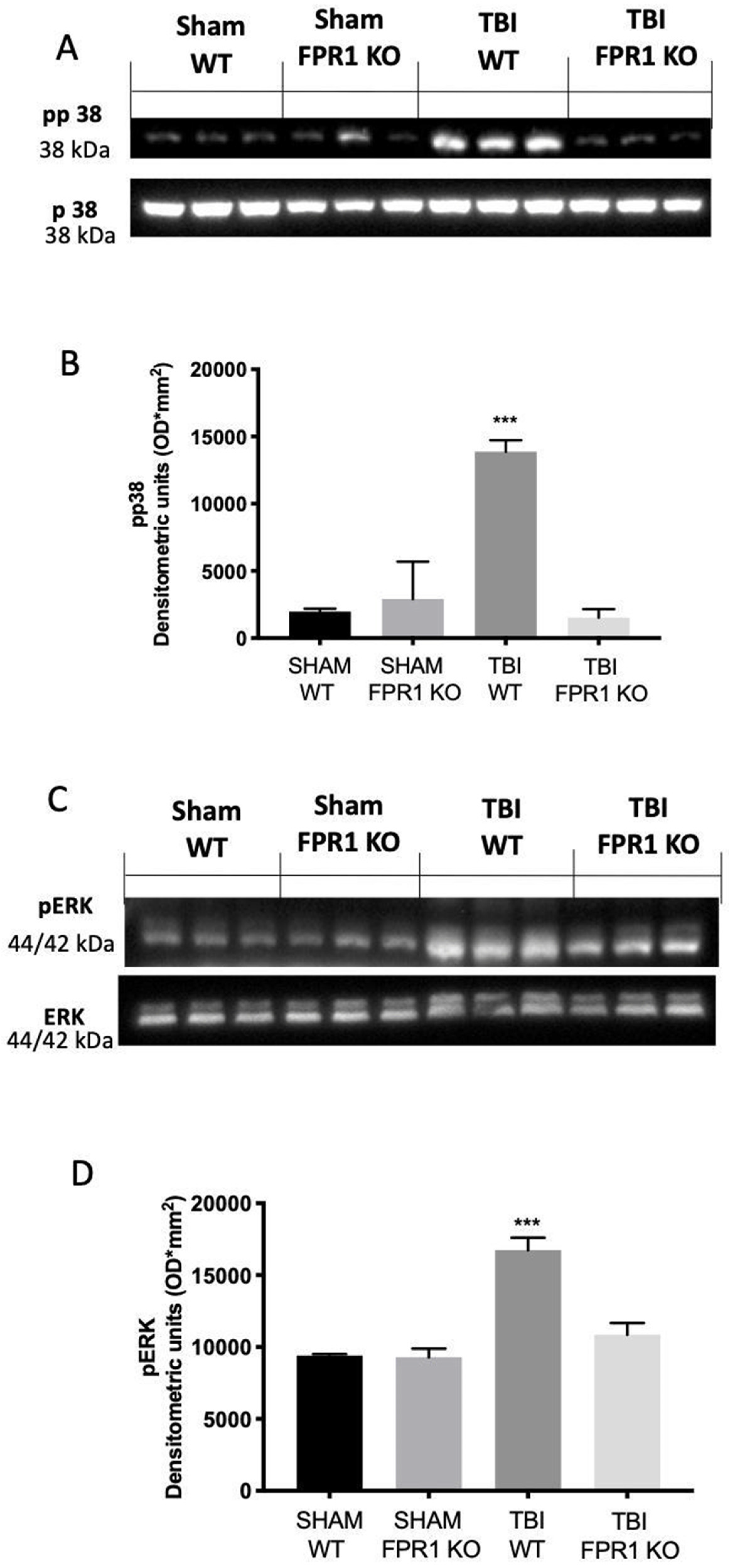
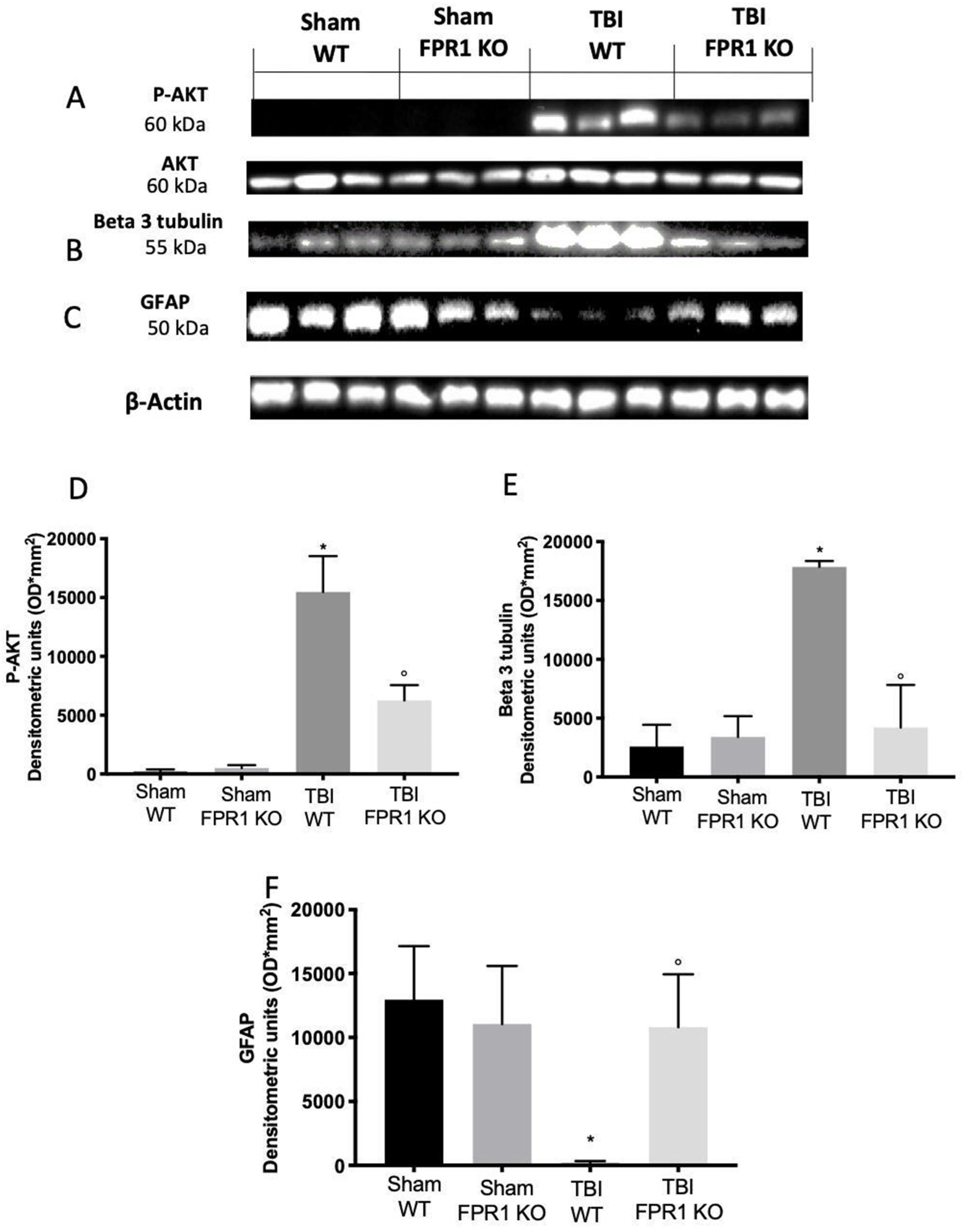
3.3. Effect of Absence of Fpr1 on MAPK Pathway 24 h Following Traumatic Brain Injury
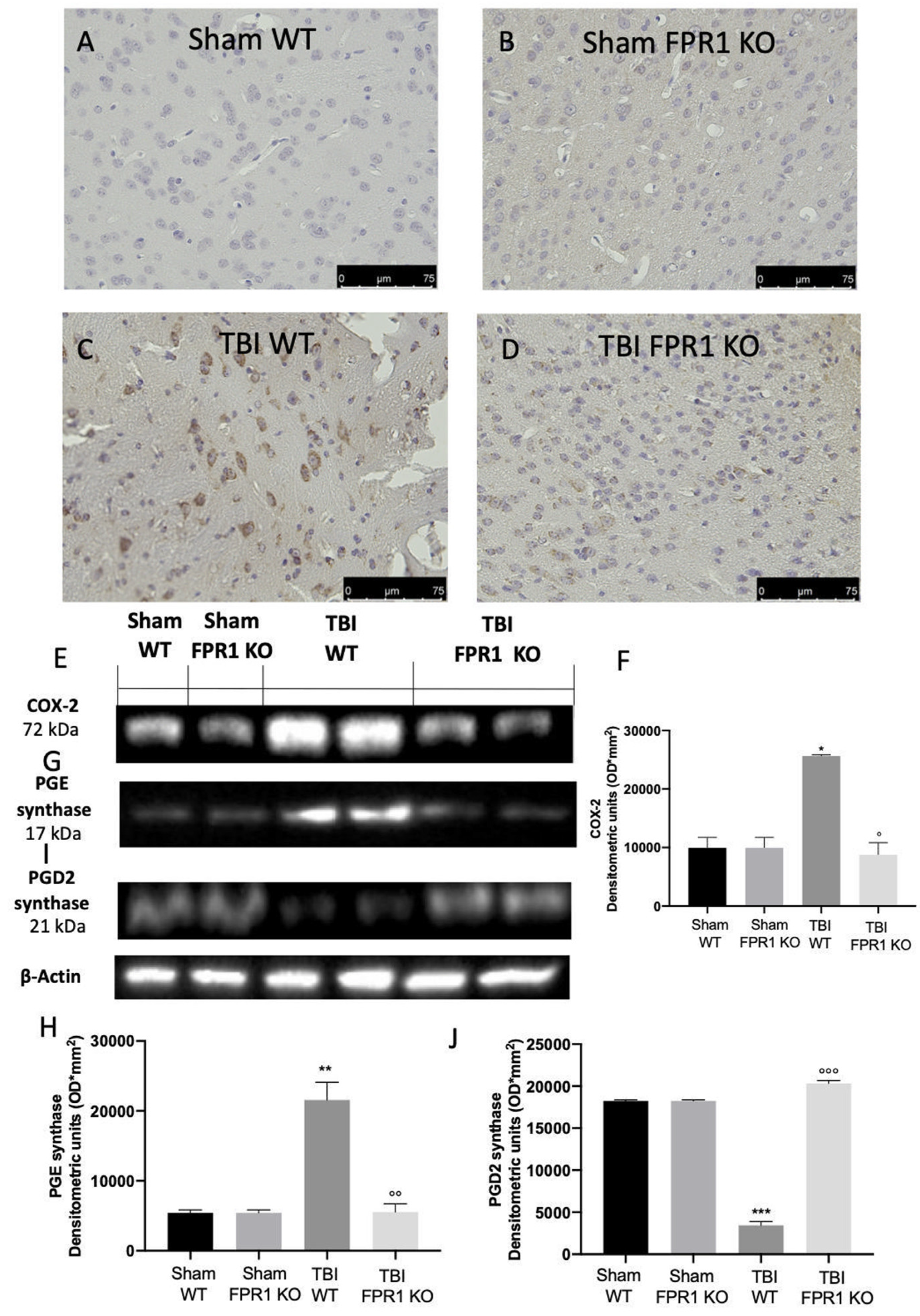
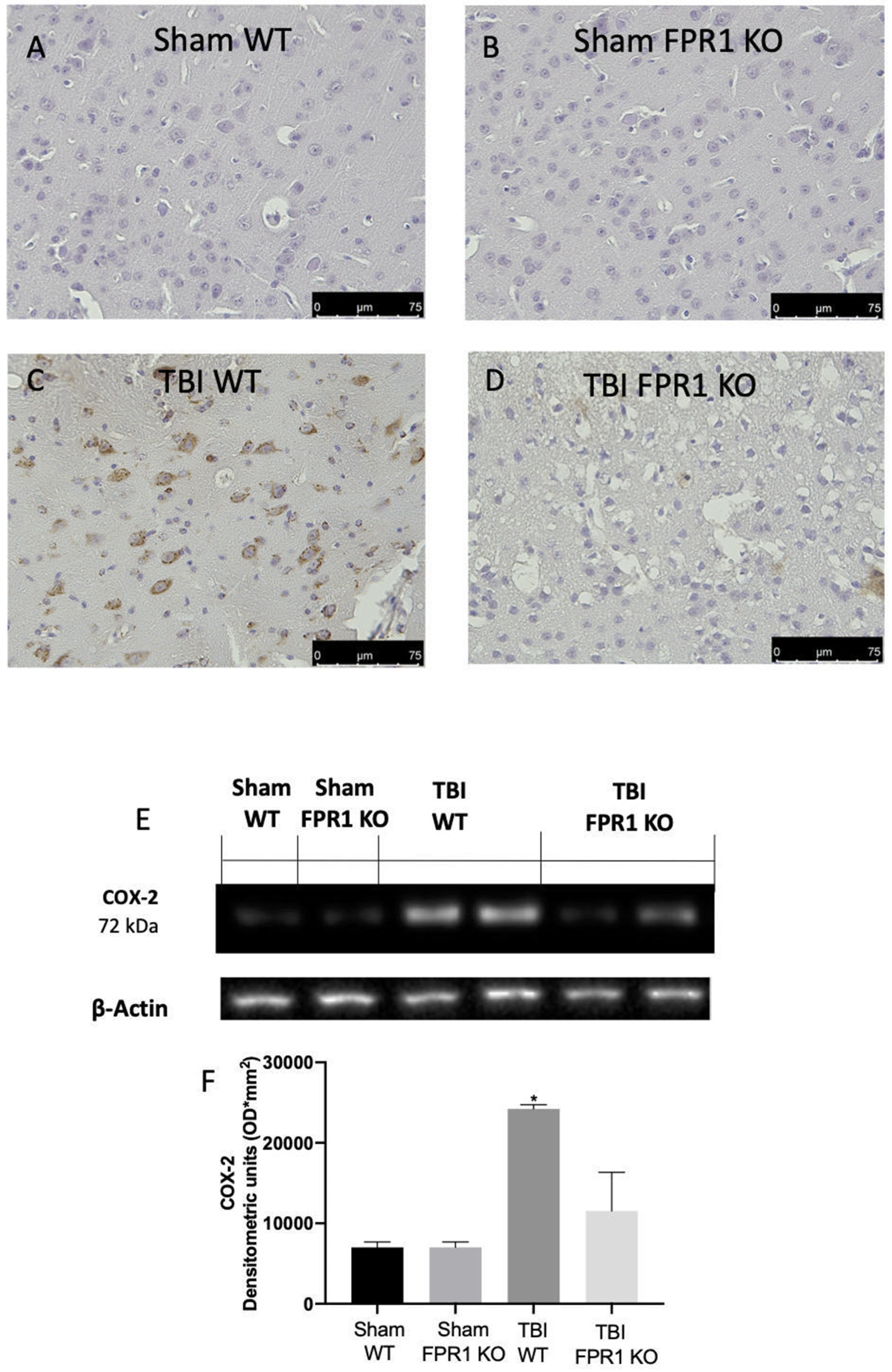
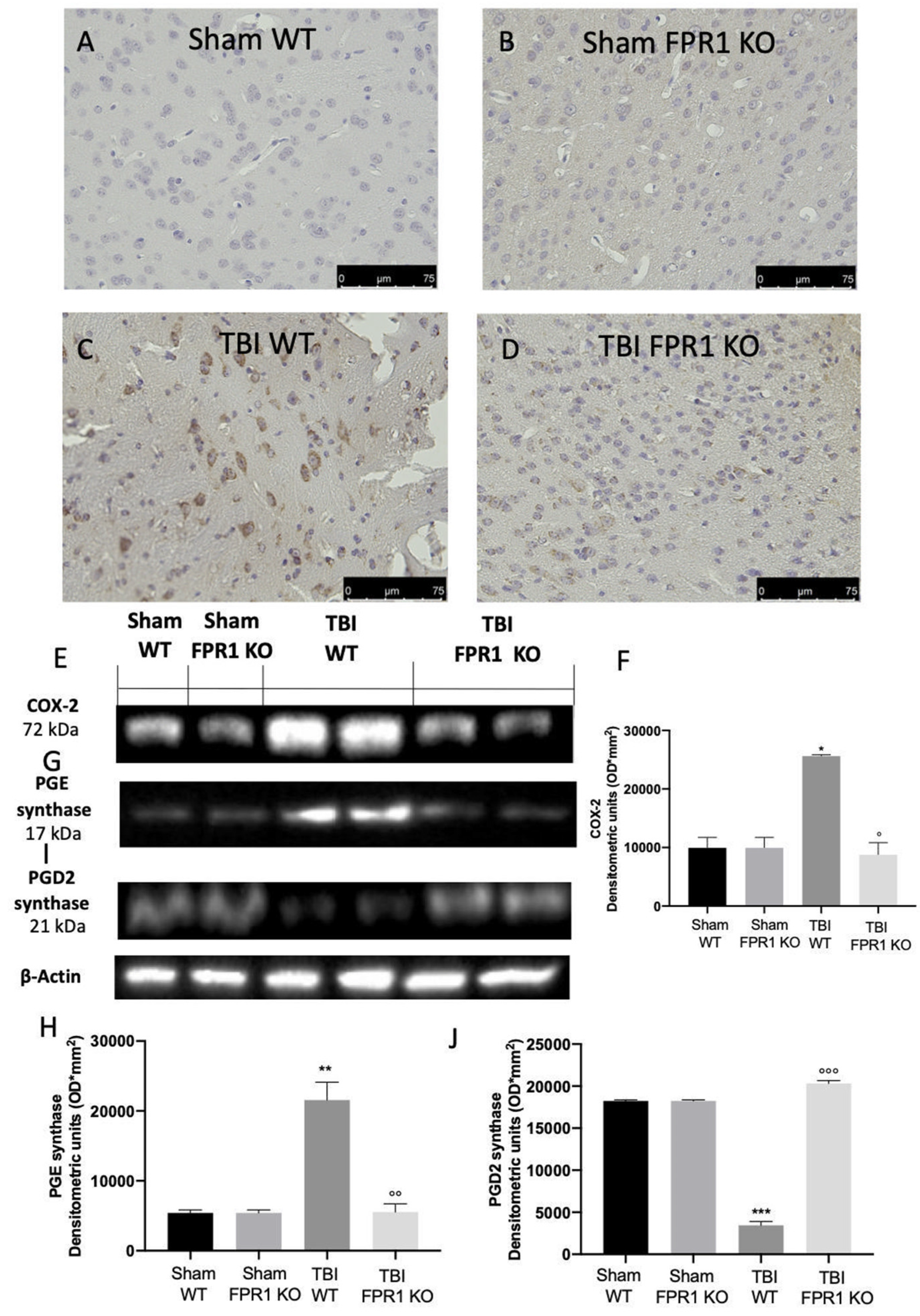
3.4. Effect of Absence of Fpr1 on COX-2 and Prostaglandin Expression 24 h Following Traumatic Brain Injury
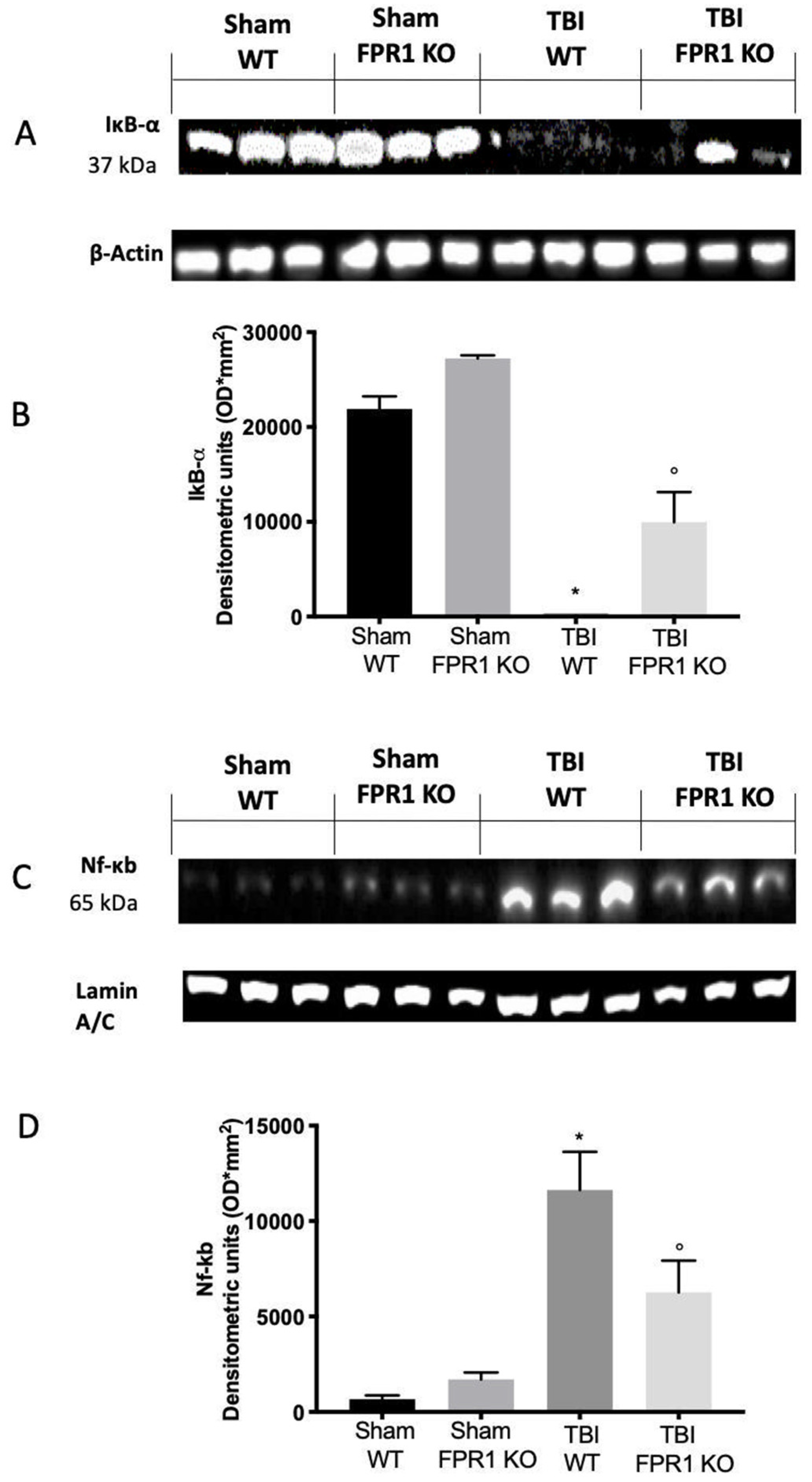
3.5. Effect of Absence of Fpr1 on IκB-α and NF-κB Expression 24 h after Traumatic Brain Injury
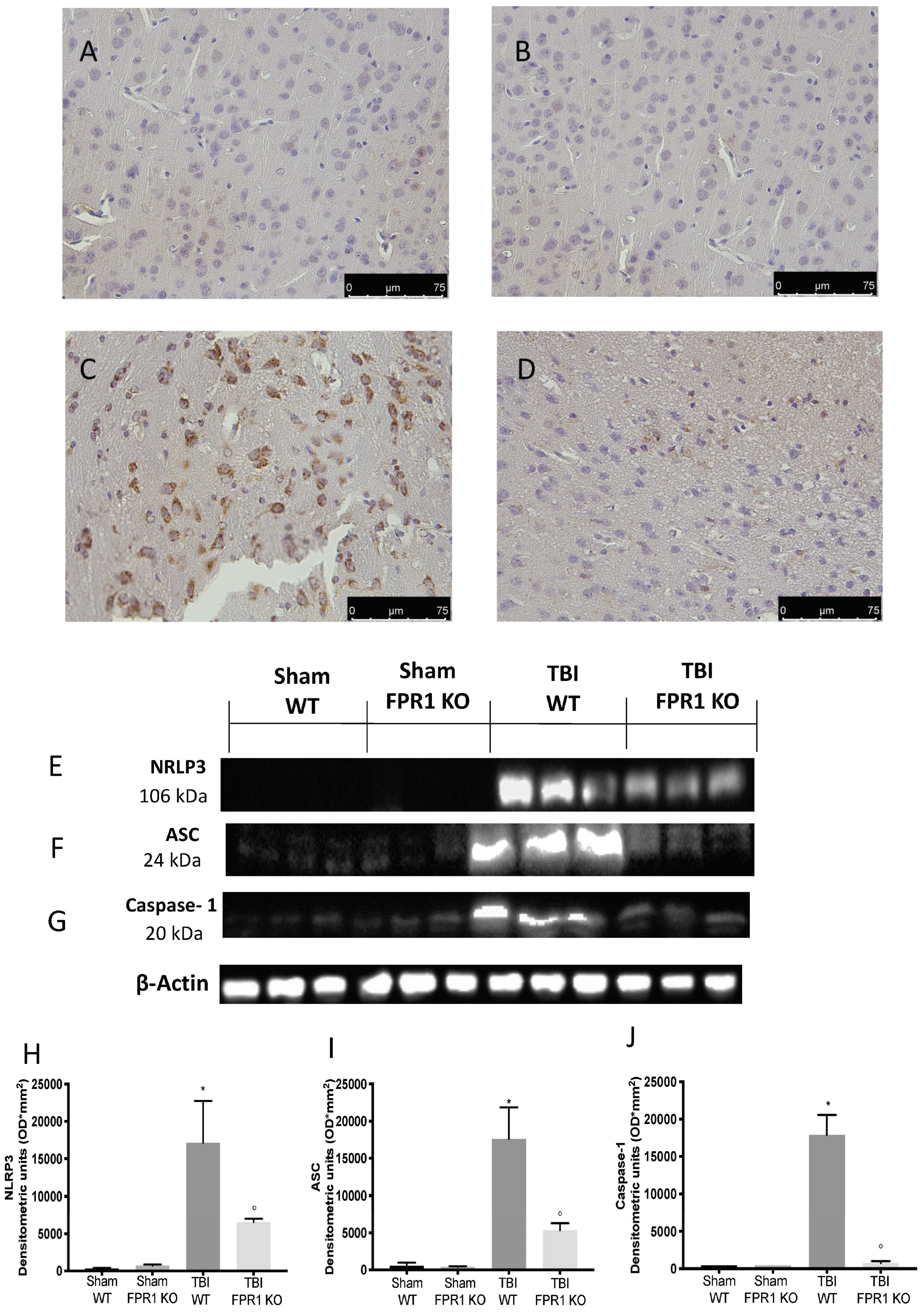
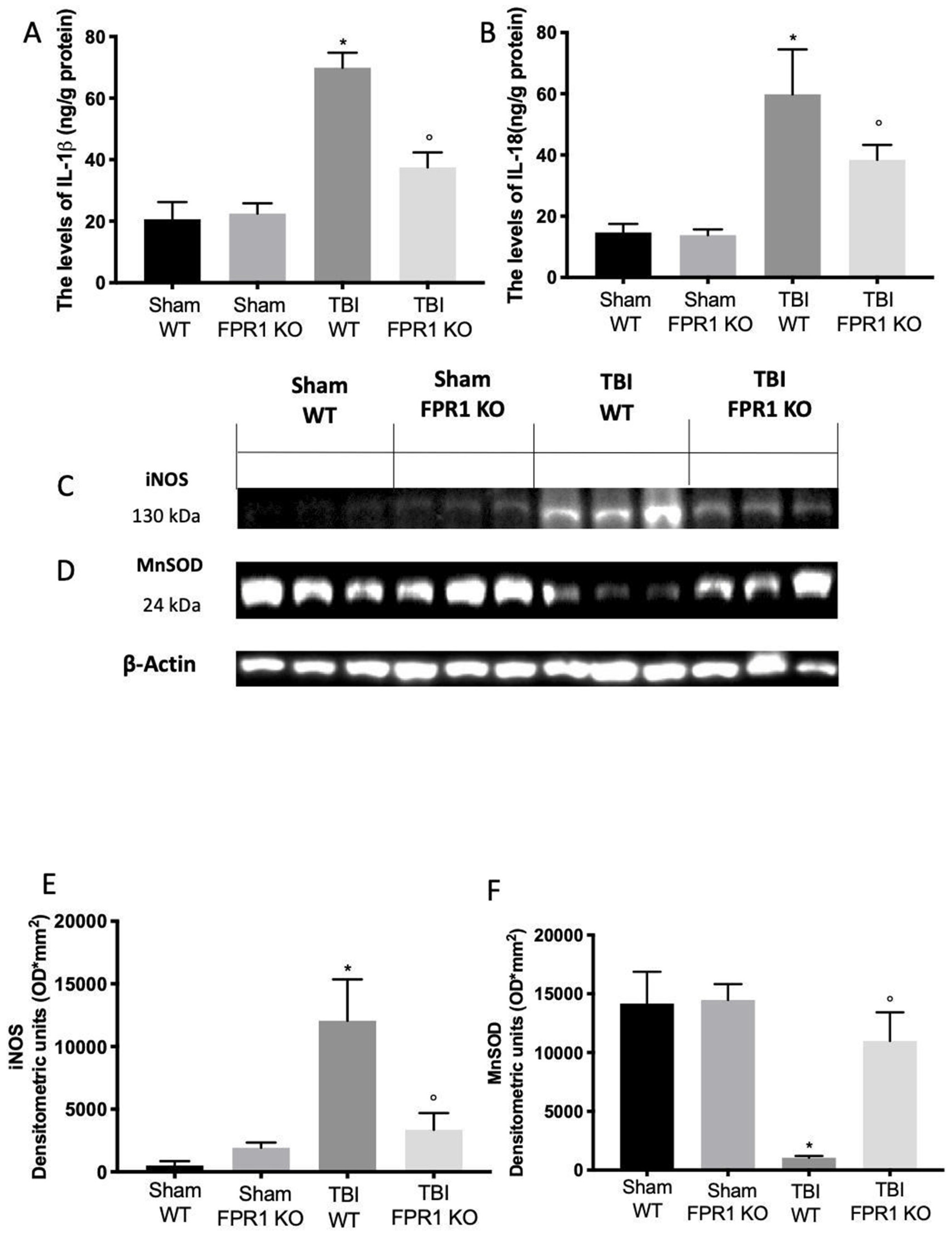
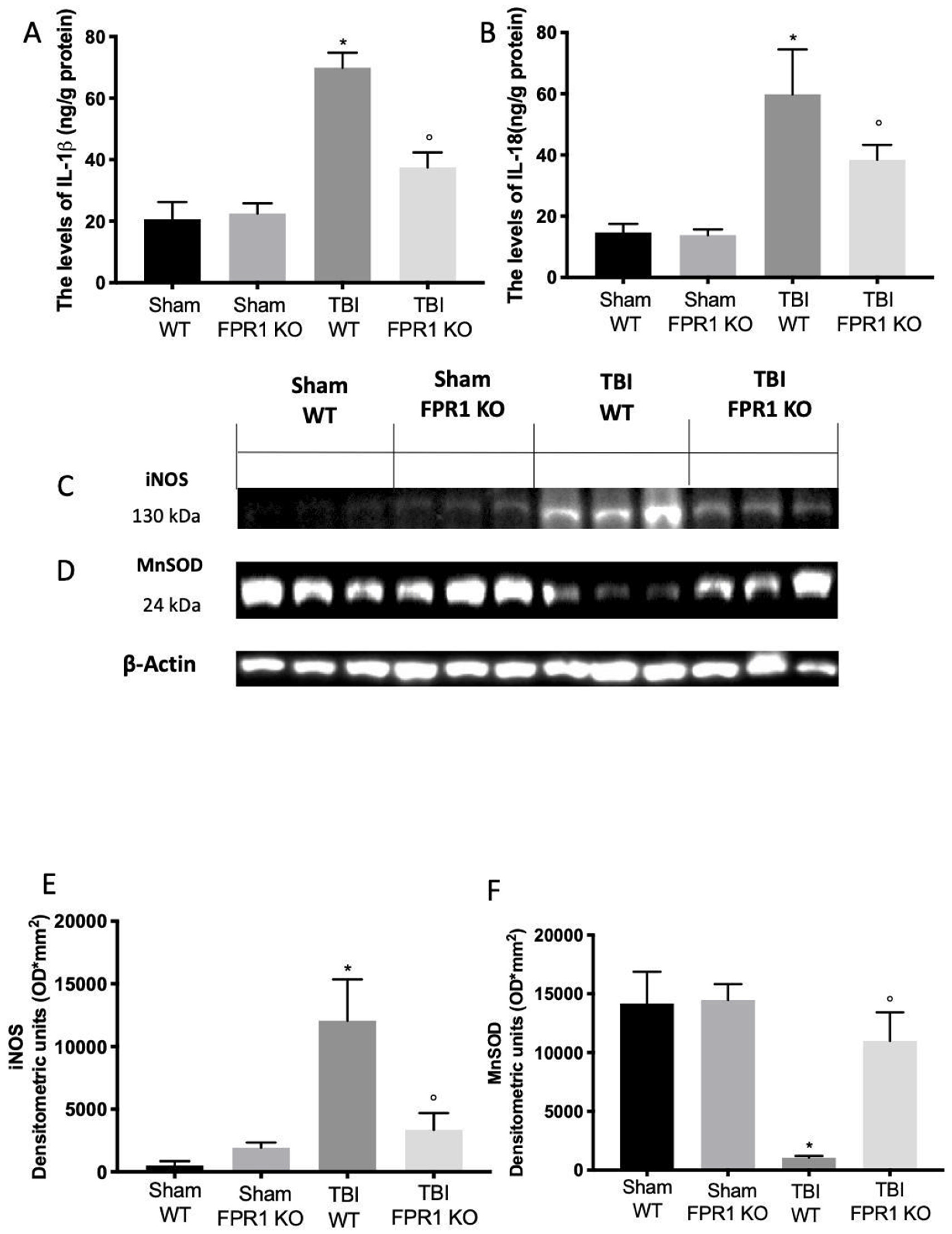
3.6. Effect of Absence of Fpr1 on Inflammasome Components 24 h after Traumatic Brain Injury
3.7. Effect of Absence of Fpr1 on Oxidative Stress Activation 24 h after Traumatic Brain Injury
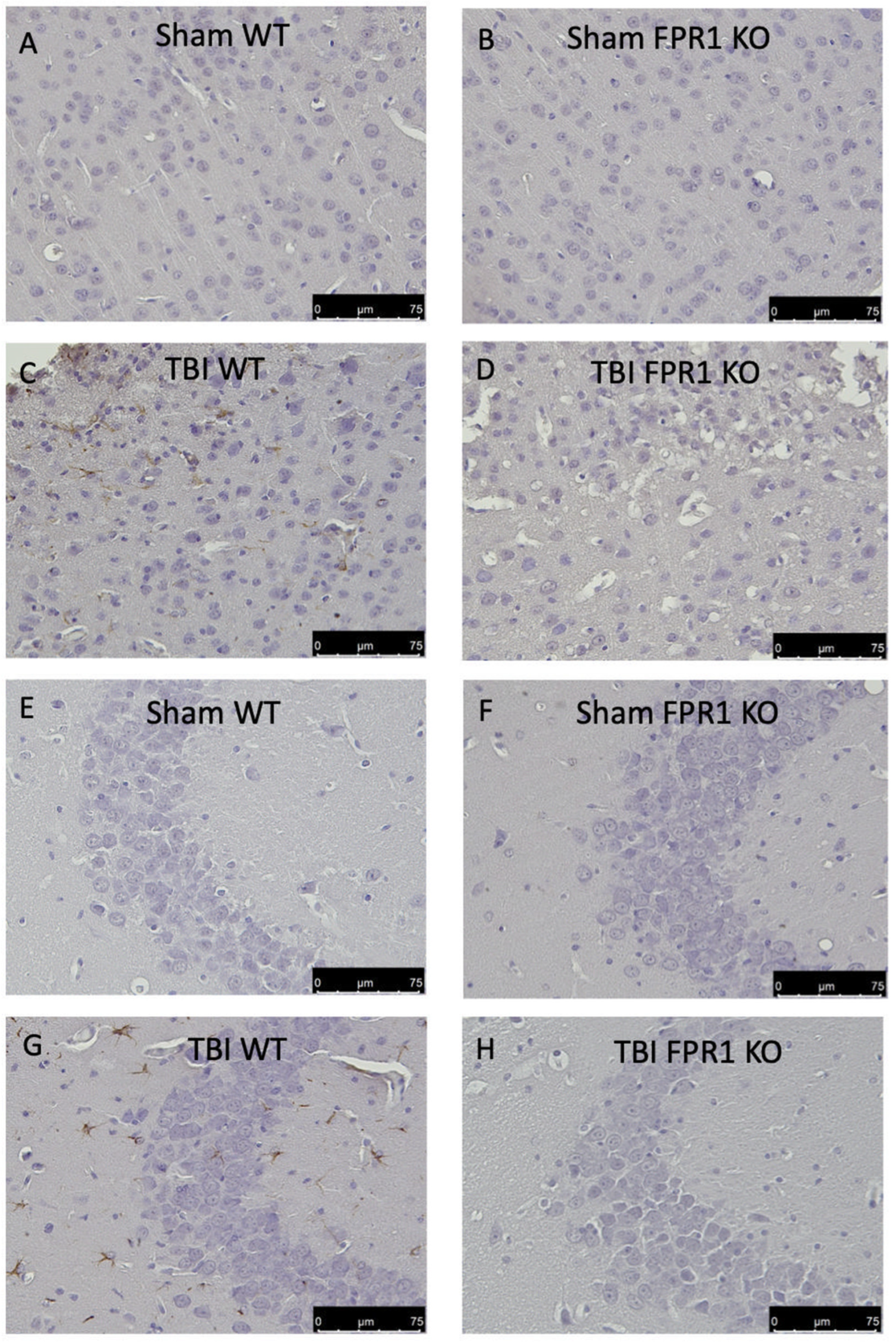
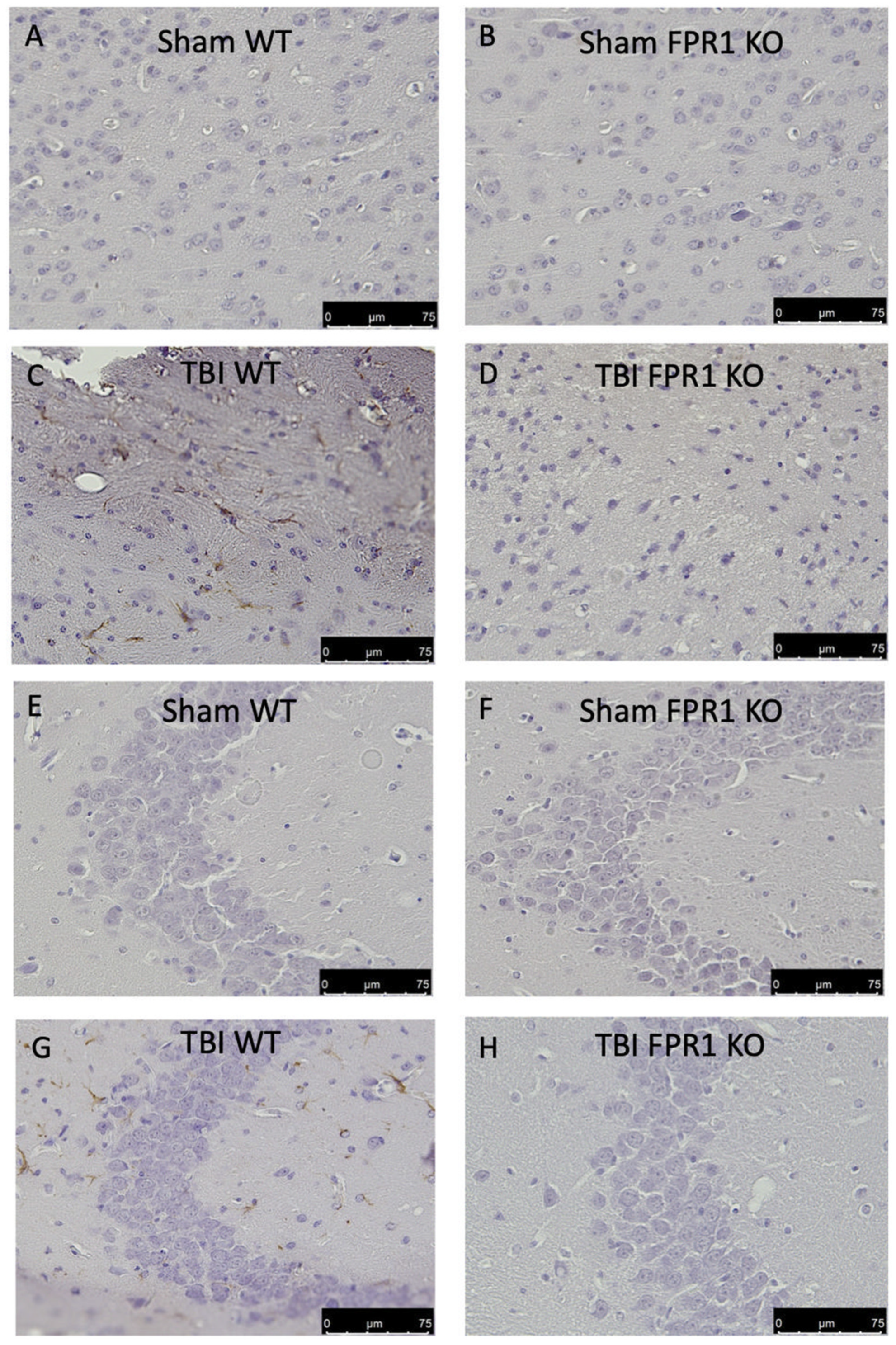
3.8. Effect of Absence of Fpr1 on Astrocytes Activation 24 h after Traumatic Brain Injury
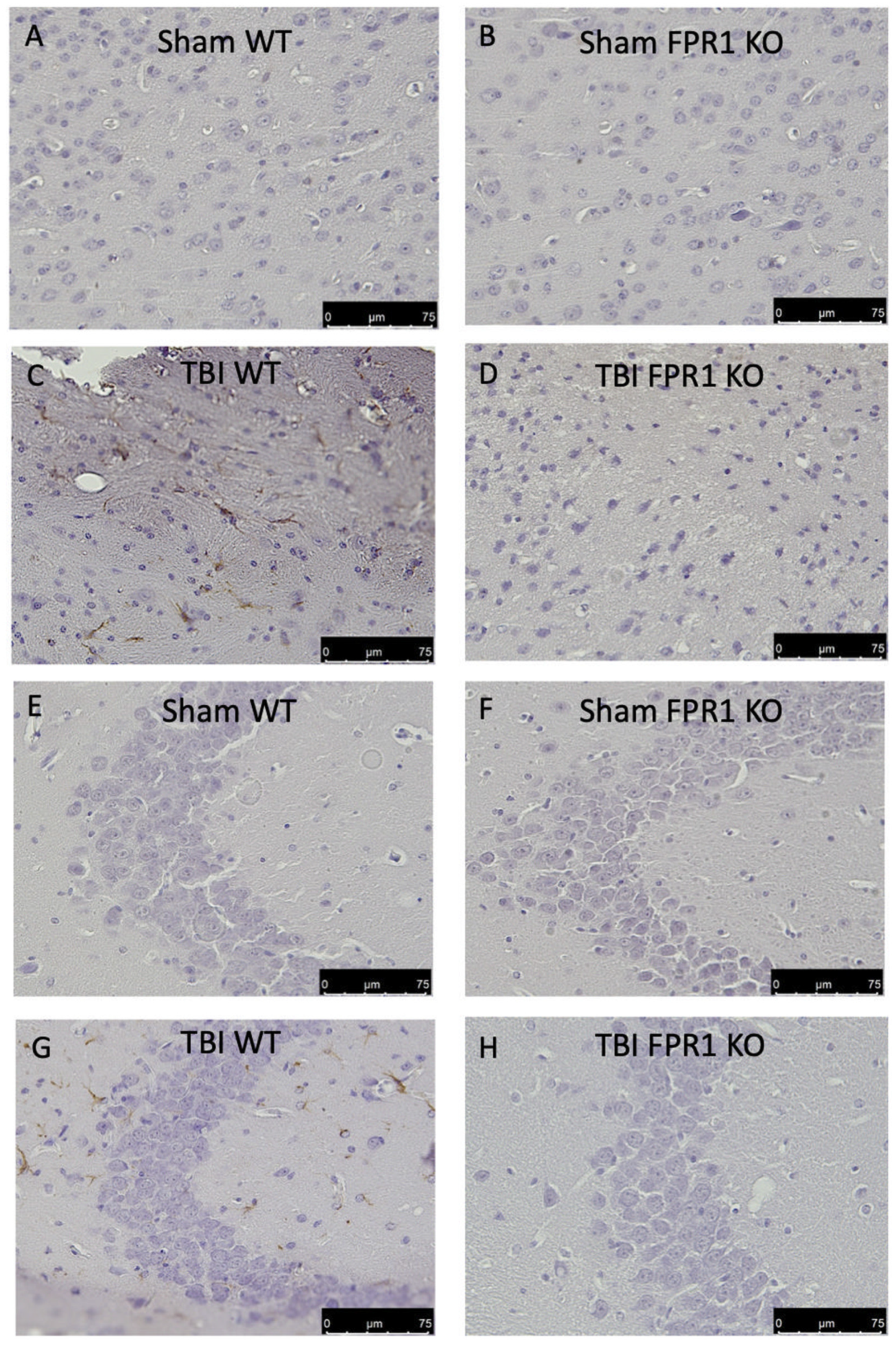
3.9. Effect of Absence of Fpr1 on Microglia Activation 24 h after Traumatic Brain Injury
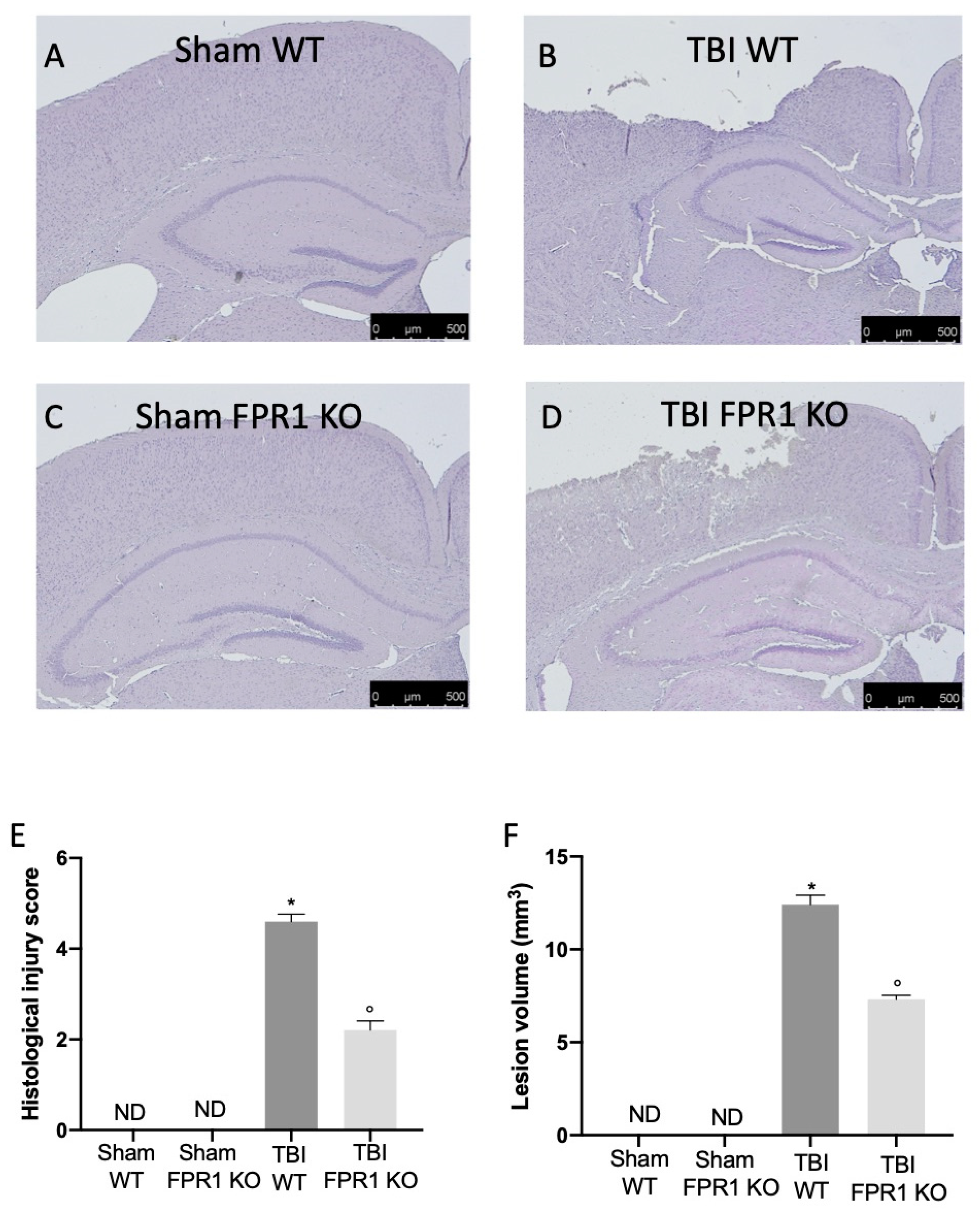
3.10. Effect of Absence of Fpr1 on Severity of Tissue Damage Four Weeks Following Traumatic Brain Injury
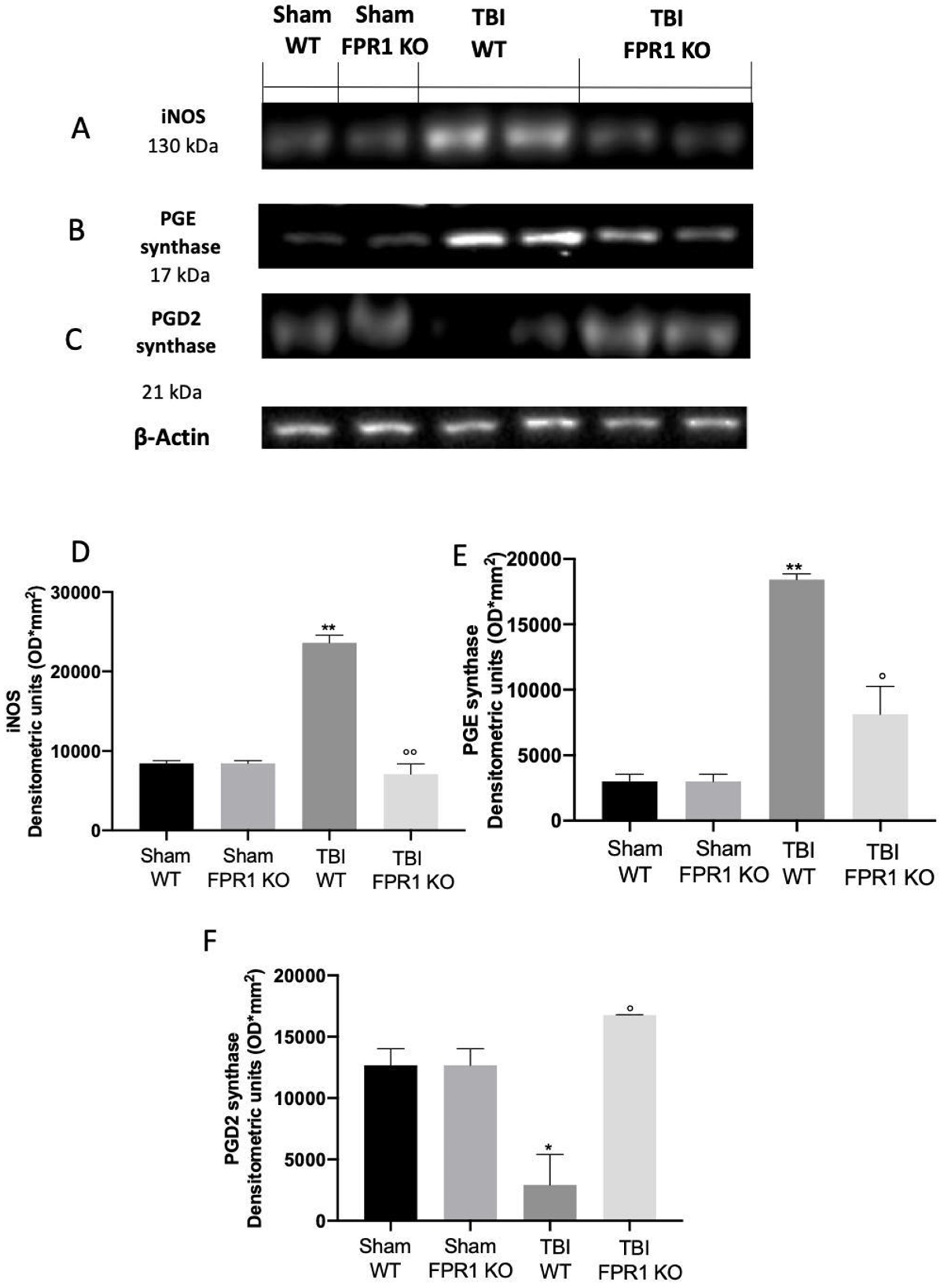
3.11. Effect of Absence of Fpr1 on iNOS, COX-2 and Prostaglandin Expression Four Weeks after Traumatic Brain Injury
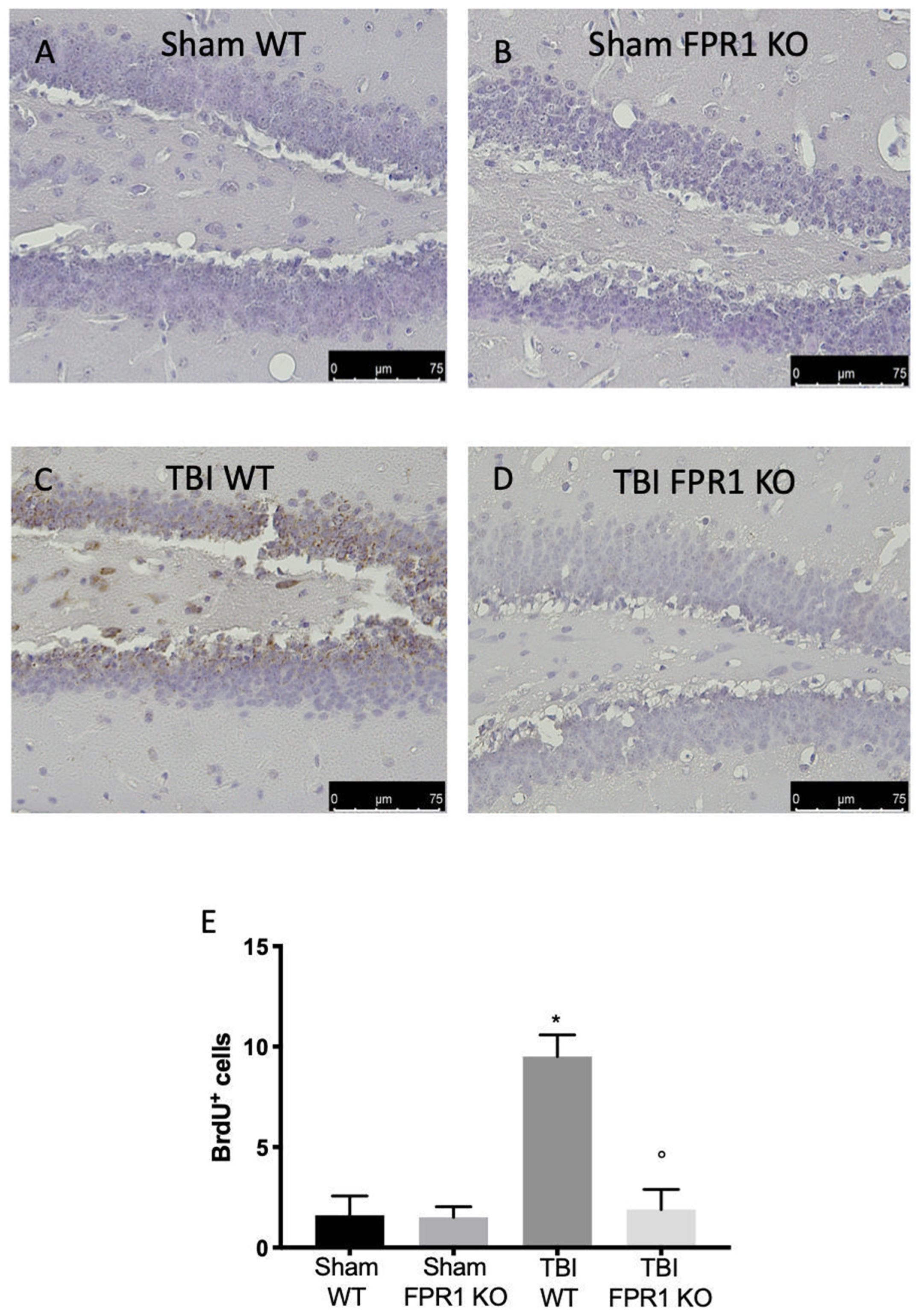
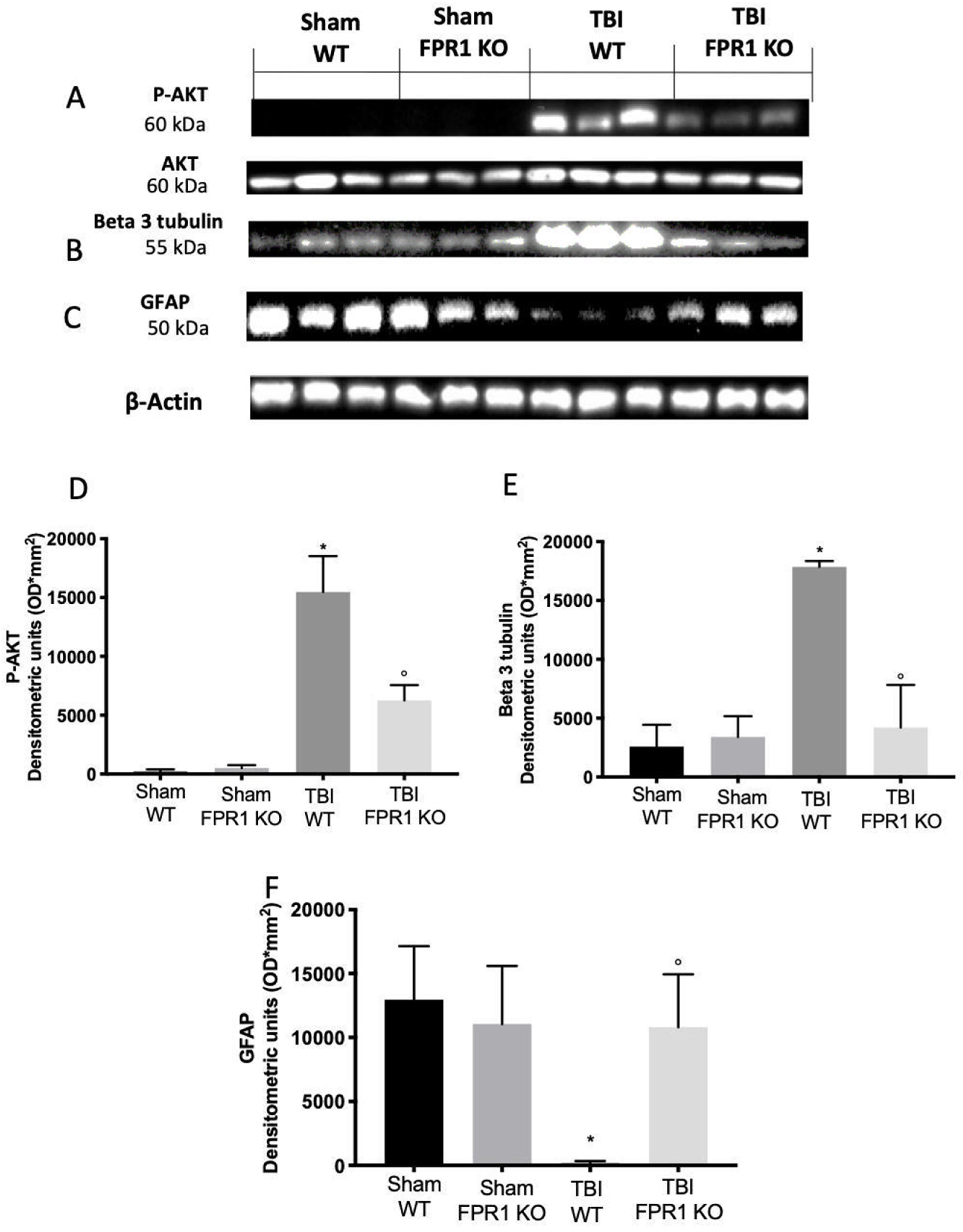
3.12. Effect of Absence of Fpr1 on Cell Proliferation Four Weeks after Traumatic Brain Injury
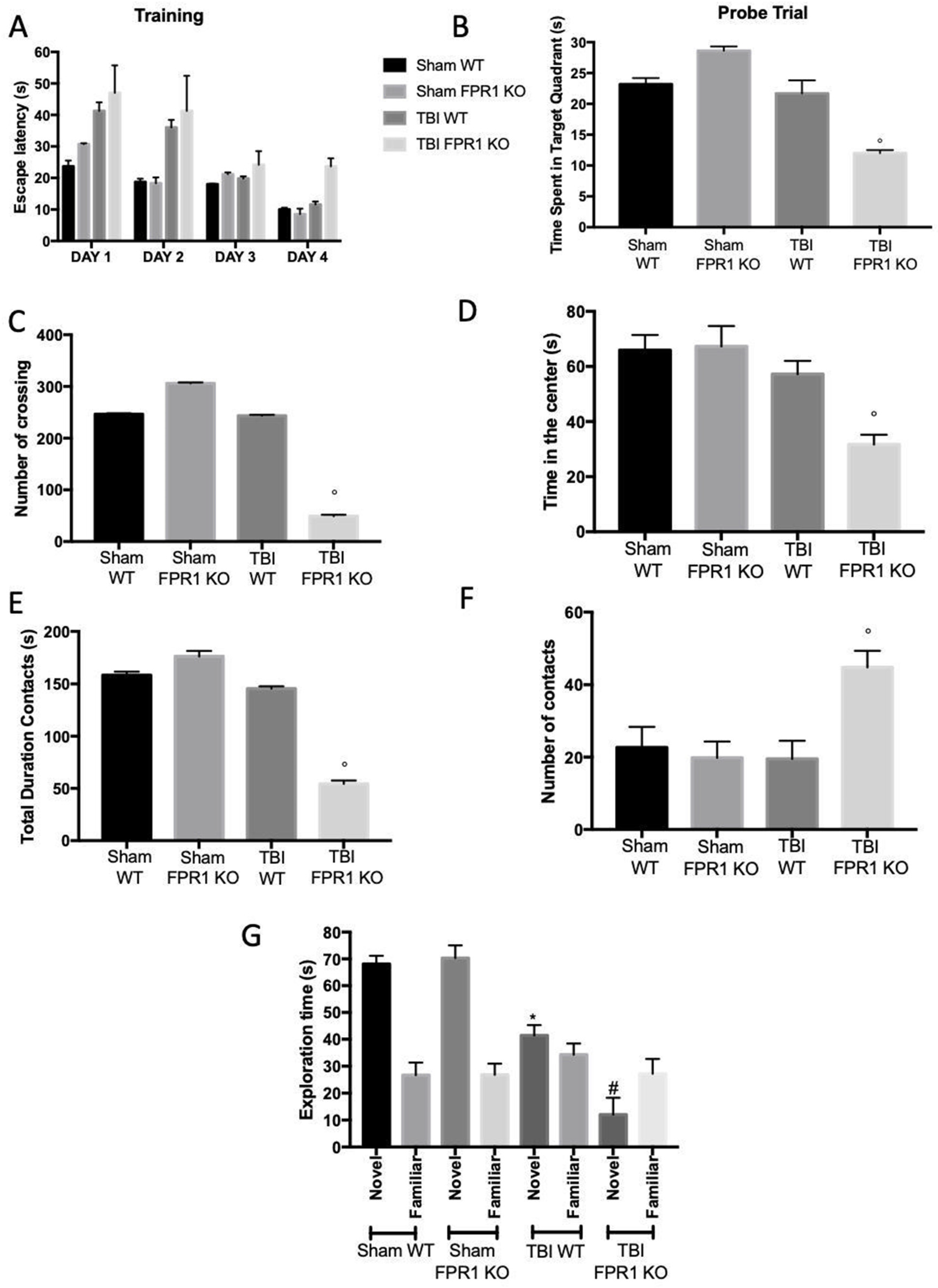
3.13. Effect of Absence of Fpr1 on Behavioral Performance Four Weeks after Traumatic Brain Injury
3.14. Effect of Absence of Fpr1 on Self-Renewal and Neurogenesis Four Weeks after Traumatic Brain Injury
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dahlgren, C.; Gabl, M.; Holdfeldt, A.; Winther, M.; Forsman, H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem. Pharmacol. 2016, 114, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef] [PubMed]
- Spurr, L.; Nadkarni, S.; Pederzoli-Ribeil, M.; Goulding, N.J.; Perretti, M.; D’Acquisto, F. Comparative analysis of Annexin A1-formyl peptide receptor 2/ALX expression in human leukocyte subsets. Int. Immunopharmacol. 2011, 11, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Le, Y.; Wang, J.M.; Oppenheim, J.J. Differential regulation of formyl peptide receptor-like 1 expression during the differentiation of monocytes to dendritic cells and macrophages. J. Immunol. 2001, 166, 4092–4098. [Google Scholar] [CrossRef] [Green Version]
- El Kebir, D.; Jozsef, L.; Filep, J.G. Opposing regulation of neutrophil apoptosis through the formyl peptide receptor-like 1/lipoxin A4 receptor: Implications for resolution of inflammation. J. Leukoc. Biol. 2008, 84, 600–606. [Google Scholar] [CrossRef]
- Yang, D.; Chen, Q.; Gertz, B.; He, R.; Phulsuksombati, M.; Ye, R.D.; Oppenheim, J.J. Human dendritic cells express functional formyl peptide receptor-like-2 (FPRL2) throughout maturation. J. Leukoc. Biol. 2002, 72, 598–607. [Google Scholar]
- Gemperle, C.; Schmid, M.; Herova, M.; Marti-Jaun, J.; Wuest, S.J.; Loretz, C.; Hersberger, M. Regulation of the formyl peptide receptor 1 (FPR1) gene in primary human macrophages. PLoS ONE 2012, 7, e50195. [Google Scholar] [CrossRef] [Green Version]
- Keitoku, M.; Kohzuki, M.; Katoh, H.; Funakoshi, M.; Suzuki, S.; Takeuchi, M.; Karibe, A.; Horiguchi, S.; Watanabe, J.; Satoh, S.; et al. FMLP actions and its binding sites in isolated human coronary arteries. J. Mol. Cell Cardiol. 1997, 29, 881–894. [Google Scholar] [CrossRef]
- Heo, S.C.; Kwon, Y.W.; Jang, I.H.; Jeong, G.O.; Yoon, J.W.; Kim, C.D.; Kwon, S.M.; Bae, Y.S.; Kim, J.H. WKYMVm-induced activation of formyl peptide receptor 2 stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells. Stem Cells 2014, 32, 779–790. [Google Scholar] [CrossRef]
- Anton, P.; O’Connell, J.; O’Connell, D.; Whitaker, L.; O’Sullivan, G.C.; Collins, J.K.; Shanahan, F. Mucosal subepithelial binding sites for the bacterial chemotactic peptide, formyl-methionyl-leucyl-phenylalanine (FMLP). Gut 1998, 42, 374–379. [Google Scholar] [CrossRef] [Green Version]
- Babbin, B.A.; Jesaitis, A.J.; Ivanov, A.I.; Kelly, D.; Laukoetter, M.; Nava, P.; Parkos, C.A.; Nusrat, A. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J. Immunol. 2007, 179, 8112–8121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, E.L.; Forouhar, F.A.; Grunnet, M.L.; Boulay, F.; Tardif, M.; Bormann, B.J.; Sodja, D.; Ye, R.D.; Woska, J.R., Jr.; Murphy, P.M. Broad immunocytochemical localization of the formylpeptide receptor in human organs, tissues, and cells. Cell Tissue Res. 1998, 292, 129–135. [Google Scholar] [CrossRef]
- Chen, K.; Iribarren, P.; Huang, J.; Zhang, L.; Gong, W.; Cho, E.H.; Lockett, S.; Dunlop, N.M.; Wang, J.M. Induction of the formyl peptide receptor 2 in microglia by IFN-gamma and synergy with CD40 ligand. J. Immunol. 2007, 178, 1759–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.H.; Le, Y.; Zhang, X.; Gong, W.; Abe, K.; Sun, R.; Van Damme, J.; Proost, P.; Wang, J.M. Up-regulation of FPR2, a chemotactic receptor for amyloid beta 1–42 (A beta 42), in murine microglial cells by TNF alpha. Neurobiol. Dis. 2002, 10, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.H.; Le, Y.; Gong, W.; Proost, P.; Van Damme, J.; Murphy, W.J.; Wang, J.M. Bacterial lipopolysaccharide selectively up-regulates the function of the chemotactic peptide receptor formyl peptide receptor 2 in murine microglial cells. J. Immunol. 2002, 168, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shi, J.X.; Yin, H.X.; Ma, C.Y.; Zhang, Q.R. The influence of subarachnoid hemorrhage on neurons: An animal model. Ann. Clin. Lab. Sci. 2005, 35, 79–85. [Google Scholar]
- Chesnut, R.M.; Marshall, L.F.; Klauber, M.R.; Blunt, B.A.; Baldwin, N.; Eisenberg, H.M.; Jane, J.A.; Marmarou, A.; Foulkes, M.A. The role of secondary brain injury in determining outcome from severe head injury. J. Trauma 1993, 34, 216–222. [Google Scholar] [CrossRef]
- Polinder, S.; Haagsma, J.A.; van Klaveren, D.; Steyerberg, E.W.; Van Beeck, E.F. Health-related quality of life after TBI: A systematic review of study design, instruments, measurement properties, and outcome. Popul. Health Metr. 2015, 13, 4. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.D.; Sullivan, P.G.; Gibson, T.R.; Pavel, K.M.; Thompson, B.M.; Scheff, S.W. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: More than a focal brain injury. J. Neurotrauma 2005, 22, 252–265. [Google Scholar] [CrossRef]
- Guevara, A.B.; Démonet, J.-F.; Polejaeva, E.; Knutson, K.M.; Wassermann, E.M.; Krueger, F.; Grafman, J. Association between long-term cognitive decline in Vietnam Veterans with TBI and caregiver attachment style. J. Head Trauma Rehabil. 2015, 30, E26–E33. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, J.M.; Wang, Y.; Munro, C.A.; Ma, S.; Yue, C.; Chen, S.; Airan, R.; Kim, P.K.; Adams, A.V.; Garcia, C. Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol. Dis. 2015, 74, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Irrera, N.; Pizzino, G.; Calò, M.; Pallio, G.; Mannino, F.; Famà, F.; Arcoraci, V.; Fodale, V.; David, A.; Francesca, C. Lack of the Nlrp3 inflammasome improves mice recovery following traumatic brain injury. Front. Pharmacol. 2017, 8, 459. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Ahmad, A.; Crupi, R.; Impellizzeri, D.; Morabito, R.; Esposito, E.; Cuzzocrea, S. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J. Endocrinol. 2013, 217, 291–301. [Google Scholar] [CrossRef]
- Zetterberg, H.; Blennow, K. Fluid biomarkers for mild traumatic brain injury and related conditions. Nat. Rev. Neurol. 2016, 12, 563. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, L.; Chen, X.; Xue, X.; Guo, Q.; Liu, M.; Zhao, J. Formylpeptide receptors promote the migration and differentiation of rat neural stem cells. Sci. Rep. 2016, 6, 25946. [Google Scholar] [CrossRef]
- Braun, H.; Schäfer, K.; Höllt, V. β III Tubulin-Expressing Neurons Reveal Enhanced Neurogenesis in Hippocampal and Cortical Structures after a Contusion Trauma in Rats. J. Neurotrauma 2002, 19, 975–983. [Google Scholar] [CrossRef]
- Bye, N.; Carron, S.; Han, X.; Agyapomaa, D.; Ng, S.Y.; Yan, E.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Neurogenesis and glial proliferation are stimulated following diffuse traumatic brain injury in adult rats. J. Neurosci. Res. 2011, 89, 986–1000. [Google Scholar] [CrossRef]
- Liberles, S.D.; Horowitz, L.F.; Kuang, D.; Contos, J.J.; Wilson, K.L.; Siltberg-Liberles, J.; Liberles, D.A.; Buck, L.B. Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc. Natl. Acad. Sci. USA 2009, 106, 9842–9847. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-L.; Schneider, E.H.; Dimitrov, E.L.; Haun, F.; Pham, T.M.; Mohammed, A.H.; Usdin, T.B.; Murphy, P.M. Reduced fear memory and anxiety-like behavior in mice lacking formylpeptide receptor 1. Behav. Genet. 2011, 41, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.L.; Lee, E.J.; Murphy, P.M. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J. Exp. Med. 1999, 189, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Cardini, S.; Dalli, J.; Fineschi, S.; Perretti, M.; Lungarella, G.; Lucattelli, M. Genetic ablation of the fpr1 gene confers protection from smoking-induced lung emphysema in mice. Am. J. Respir. Cell Mol. Biol. 2012, 47, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Peritore, A.F.; Crupi, R.; Scuto, M.; Gugliandolo, E.; Siracusa, R.; Impellizzeri, D.; Cordaro, M.; D’Amico, R.; Fusco, R.; Di Paola, R.; et al. The Role of Annexin A1 and Formyl Peptide Receptor 2/3 Signaling in Chronic Corticosterone-Induced Depression-Like behaviors and Impairment in Hippocampal-Dependent Memory. CNS Neurol. Disord. Drug Targets 2020, 19, 27–43. [Google Scholar] [CrossRef]
- Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Neuroprotective Effect of Artesunate in Experimental Model of Traumatic Brain Injury. Front. Neurol. 2018, 9, 590. [Google Scholar] [CrossRef] [Green Version]
- Impellizzeri, D.; Cordaro, M.; Bruschetta, G.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. N-Palmitoylethanolamine-Oxazoline as a new therapeutic strategy to control neuroinflammation: Neuroprotective effects in experimental models of spinal cord and brain injury. J. Neurotrauma 2017, 34, 2609–2623. [Google Scholar] [CrossRef]
- Di Paola, R.; Fusco, R.; Gugliandolo, E.; Crupi, R.; Evangelista, M.; Granese, R.; Cuzzocrea, S. Co-micronized palmitoylethanolamide/polydatin treatment causes endometriotic lesion regression in a rodent model of surgically induced endometriosis. Front. Pharmacol. 2016, 7, 382. [Google Scholar] [CrossRef] [Green Version]
- Siracusa, R.; Impellizzeri, D.; Cordaro, M.; Crupi, R.; Esposito, E.; Petrosino, S.; Cuzzocrea, S. Anti-Inflammatory and neuroprotective effects of Co-UltraPEALut in a mouse model of vascular dementia. Front. Neurol. 2017, 8, 233. [Google Scholar] [CrossRef]
- Parrini, M.; Ghezzi, D.; Deidda, G.; Medrihan, L.; Castroflorio, E.; Alberti, M.; Baldelli, P.; Cancedda, L.; Contestabile, A. Aerobic exercise and a BDNF-mimetic therapy rescue learning and memory in a mouse model of Down syndrome. Sci. Rep. 2017, 7, 16825. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Hervias, I.; Ricobaraza, A.; Puerta, E.; Perez-Roldan, J.M.; Garcia-Barroso, C.; Franco, R.; Aguirre, N.; Garcia-Osta, A. Sildenafil restores cognitive function without affecting beta-amyloid burden in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 2029–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaro, M.; Impellizzeri, D.; Siracusa, R.; Gugliandolo, E.; Fusco, R.; Inferrera, A.; Esposito, E.; Di Paola, R.; Cuzzocrea, S. Effects of a co-micronized composite containing palmitoylethanolamide and polydatin in an experimental model of benign prostatic hyperplasia. Toxicol. Appl. Pharmacol. 2017, 329, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Impellizzeri, D.; Fusco, R.; Cordaro, M.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. Ultramicronized palmitoylethanolamide (PEA-um®) in the treatment of idiopathic pulmonary fibrosis. Pharmacol. Res. 2016, 111, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Clark, R.S.; Schiding, J.K.; Kaczorowski, S.L.; Marion, D.W.; Kochanek, P.M. Neutrophil accumulation after traumatic brain injury in rats: Comparison of weight drop and controlled cortical impact models. J. Neurotrauma 1994, 11, 499–506. [Google Scholar] [CrossRef]
- Yen, T.L.; Chang, C.C.; Chung, C.L.; Ko, W.C.; Yang, C.H.; Hsieh, C.Y. Neuroprotective Effects of Platonin, a Therapeutic Immunomodulating Medicine, on Traumatic Brain Injury in Mice after Controlled Cortical Impact. Int. J. Mol. Sci. 2018, 19, 1100. [Google Scholar] [CrossRef] [Green Version]
- Fusco, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; et al. N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. Int. J. Mol. Sci. 2019, 20, 4845. [Google Scholar] [CrossRef] [Green Version]
- D’amico, R.; Fusco, R.; Gugliandolo, E.; Cordaro, M.; Siracusa, R.; Impellizzeri, D.; Peritore, A.F.; Crupi, R.; Cuzzocrea, S.; Di Paola, R. Effects of a new compound containing Palmitoylethanolamide and Baicalein in myocardial ischaemia/reperfusion injury in vivo. Phytomedicine 2019, 54, 27–42. [Google Scholar] [CrossRef]
- Wang, X.; Seekaew, P.; Gao, X.; Chen, J. Traumatic Brain Injury Stimulates Neural Stem Cell Proliferation via Mammalian Target of Rapamycin Signaling Pathway Activation. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, R.; Fusco, R.; Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Impellizzeri, D.; Perretti, M.; Cuzzocrea, S. Formyl peptide receptor 1 signalling promotes experimental colitis in mice. Pharmacol. Res. 2019, 141, 591–601. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Esposito, E.; Cuzzocrea, S. A controlled cortical impact preclinical model of traumatic brain injury. In Neurotrophic Factors; Springer: Berlin/Heidelberg, Germany, 2018; pp. 385–391. [Google Scholar]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Issekutz, A.C.; Issekutz, T.B. Cellular and Vascular Phenomena in Inflammation. Method Enzymol. 1988, 162, 301–320. [Google Scholar]
- Kochanek, P.M.; Hallenbeck, J.M. Polymorphonuclear Leukocytes and Monocytes/Macrophages in the Pathogenesis of Cerebral-Ischemia and Stroke. Stroke 1992, 23, 1367–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, R.; D’amico, R.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Absence of formyl peptide receptor 1 causes endometriotic lesion regression in a mouse model of surgically-induced endometriosis. Oncotarget 2018, 9, 31355. [Google Scholar] [CrossRef] [Green Version]
- Sengelov, H.; Boulay, F.; Kjeldsen, L.; Borregaard, N. Subcellular localization and translocation of the receptor for N-formylmethionyl-leucyl-phenylalanine in human neutrophils. Biochem. J. 1994, 299, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Cowland, J.B.; Borregaard, N. The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J. Leukoc. Biol. 1999, 66, 989–995. [Google Scholar] [CrossRef]
- Hwang, T.-L.; Wang, C.-C.; Kuo, Y.-H.; Huang, H.-C.; Wu, Y.-C.; Kuo, L.-M.; Wu, Y.-H. The hederagenin saponin SMG-1 is a natural FMLP receptor inhibitor that suppresses human neutrophil activation. Biochem. Pharmacol. 2010, 80, 1190–1200. [Google Scholar] [CrossRef]
- Dang, P.M.-C.; Stensballe, A.; Boussetta, T.; Raad, H.; Dewas, C.; Kroviarski, Y.; Hayem, G.; Jensen, O.N.; Gougerot-Pocidalo, M.-A.; El-Benna, J. A specific p47 phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Investig. 2006, 116, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Hsu, C.Y.; Liu, T.H.; Hogan, E.L.; Perot, P.L.; Tai, H.H. Leukotriene-B4 Release and Polymorphonuclear Cell Infiltration in Spinal-Cord Injury. J. Neurochem. 1990, 55, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Wang, X.Y.; Jung, T.C.; Sumii, T.; Singhal, A.B.; Fini, M.E.; Dixon, C.E.; Alessandrini, A.; Lo, E.H. Mitogen-activated protein kinase inhibition in traumatic brain injury: In vitro and in vivo effects. J. Cereb. Blood Flow Metab. 2002, 22, 444–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouliot, M.; Fiset, M.E.; Masse, M.; Naccache, P.H.; Borgeat, P. Adenosine up-regulates cyclooxygenase-2 in human granulocytes: Impact on the balance of eicosanoid generation. J. Immunol. 2002, 169, 5279–5286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojo, H.; Borlongan, C.V.; Mabuchi, T. Genetic and Histological Alterations Reveal Key Role of Prostaglandin Synthase and Cyclooxygenase 1 and 2 in Traumatic Brain Injury-Induced Neuroinflammation in the Cerebral Cortex of Rats Exposed to Moderate Fluid Percussion Injury. Cell Transplant. 2017, 26, 1301–1313. [Google Scholar] [CrossRef]
- Mattson, M.; Meffert, M. Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006, 13, 852. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Li, Q.; Shi, W. Hyperbaric oxygen alleviates the activation of NLRP-3-inflammasomes in traumatic brain injury. Mol. Med. Rep. 2017, 16, 3922–3928. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-D.; Li, W.; Chen, Z.-R.; Hu, Y.-C.; Zhang, D.-D.; Shen, W.; Zhou, M.-L.; Zhu, L.; Hang, C.-H. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem. Res. 2013, 38, 2072–2083. [Google Scholar] [CrossRef]
- Ismael, S.; Nasoohi, S.; Ishrat, T. MCC950, the selective NLRP3 inflammasome inhibitor protects mice against traumatic brain injury. J. Neurotrauma 2018, 35, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP 3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Stover, J.F.; Schöning, B.; Beyer, T.F.; Woiciechowsky, C.; Unterberg, A.W. Temporal profile of cerebrospinal fluid glutamate, interleukin-6, and tumor necrosis factor-α in relation to brain edema and contusion following controlled cortical impact injury in rats. Neurosci. Lett. 2000, 288, 25–28. [Google Scholar] [CrossRef]
- Rothwell, N. Interleukin-1 and neuronal injury: Mechanisms, modification, and therapeutic potential. Brain Behav. Immun. 2003, 17, 152–157. [Google Scholar] [CrossRef]
- Bowie, A.; O’Neill, L.A. Oxidative stress and nuclear factor-κB activation: A reassessment of the evidence in the light of recent discoveries. Biochem. Pharmacol. 2000, 59, 13–23. [Google Scholar] [CrossRef]
- Orihara, Y.; Ikematsu, K.; Tsuda, R.; Nakasono, I. Induction of nitric oxide synthase by traumatic brain injury. Forensic Sci. Int. 2001, 123, 142–149. [Google Scholar] [CrossRef]
- Gahm, C.; Holmin, S.; Wiklund, P.N.; Brundin, L.; Mathiesen, T. Neuroprotection by selective inhibition of inducible nitric oxide synthase after experimental brain contusion. J. Neurotrauma 2006, 23, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Morganti-Kossmann, M.C.; Satgunaseelan, L.; Bye, N.; Kossmann, T. Modulation of immune response by head injury. Injury 2007, 38, 1392–1400. [Google Scholar] [CrossRef]
- Kernie, S.G.; Erwin, T.M.; Parada, L.F. Brain remodeling due to neuronal and astrocytic proliferation after controlled cortical injury in mice. J. Neurosci. Res. 2001, 66, 317–326. [Google Scholar] [CrossRef]
- Zhao, M.; Liang, F.; Xu, H.; Yan, W.; Zhang, J. Methylene blue exerts a neuroprotective effect against traumatic brain injury by promoting autophagy and inhibiting microglial activation. Mol. Med. Rep. 2016, 13, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Loane, D.J.; Stoica, B.A.; Tchantchou, F.; Kumar, A.; Barrett, J.P.; Akintola, T.; Xue, F.; Conn, P.J.; Faden, A.I. Novel mGluR5 positive allosteric modulator improves functional recovery, attenuates neurodegeneration, and alters microglial polarization after experimental traumatic brain injury. Neurotherapeutics 2014, 11, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, T.; Morganti-Kossmann, M.C. The role of markers of inflammation in traumatic brain injury. Front. Neurol. 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gao, X.; Michalski, S.; Zhao, S.; Chen, J. Traumatic brain injury severity affects neurogenesis in adult mouse hippocampus. J. Neurotrauma 2016, 33, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.K.; Jang, Y.H.; Yoo, H.J.; Kang, D.W.; Park, M.H.; Kim, M.K.; Song, J.H.; Kim, S.D.; Min, G.; You, H.K. N-formyl-methionyl-leucyl-phenylalanine (fMLP) promotes osteoblast differentiation via the N-formyl peptide receptor 1-mediated signaling pathway in human mesenchymal stem cells from bone marrow. J. Biol. Chem. 2011, 286, 17133–17143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, A.; Painter, R.G.; Lanson, N.A.; Wang, G. Functional expression of N-formyl peptide receptors in human bone marrow-derived mesenchymal stem cells. Stem Cells 2007, 25, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-K.; Park, Y.J.; Kim, J.H.; Ryu, S.H.; Bae, Y.-S. Expression and functional role of formyl peptide receptor in human bone marrow-derived mesenchymal stem cells. FEBS Lett. 2007, 581, 1917–1922. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, G.; Chen, X.X.; Xue, X.; Guo, Q.N.; Liu, M.Y.; Zhao, J.H. Formyl peptide receptors promotes neural differentiation in mouse neural stem cells by ROS generation and regulation of PI3KAKT signaling. Sci. Rep. 2017, 7, 206. [Google Scholar] [CrossRef]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef] [Green Version]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fusco, R.; Gugliandolo, E.; Siracusa, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Evangelista, M.; Peli, A.; Peritore, A.F.; Impellizzeri, D.; et al. Formyl Peptide Receptor 1 Signaling in Acute Inflammation and Neural Differentiation Induced by Traumatic Brain Injury. Biology 2020, 9, 238. https://doi.org/10.3390/biology9090238
Fusco R, Gugliandolo E, Siracusa R, Scuto M, Cordaro M, D’Amico R, Evangelista M, Peli A, Peritore AF, Impellizzeri D, et al. Formyl Peptide Receptor 1 Signaling in Acute Inflammation and Neural Differentiation Induced by Traumatic Brain Injury. Biology. 2020; 9(9):238. https://doi.org/10.3390/biology9090238
Chicago/Turabian StyleFusco, Roberta, Enrico Gugliandolo, Rosalba Siracusa, Maria Scuto, Marika Cordaro, Ramona D’Amico, Maurizio Evangelista, Angelo Peli, Alessio Filippo Peritore, Daniela Impellizzeri, and et al. 2020. "Formyl Peptide Receptor 1 Signaling in Acute Inflammation and Neural Differentiation Induced by Traumatic Brain Injury" Biology 9, no. 9: 238. https://doi.org/10.3390/biology9090238
APA StyleFusco, R., Gugliandolo, E., Siracusa, R., Scuto, M., Cordaro, M., D’Amico, R., Evangelista, M., Peli, A., Peritore, A. F., Impellizzeri, D., Crupi, R., Cuzzocrea, S., & Di Paola, R. (2020). Formyl Peptide Receptor 1 Signaling in Acute Inflammation and Neural Differentiation Induced by Traumatic Brain Injury. Biology, 9(9), 238. https://doi.org/10.3390/biology9090238