
Genomic Analysis of 18th-Century Kazakh Individuals and Their Oral Microbiome

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ancient DNA Extraction
2.2. Processing of Sequencing Data and Mapping to the Human Genome
2.3. Assessing the Authenticity of the Human Genomic Data
2.4. Assessing the Genetic Sex and Degree of Contamination
2.5. Mitochondrial DNA and Y-Chromosome Haplogroup Determination
2.6. Kinship Analysis
2.7. Analysing Population Structure via Clustering Algorithms
2.8. Ancestry Modelling
2.9. Processing of Sequencing Data and Mapping of Ancient Microbes
2.10. Assessing the Authenticity of the Ancient Microbial DNA
2.11. Phylogenetic Analysis of T. forsythia
2.12. Phylogenetic Dataset Filtering
2.13. Tip Calibrated Dating
2.14. Phylogenetic Analysis of T. denticola and P. gingivalis
2.15. T. forsythia Pan-Genome Analysis
3. Results
3.1. Human DNA Analysis
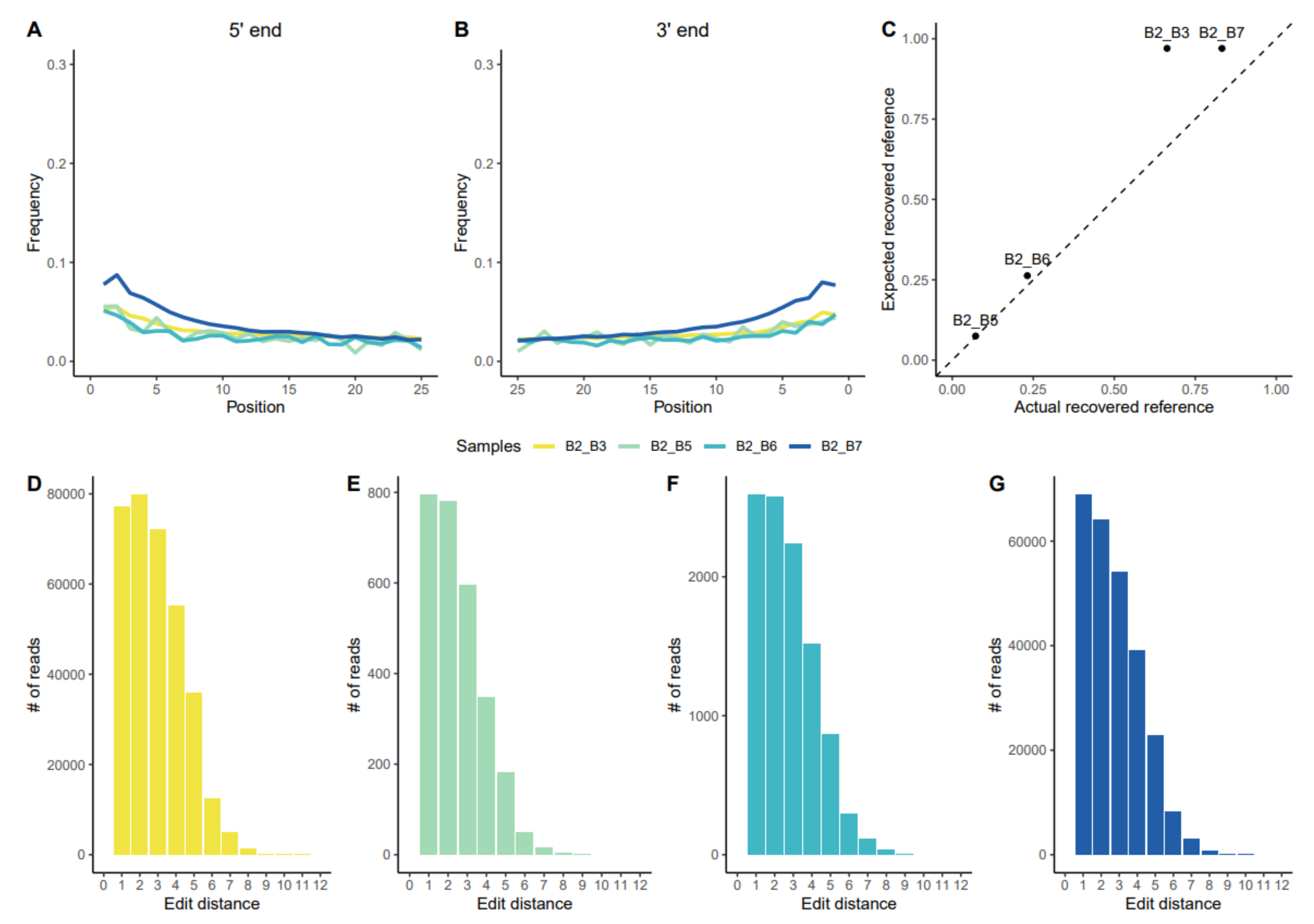
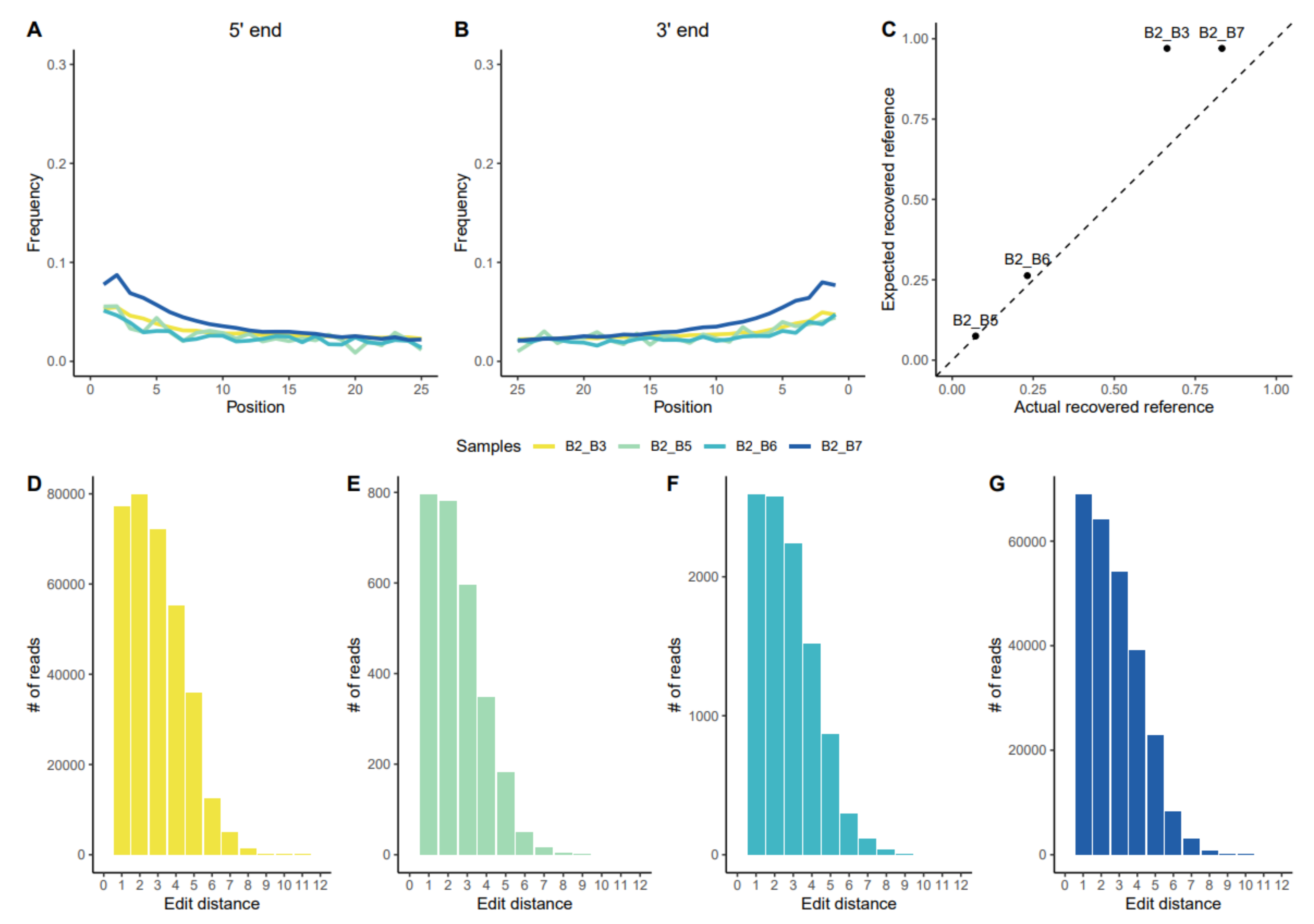
3.1.1. Assessing aDNA Reliability
3.1.2. Uniparental Markers and Kinship Relationships
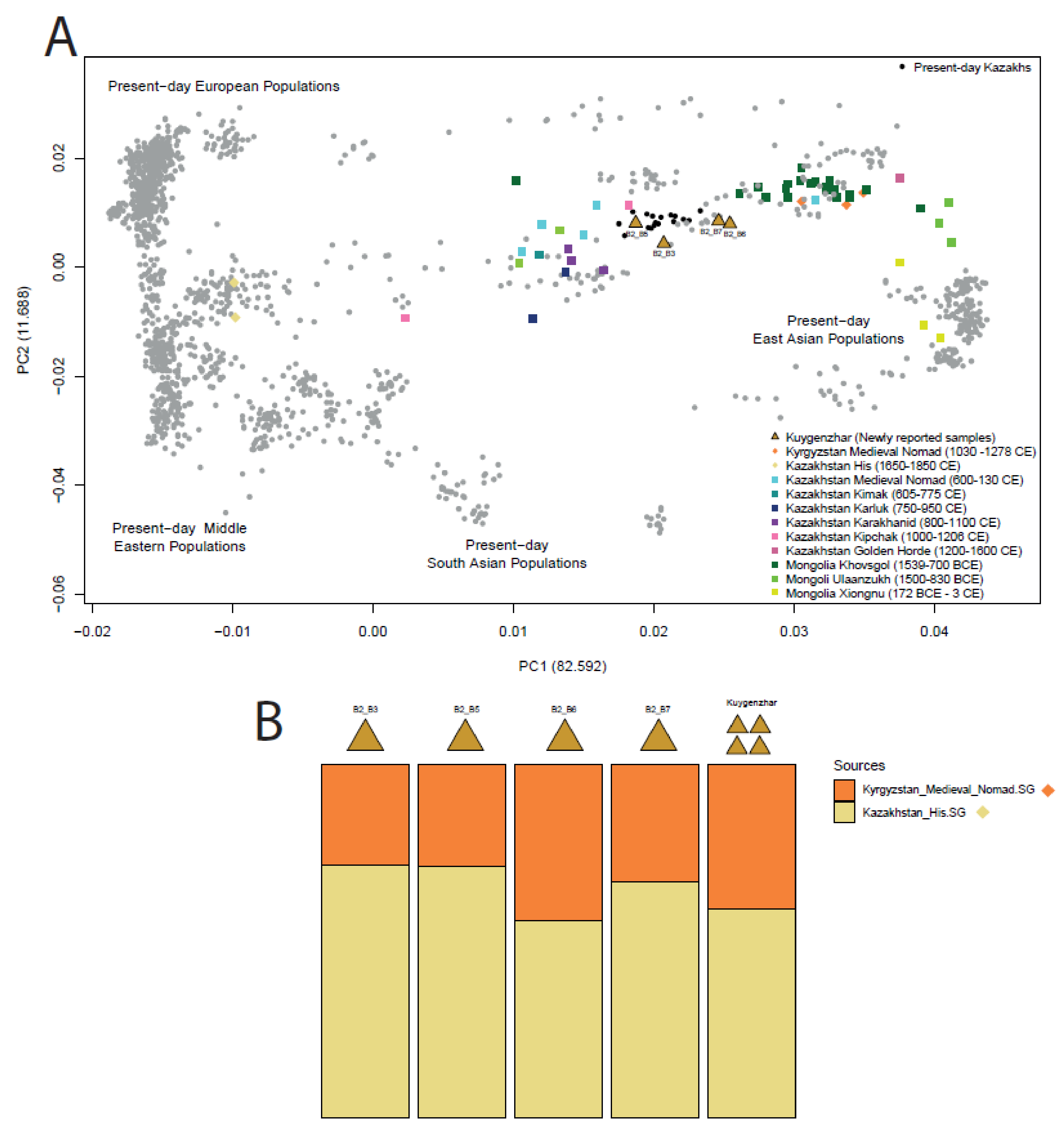
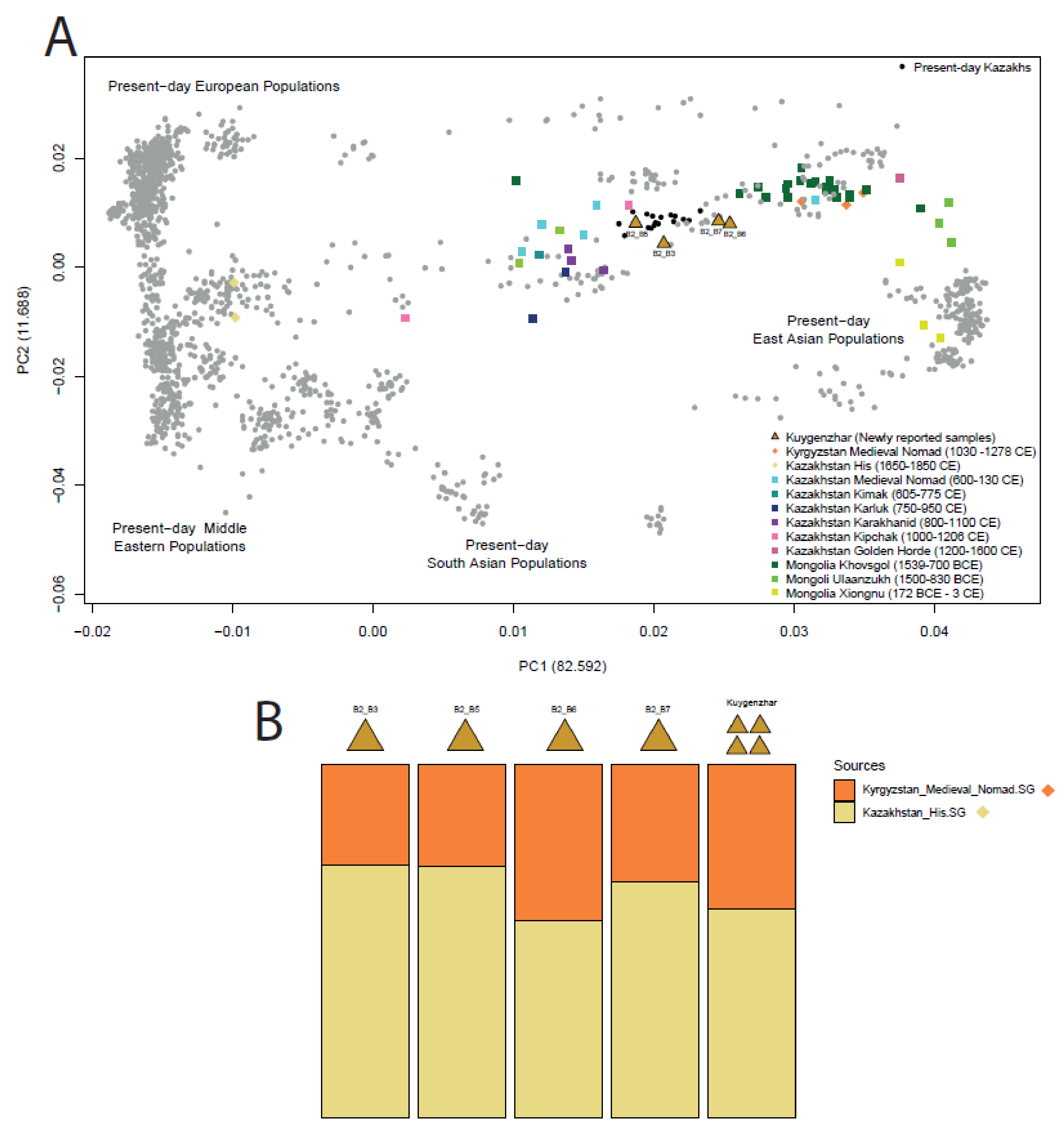
3.1.3. Human Genetic Data Exploration
3.1.4. f-Statistics Ancestry Modelling
3.2. Microbiome Analysis
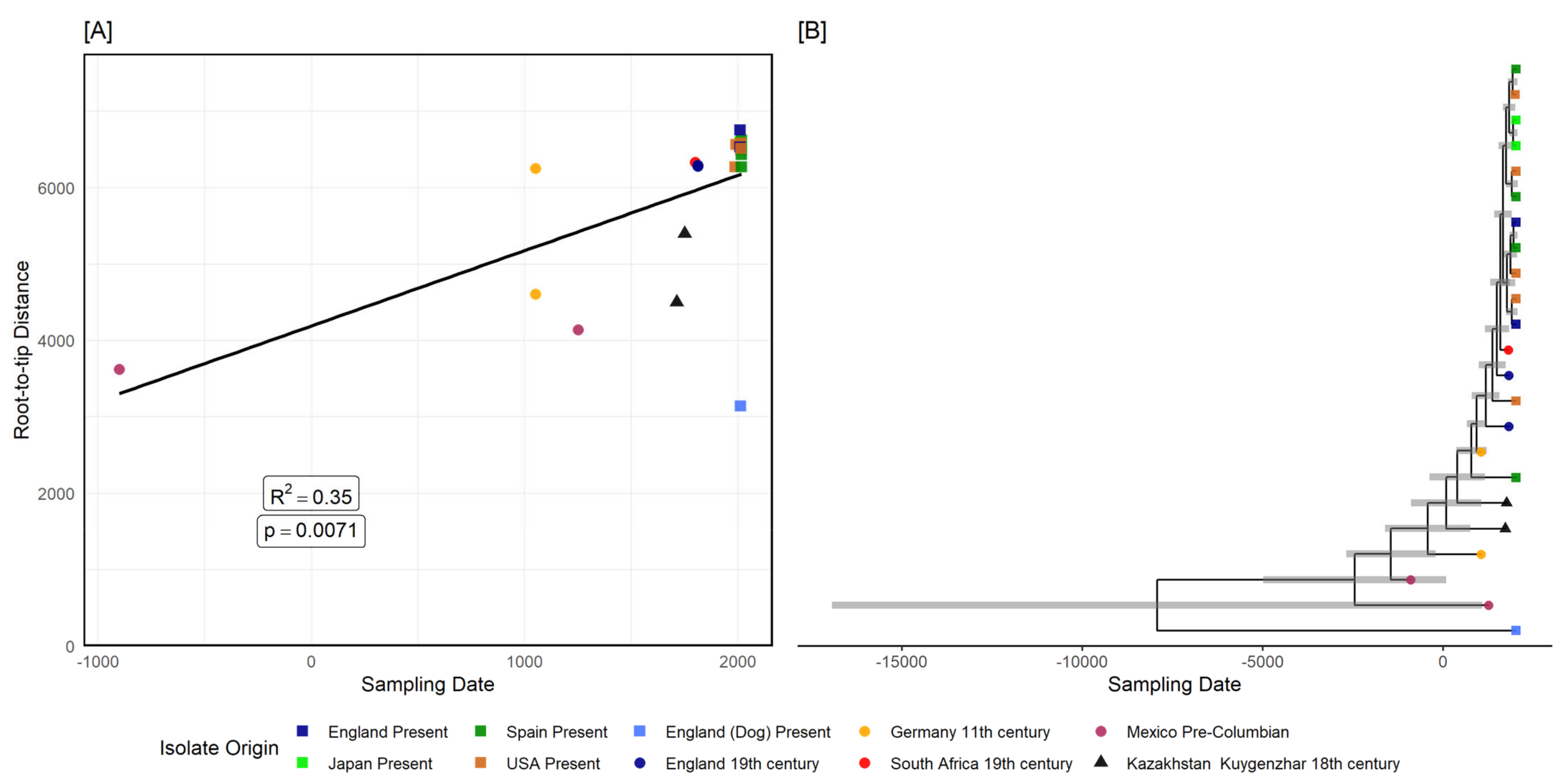
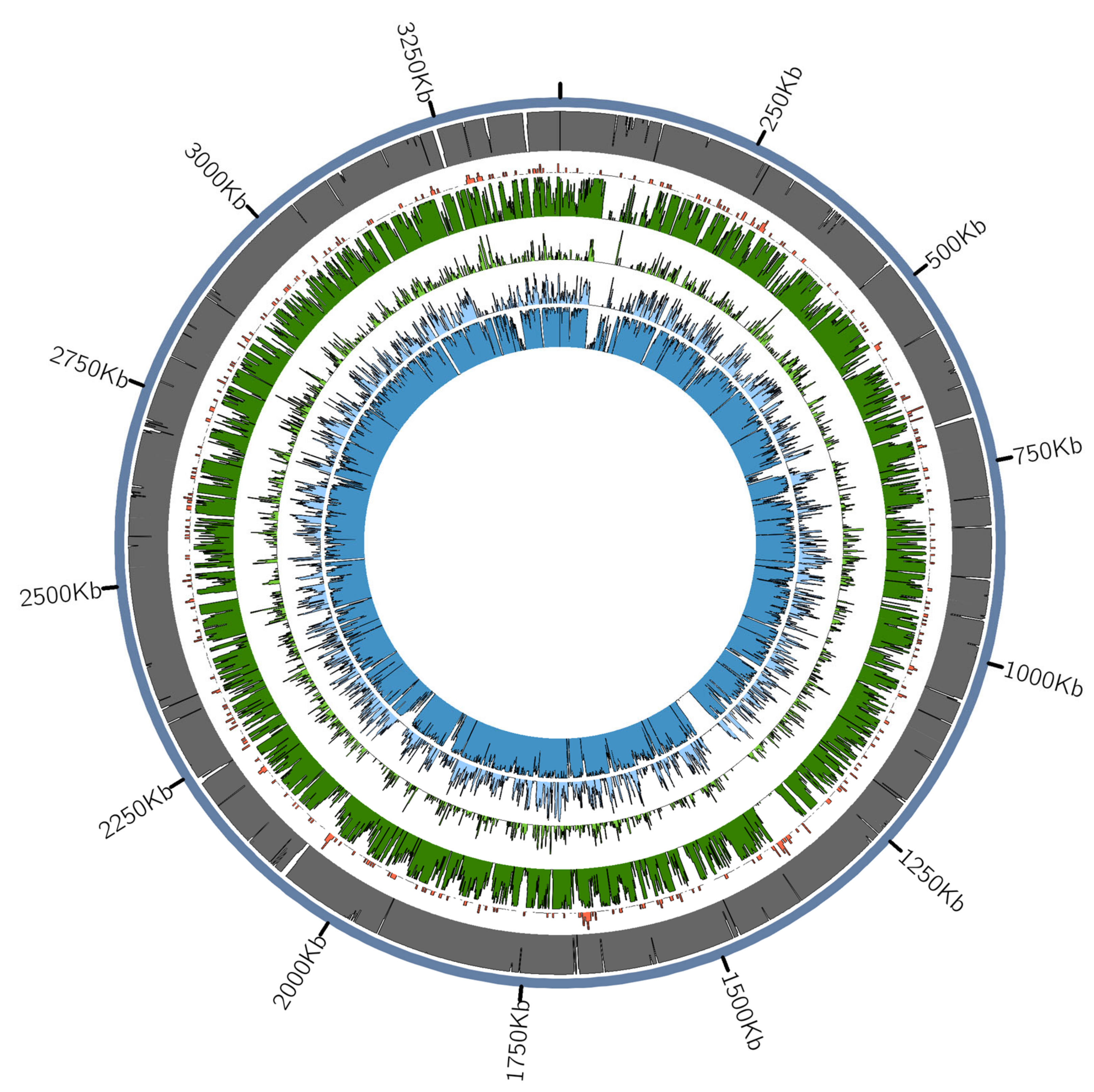
3.2.1. Red Complex Bacteria Detection and Authentication
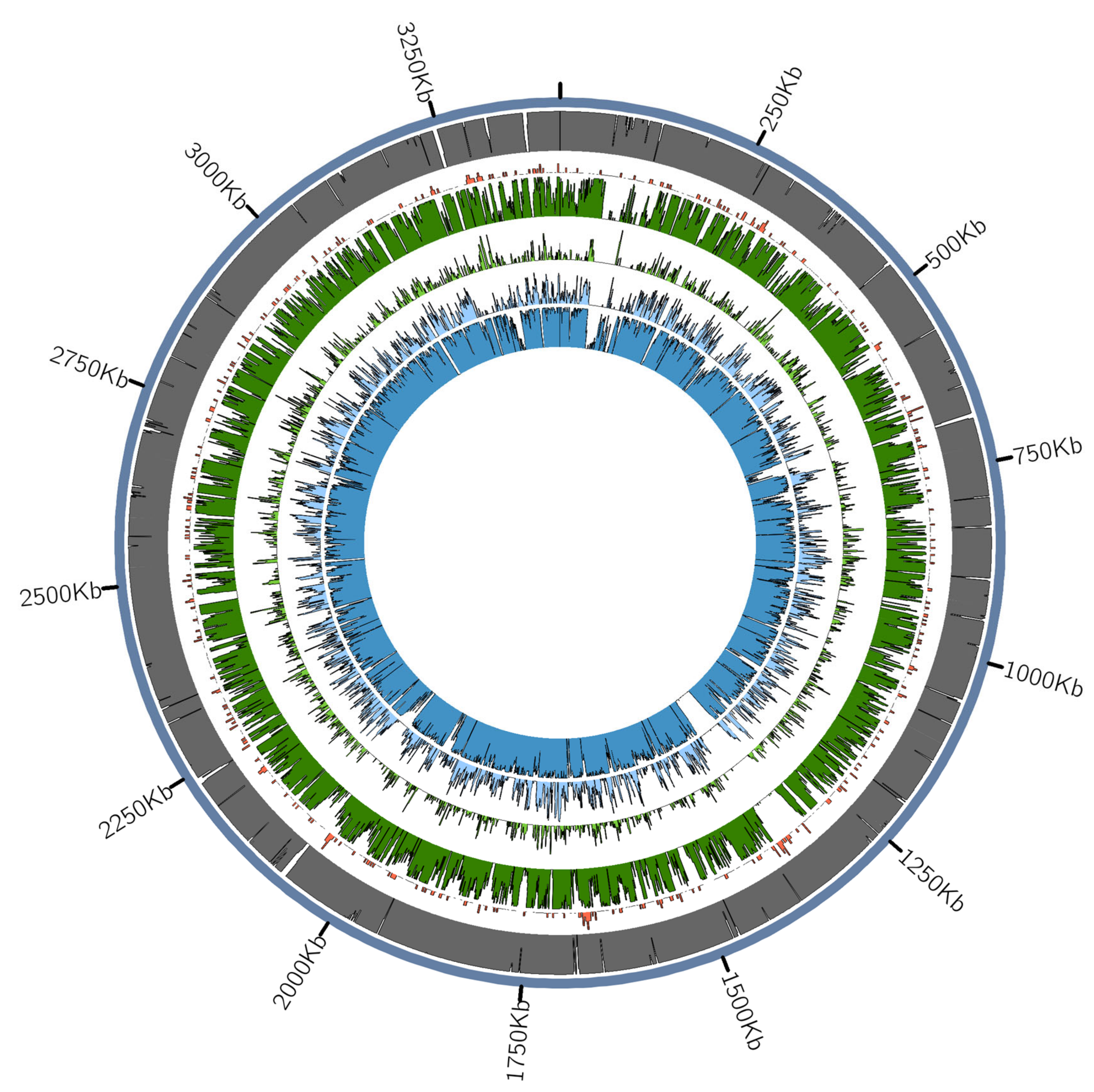
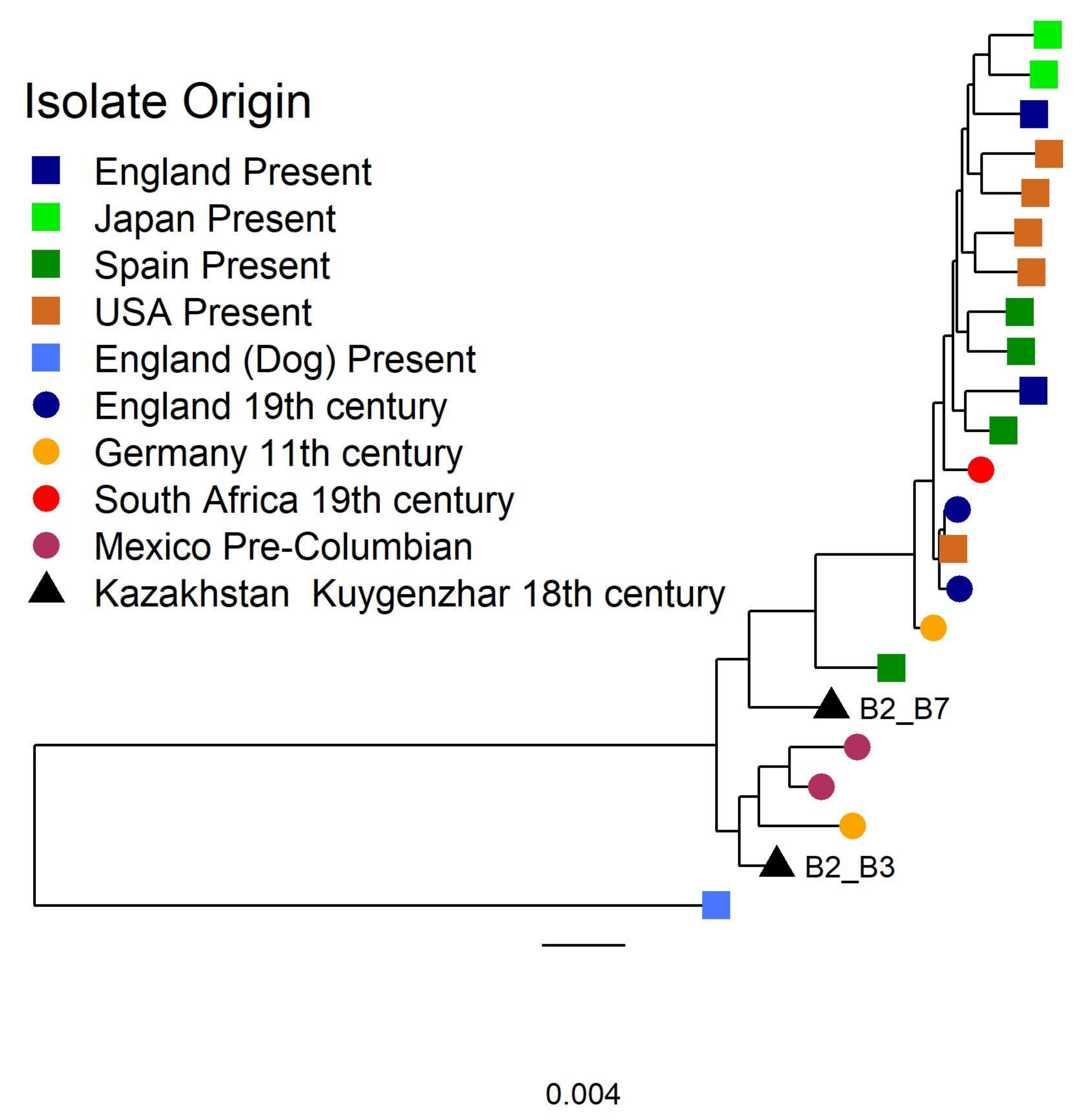
3.2.2. Tannerella Forsythia Phylogeny and Clade Dating
3.2.3. Presence of Virulence or Lifestyle-Related Tannerella Genes
3.2.4. T. denticola and P. gingivalis Trees
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathieson, I.; Lazaridis, I.; Rohland, N.; Mallick, S.; Patterson, N.; Roodenberg, S.A.; Harney, E.; Stewardson, K.; Fernandes, D.; Novak, M.; et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 2015, 528, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Allentoft, M.E.; Sikora, M.; Sjögren, K.G.; Rasmussen, S.; Rasmussen, M.; Stenderup, J.; Damgaard, P.B.; Schroeder, H.; Ahlström, T.; Vinner, L.; et al. Population genomics of Bronze Age Eurasia. Nature 2015, 522, 167–172. [Google Scholar] [CrossRef]
- Lazaridis, I.; Nadel, D.; Rollefson, G.; Merrett, D.C.; Rohland, N.; Mallick, S.; Fernandes, D.; Novak, M.; Gamarra, B.; Sirak, K.; et al. Genomic insights into the origin of farming in the ancient Near East. Nature 2016, 536, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, V.M.; Patterson, N.; Moorjani, P.; Rohland, N.; Bernardos, R.; Mallick, S.; Lazaridis, I.; Nakatsuka, N.; Olalde, I.; Lipson, M.; et al. The formation of human populations in South and Central Asia. Science 2019, 365, eaat7487. [Google Scholar] [CrossRef]
- Gnecchi-Ruscone, G.A.; Khussainova, E.; Kahbatkyzy, N.; Musralina, L.; Spyrou, M.A.; Bianco, R.A.; Radzeviciute, R.; Martins, N.F.G.; Freund, C.; Iksan, O.; et al. Ancient genomic time transect from the Central Asian Steppe unravels the history of the Scythians. Sci. Adv. 2021, 7, eabe4414. [Google Scholar] [CrossRef] [PubMed]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Mallick, S.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Barros Damgaard, P.; Martiniano, R.; Kamm, J.; Víctor Moreno-Mayar, J.; Kroonen, G.; Peyrot, M.; Barjamovic, G.; Rasmussen, S.; Zacho, C.; Baimukhanov, N.; et al. The first horse herders and the impact of early Bronze Age steppe expansions into Asia. Science 2018, 360, eaar7711. [Google Scholar] [CrossRef] [Green Version]
- De Barros Damgaard, P.; Marchi, N.; Rasmussen, S.; Peyrot, M.; Renaud, G.; Korneliussen, T.; Moreno-Mayar, J.V.; Pedersen, M.W.; Goldberg, A.; Usmanova, E.; et al. 137 ancient human genomes from across the Eurasian steppes. Nature 2018, 557, 369–374. [Google Scholar] [CrossRef]
- González-Ruiz, M.; Santos, C.; Jordana, X.; Simón, M.; Lalueza-Fox, C.; Gigli, E.; Aluja, M.P.; Malgosa, A. Tracing the Origin of the East-West Population Admixture in the Altai Region (Central Asia). PLoS ONE 2012, 7, e48904. [Google Scholar] [CrossRef] [Green Version]
- Lalueza-Fox, C.; Sampietro, M.L.; Gilbert, M.T.P.; Castri, L.; Facchini, F.; Pettener, D.; Bertranpetit, J. Unravelling migrations in the steppe: Mitochondrial DNA sequences from ancient Central Asians. Proc. R. Soc. B Biol. Sci. 2004, 271, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Seidualy, M.; Blazyte, A.; Jeon, S.; Bhak, Y.; Jeon, Y.; Kim, J.; Eriksson, A.; Bolser, D.; Yoon, C.; Manica, A.; et al. Decoding a highly mixed Kazakh genome. Hum. Genet. 2020, 139, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Esenova, S. Soviet Nationality, Identity, and Ethnicity in Central Asia: Historic Narratives and Kazakh Ethnic Identity. J. Muslim Minor. Aff. 2002, 22, 11–38. [Google Scholar] [CrossRef]
- Helmi, M.F.; Huang, H.; Goodson, J.M.; Hasturk, H.; Tavares, M.; Natto, Z.S. Prevalence of periodontitis and alveolar bone loss in a patient population at Harvard School of Dental Medicine. BMC Oral Health 2019, 19, 254. [Google Scholar] [CrossRef] [Green Version]
- Könönen, E.; Gursoy, M.; Gursoy, U. Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues. J. Clin. Med. 2019, 8, 1135. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Lopez, M.; Villa-Islas, V.; Rocha Arriaga, C.; Villaseñor-Altamirano, A.B.; Guzmán-Solís, A.; Sandoval-Velasco, M.; Wesp, J.K.; Alcantara, K.; López-Corral, A.; Gómez-Valdés, J.; et al. Paleogenomic insights into the red complex bacteria Tannerella forsythia in Pre-Hispanic and Colonial individuals from Mexico. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190580. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, B.; Wȩsierska, A.; Zalewska, K.; Sokołowska, M.M.; Bursiewicz, W.; Socha, M.; Ozorowski, M.; Pawlak-Osińska, K.; Wiciński, M. The role of Tannerella forsythia and Porphyromonas gingivalis in pathogenesis of esophageal cancer. Infect. Agents Cancer 2019, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Castrillon, C.A.; Hincapie, J.P.; Yepes, F.L.; Roldan, N.; Moreno, S.M.; Contreras, A.; Botero, J.E. Occurrence of red complex microorganisms and Aggregatibacter actinomycetemcomitans in patients with diabetes. J. Investig. Clin. Dent. 2015, 6, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Casanova, L.; Hughes, F.J.; Preshaw, P.M. Diabetes and periodontal disease: A two-way relationship. Br. Dent. J. 2014, 217, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, D.; Inquimbert, C.; Ottolenghi, L.; Carrouel, F. Periodontal pathogens as risk factors of cardiovascular diseases, diabetes, rheumatoid arthritis, cancer, and chronic obstructive pulmonary disease—Is there cause for consideration? Microorganisms 2019, 7, 424. [Google Scholar] [CrossRef] [Green Version]
- Nagao, Y.; Tanigawa, T. Red complex periodontal pathogens are risk factors for liver cirrhosis. Biomed. Rep. 2019, 11, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Sampaio-Maia, B.; Caldas, I.M.; Pereira, M.L.; Pérez-Mongiovi, D.; Araujo, R. Chapter Four—The Oral Microbiome in Health and Its Implication in Oral and Systemic Diseases. Adv. Appl. Microbiol. 2016, 97, 171–210. [Google Scholar] [CrossRef]
- Spyrou, M.A.; Bos, K.I.; Herbig, A.; Krause, J. Ancient pathogen genomics as an emerging tool for infectious disease research. Nat. Rev. Genet. 2019, 20, 323–340. [Google Scholar] [CrossRef]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carøe, C.; Gopalakrishnan, S.; Vinner, L.; Mak, S.S.T.; Sinding, M.H.S.; Samaniego, J.A.; Wales, N.; Sicheritz-Pontén, T.; Gilbert, M.T.P. Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 2018, 9, 410–419. [Google Scholar] [CrossRef]
- Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Institute, B. Picard. Available online: http://picard.sourceforge.net (accessed on 6 July 2021).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; Von Haeseler, A.; Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brotherton, P.; Endicott, P.; Sanchez, J.J.; Beaumont, M.; Barnett, R.; Austin, J.; Cooper, A. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 2007, 35, 5717–5728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S.; Krause, J.; Jakobsson, M. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [Green Version]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. MapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, P.; Storå, J.; Götherström, A.; Jakobsson, M. Accurate sex identification of ancient human remains using DNA shotgun sequencing. J. Archaeol. Sci. 2013, 40, 4477–4482. [Google Scholar] [CrossRef]
- Renaud, G.; Slon, V.; Duggan, A.T.; Kelso, J. Schmutzi: Estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome Biol. 2015, 16, 224. [Google Scholar] [CrossRef] [Green Version]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Kloss-Brandstätter, A.; Pacher, D.; Schönherr, S.; Weissensteiner, H.; Binna, R.; Specht, G.; Kronenberg, F. HaploGrep: A fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum. Mutat. 2011, 32, 25–32. [Google Scholar] [CrossRef]
- Van Oven, M. PhyloTree Build 17: Growing the human mitochondrial DNA tree. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e392–e394. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, J.M.M.; Jakobsson, M.; Günther, T. Estimating genetic kin relationships in prehistoric populations. PLoS ONE 2018, 13, e0195491. [Google Scholar] [CrossRef] [Green Version]
- Schiffels, S. Sequencetools. 2018. Available online: https://github.com/stschiff/sequenceTools (accessed on 6 July 2021).
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, 2074–2093. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, I.; Patterson, N.; Mittnik, A.; Renaud, G.; Mallick, S.; Kirsanow, K.; Sudmant, P.H.; Schraiber, J.G.; Castellano, S.; Lipson, M.; et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 2014, 513, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Behr, A.A.; Liu, K.Z.; Liu-Fang, G.; Nakka, P.; Ramachandran, S. Pong: Fast analysis and visualization of latent clusters in population genetic data. Bioinformatics 2016, 32, 2817–2823. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.; Thangaraj, K.; Patterson, N.; Price, A.L.; Singh, L. Reconstructing Indian population history. Nature 2009, 461, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient admixture in human history. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef] [Green Version]
- Peter, B.M. Admixture, population structure, and f-statistics. Genetics 2016, 202, 1485–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, C.; Wang, K.; Wilkin, S.; Taylor, W.T.T.; Miller, B.K.; Bemmann, J.H.; Stahl, R.; Chiovelli, C.; Knolle, F.; Ulziibayar, S.; et al. A Dynamic 6000-Year Genetic History of Eurasia’s Eastern Steppe. Cell 2020, 183, 890–904.e29. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwängler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of Ice Age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Järve, M.; Saag, L.; Scheib, C.L.; Pathak, A.K.; Montinaro, F.; Pagani, L.; Flores, R.; Guellil, M.; Saag, L.; Tambets, K.; et al. Shifts in the Genetic Landscape of the Western Eurasian Steppe Associated with the Beginning and End of the Scythian Dominance. Curr. Biol. 2019, 29, 2430–2441.e10. [Google Scholar] [CrossRef]
- Jeong, C.; Wilkin, S.; Amgalantugs, T.; Bouwman, A.S.; Taylor, W.T.T.; Hagan, R.W.; Bromage, S.; Tsolmon, S.; Trachsel, C.; Grossmann, J.; et al. Bronze Age population dynamics and the rise of dairy pastoralism on the eastern Eurasian steppe. Proc. Natl. Acad. Sci. USA 2018, 115, E11248–E11255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.; Allentoft, M.E.; Nielsen, K.; Orlando, L.; Sikora, M.; Sjögren, K.G.; Pedersen, A.G.; Schubert, M.; Van Dam, A.; Kapel, C.M.O.; et al. Early Divergent Strains of Yersinia pestis in Eurasia 5,000 Years Ago. Cell 2015, 163, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Core R Team. A Language and Environment for Statistical Computing; R Foundation for Statical Computing: Vienna, Austria, 2019; Available online: https://www.R--project.org (accessed on 6 July 2021).
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Rieux, A.; Balloux, F. Inferences from tip-calibrated phylogenies: A review and a practical guide. Mol. Ecol. 2016, 25, 1911–1924. [Google Scholar] [CrossRef] [Green Version]
- Didelot, X.; Wilson, D.J.; Bryant, D.; Quail, M.; Cockfield, J. ClonalFrameML: Efficient Inference of Recombination in Whole Bacterial Genomes. PLoS Comput. Biol. 2015, 11, e1004041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crispell, J.; Balaz, D.; Gordon, S.V. Homoplasyfinder: A simple tool to identify homoplasies on a phylogeny. Microb. Genom. 2019, 5, e000245. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M. Toward Defining the Course of Evolution: Minimum Change for a Specific Tree Topology. Syst. Zool. 1971, 20, 406. [Google Scholar] [CrossRef]
- Didelot, X.; Croucher, N.J.; Bentley, S.D.; Harris, S.R.; Wilson, D.J. Bayesian inference of ancestral dates on bacterial phylogenetic trees. Nucleic Acids Res. 2018, 46, e134. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.A.F.; Velsko, I.M.; Aron, F.; Posth, C.; Hofman, C.A.; Austin, R.M.; Parker, C.E.; Mann, A.E.; Nagele, K.; Arthur, K.W.; et al. The evolution and changing ecology of the African hominid oral microbiome. Proc. Natl. Acad. Sci. USA 2021, 118, e2021655118. [Google Scholar] [CrossRef]
- Warinner, C.; Rodrigues, J.F.M.; Vyas, R.; Trachsel, C.; Shved, N.; Grossmann, J.; Radini, A.; Hancock, Y.; Tito, R.Y.; Fiddyment, S.; et al. Pathogens and host immunity in the ancient human oral cavity. Nat. Genet. 2014, 46, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Velsko, I.M.; Fellows Yates, J.A.; Aron, F.; Hagan, R.W.; Frantz, L.A.F.; Loe, L.; Martinez, J.B.R.; Chaves, E.; Gosden, C.; Larson, G.; et al. Microbial differences between dental plaque and historic dental calculus are related to oral biofilm maturation stage. Microbiome 2019, 7, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwickl, N.F.; Stralis-Pavese, N.; Schäffer, C.; Dohm, J.C.; Himmelbauer, H. Comparative genome characterization of the periodontal pathogen Tannerella forsythia. BMC Genom. 2020, 21, 150. [Google Scholar] [CrossRef]
- Comas, D.; Plaza, S.; Wells, R.S.; Yuldaseva, N.; Lao, O.; Calafell, F.; Bertranpetit, J. Admixture, migrations, and dispersals in Central Asia: Evidence from maternal DNA lineages. Eur. J. Hum. Genet. 2004, 12, 495–504. [Google Scholar] [CrossRef]
- Balanovsky, O.; Zhabagin, M.; Agdzhoyan, A.; Chukhryaeva, M.; Zaporozhchenko, V.; Utevska, O.; Highnam, G.; Sabitov, Z.; Greenspan, E.; Dibirova, K.; et al. Deep phylogenetic analysis of haplogroup G1 provides estimates of SNP and STR mutation rates on the human Y-chromosome and reveals migrations of iranic speakers. PLoS ONE 2015, 10, e0122968. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.H.; Yan, S.; Lu, Y.; Wen, S.Q.; Huang, Y.Z.; Wang, L.X.; Li, S.L.; Yang, Y.J.; Wang, X.F.; Zhang, C.; et al. Whole-sequence analysis indicates that the y chromosome C2∗-Star Cluster traces back to ordinary Mongols, rather than Genghis Khan/631/208/457/631/208/514 article. Eur. J. Hum. Genet. 2018, 26, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cai, D.; Zhang, Y.; Zhu, H.; Zhou, H. Ancient DNA reveals two paternal lineages C2a1a1b1a/F3830 and C2b1b/F845 in past nomadic peoples distributed on the Mongolian Plateau. Am. J. Phys. Anthropol. 2020, 172, 402–411. [Google Scholar] [CrossRef]
- Ioannidis, I.; Sakellari, D.; Spala, A.; Arsenakis, M.; Konstantinidis, A. Prevalence of tetM, tetQ, nim and blaTEM genes in the oral cavities of Greek subjects: A pilot study. J. Clin. Periodontol. 2009, 36, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, H.C.; Lee, S.W.S. Characterization of antibiotic resistance determinants in oral biofilms. J. Microbiol. 2011, 49, 595. [Google Scholar] [CrossRef]
- Ksiazek, M.; Mizgalska, D.; Eick, S.; Thøgersen, I.B.; Enghild, J.J.; Potempa, J. KLIKK proteases of Tannerella forsythia: Putative virulence factors with a unique domain structure. Front. Microbiol. 2015, 6, 312. [Google Scholar] [CrossRef]
- Philips, A.; Stolarek, I.; Handschuh, L.; Nowis, K.; Juras, A.; Trzciński, D.; Nowaczewska, W.; Wrzesińska, A.; Potempa, J.; Potempa, J.; et al. Analysis of oral microbiome from fossil human remains revealed the significant differences in virulence factors of modern and ancient Tannerella forsythia. BMC Genom. 2020, 21, 402. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Song, K.P.; Ong, G. Bacteroides forsythus prtH genotype in periodontitis patients: Occurrence and association with periodontal disease. J. Periodontal Res. 2001, 36, 398–403. [Google Scholar] [CrossRef]
- Hamlet, S.M.; Ganashan, N.; Cullinan, M.P.; Westerman, B.; Palmer, J.E.; Seymour, G.J. A 5-Year Longitudinal Study of Tannerella forsythia prtH Genotype: Association with Loss of Attachment. J. Periodontol. 2008, 79, 144–149. [Google Scholar] [CrossRef]
- Shimotahira, N.; Oogai, Y.; Kawada-Matsuo, M.; Yamada, S.; Fukutsuji, K.; Nagano, K.; Yoshimura, F.; Noguchi, K.; Komatsuzawa, H. The surface layer of Tannerella forsythia contributes to serum resistance and oral bacterial coaggregation. Infect. Immun. 2013, 81, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kılınç, G.M.; Kashuba, N.; Koptekin, D.; Bergfeldt, N.; Dönertaş, H.M.; Rodríguez-Varela, R.; Shergin, D.; Ivanov, G.; Kichigin, D.; Pestereva, K.; et al. Human population dynamics and Yersinia pestis in ancient northeast Asia. Sci. Adv. 2021, 7, eabc4587. [Google Scholar] [CrossRef]
- Heyer, E.; Balaresque, P.; Jobling, M.A.; Quintana-Murci, L.; Chaix, R.; Segurel, L.; Aldashev, A.; Hegay, T. Genetic diversity and the emergence of ethnic groups in Central Asia. BMC Genet. 2009, 10, 49. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Ramond, J.B.; Rifkin, R.F.; Underdown, S.J. Migrating microbes: What pathogens can tell us about population movements and human evolution. Ann. Hum. Biol. 2017, 44, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coggeshall, M.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, M.; Suzuki, M.; Umeda, M.; Ishikawa, I.; Benno, Y. Reclassification of Bacteroides forsythus (Tanner et al. 1986) as Tannerella forsythensis corrig., gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, L.S.; Dobney, K.; Cooper, A. Ancient DNA analysis of dental calculus. J. Hum. Evol. 2015, 79, 119–124. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
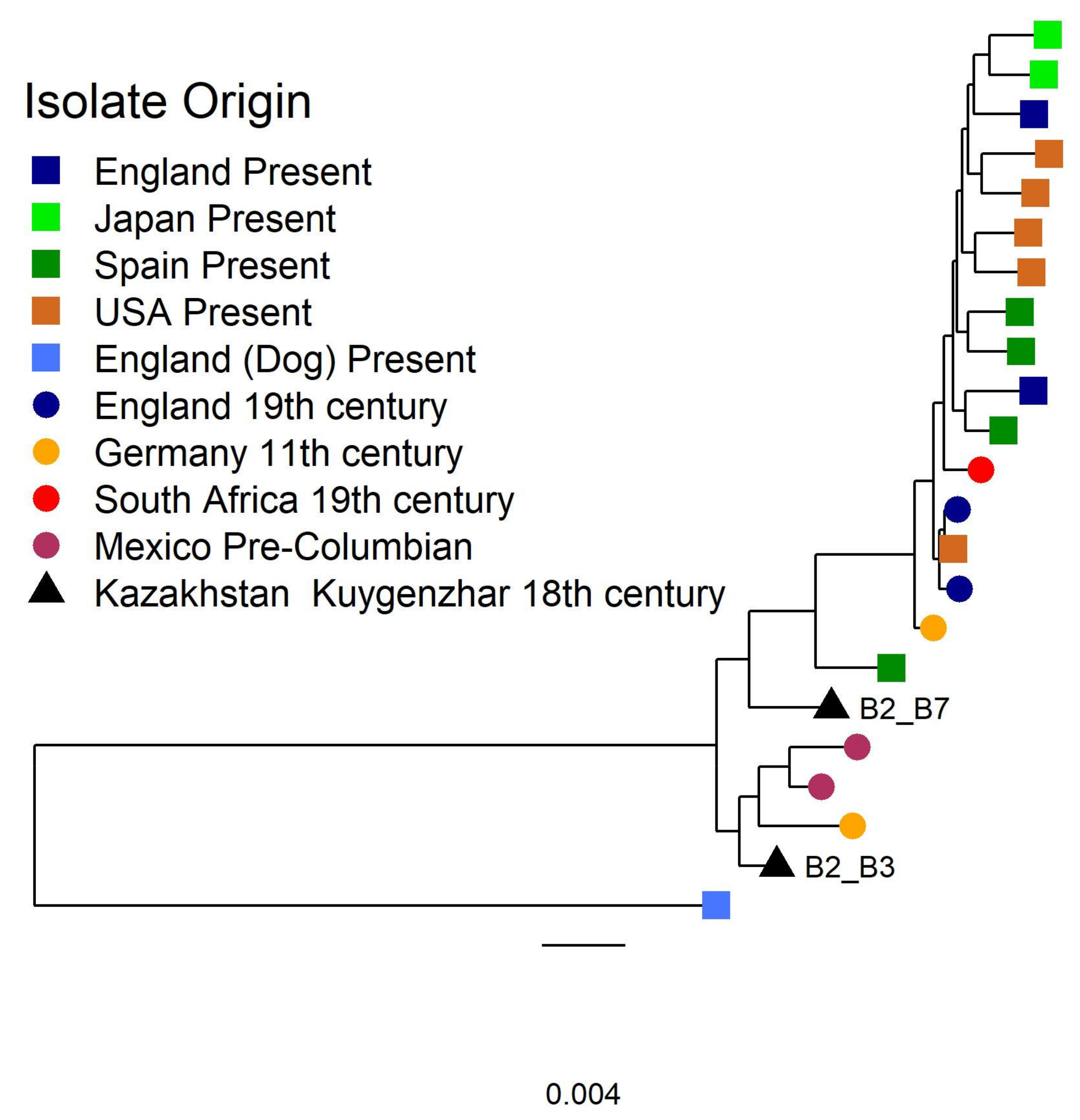{kind=link}
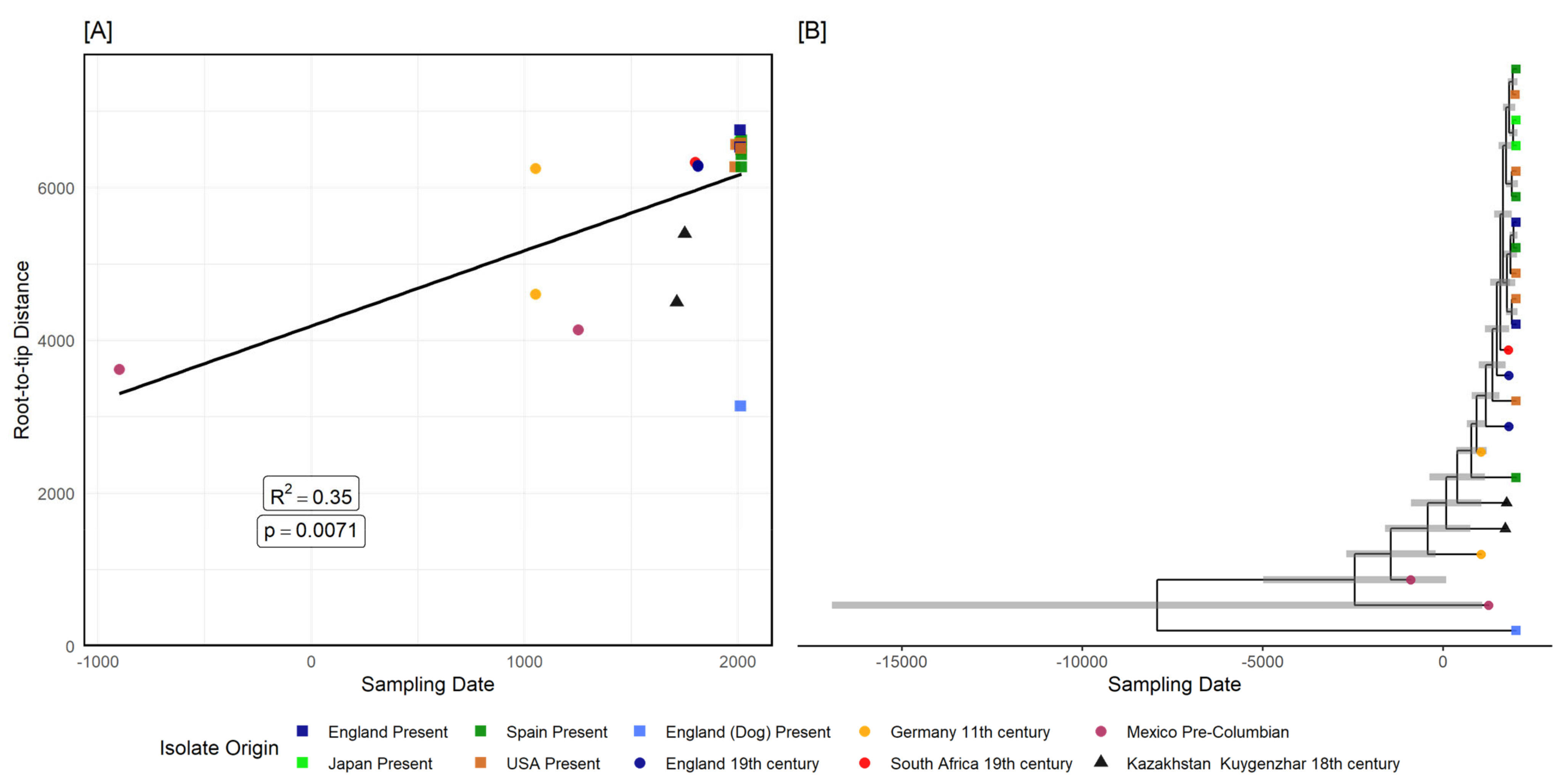{kind=link}
| Nuclear DNA | Number of Sequenced Paired Reads | Mapped Reads | Reads with a Mapping Quality > 30 | Average Coverage | Covered Positions (%) |
|---|---|---|---|---|---|
| B2_B1 | 51,212,164 | 6715 | 5140 | 0.0002x | 0.02% |
| B2_B3 | 86,182,644 | 13,108,305 | 11,448,368 | 0.3977x | 31.59% |
| B2_B5 | 82,504,751 | 16,069,879 | 13,838,347 | 0.4788x | 36.18% |
| B2_B6 | 54,440,203 | 13,936,061 | 11,947,290 | 0.3379x | 26.49% |
| B2_B7 | 40,649,429 | 3,347,196 | 2,961,184 | 0.0973x | 9.12% |
| Mitochondrial DNA | |||||
| B1_B2 | 51,212,164 | 41 | 39 | 0.2187x | 19.05% |
| B2_B3 | 86,182,644 | 8799 | 6847 | 43.8743x | 99.99% |
| B2_B5 | 82,504,751 | 18,689 | 12,439 | 82.3307x | 100% |
| B2_B6 | 54,440,203 | 7853 | 6212 | 33.8299x | 99.98% |
| B2_B7 | 40,649,429 | 2215 | 1991 | 11.0503x | 99.79% |
| Sample Name | Human Free Reads | Mapped Reads | Reads with a Mapping Quality > 30 | Average Coverage | Covered Positions (%) |
|---|---|---|---|---|---|
| B2_B1 | 148,214,698 | 273 | 132 | 0.003x | 0.31% |
| B2_B3 | 87,420,490 | 537,695 | 391,726 | 11.416x | 66.29% |
| B2_B5 | 84,107,411 | 4275 | 3366 | 0.081x | 7.23% |
| B2_B6 | 114,507,988 | 15,450 | 12,304 | 0.328x | 23.18% |
| B2_B7 | 117,513,937 | 428,178 | 318,145 | 8.889x | 83.23% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, A.E.; de-Dios, T.; Carrión, P.; Bonora, G.L.; Llovera, L.; Cilli, E.; Lizano, E.; Khabdulina, M.K.; Tleugabulov, D.T.; Olalde, I.; et al. Genomic Analysis of 18th-Century Kazakh Individuals and Their Oral Microbiome. Biology 2021, 10, 1324. https://doi.org/10.3390/biology10121324
White AE, de-Dios T, Carrión P, Bonora GL, Llovera L, Cilli E, Lizano E, Khabdulina MK, Tleugabulov DT, Olalde I, et al. Genomic Analysis of 18th-Century Kazakh Individuals and Their Oral Microbiome. Biology. 2021; 10(12):1324. https://doi.org/10.3390/biology10121324
Chicago/Turabian StyleWhite, Anna E., Toni de-Dios, Pablo Carrión, Gian Luca Bonora, Laia Llovera, Elisabetta Cilli, Esther Lizano, Maral K. Khabdulina, Daniyar T. Tleugabulov, Iñigo Olalde, and et al. 2021. "Genomic Analysis of 18th-Century Kazakh Individuals and Their Oral Microbiome" Biology 10, no. 12: 1324. https://doi.org/10.3390/biology10121324
APA StyleWhite, A. E., de-Dios, T., Carrión, P., Bonora, G. L., Llovera, L., Cilli, E., Lizano, E., Khabdulina, M. K., Tleugabulov, D. T., Olalde, I., Marquès-Bonet, T., Balloux, F., Pettener, D., van Dorp, L., Luiselli, D., & Lalueza-Fox, C. (2021). Genomic Analysis of 18th-Century Kazakh Individuals and Their Oral Microbiome. Biology, 10(12), 1324. https://doi.org/10.3390/biology10121324