
Merkel Cell Carcinoma: From Pathobiology to Clinical Management




 and
and 
Simple Summary
Abstract
1. Introduction
2. Epidemiology
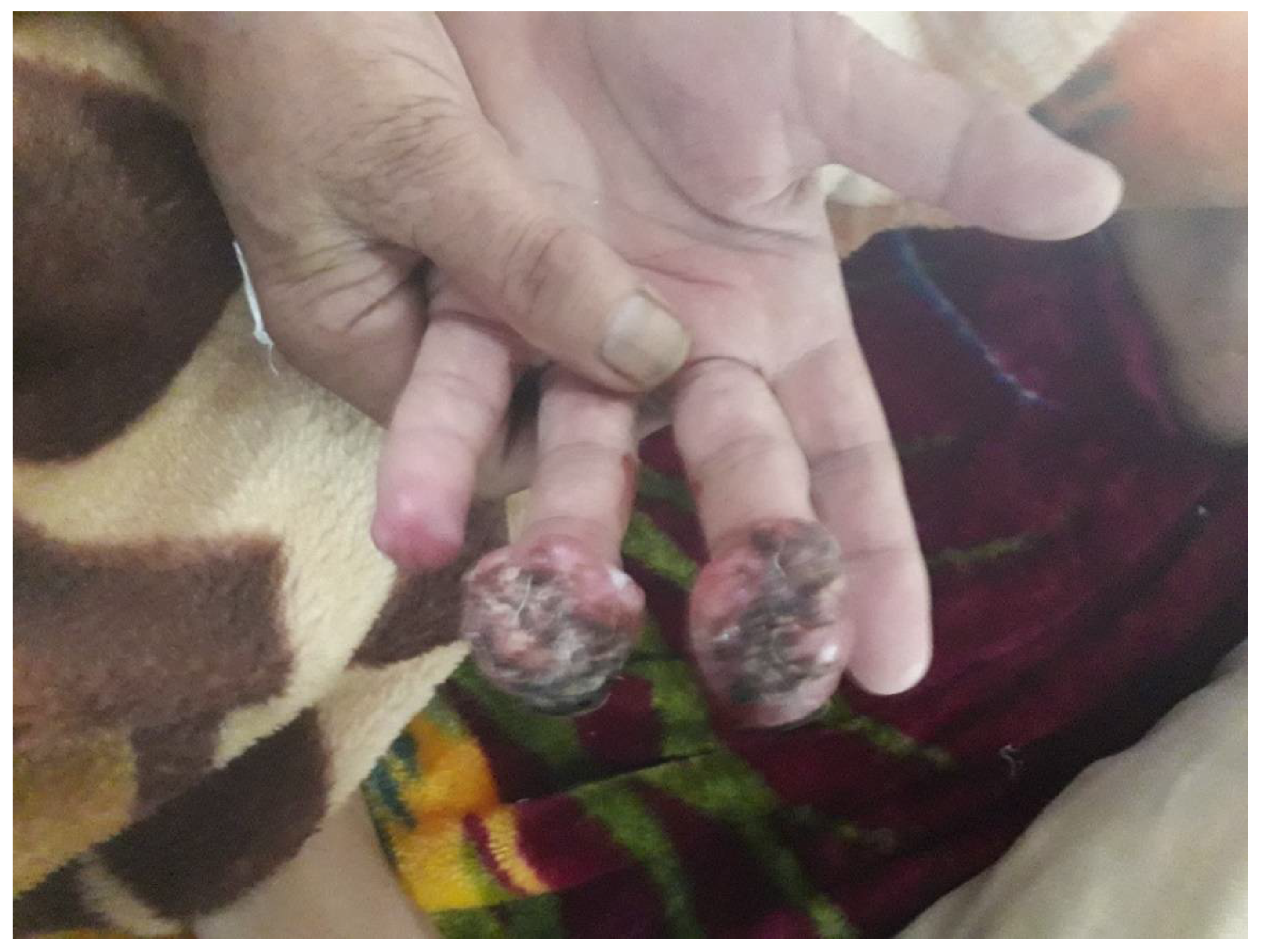
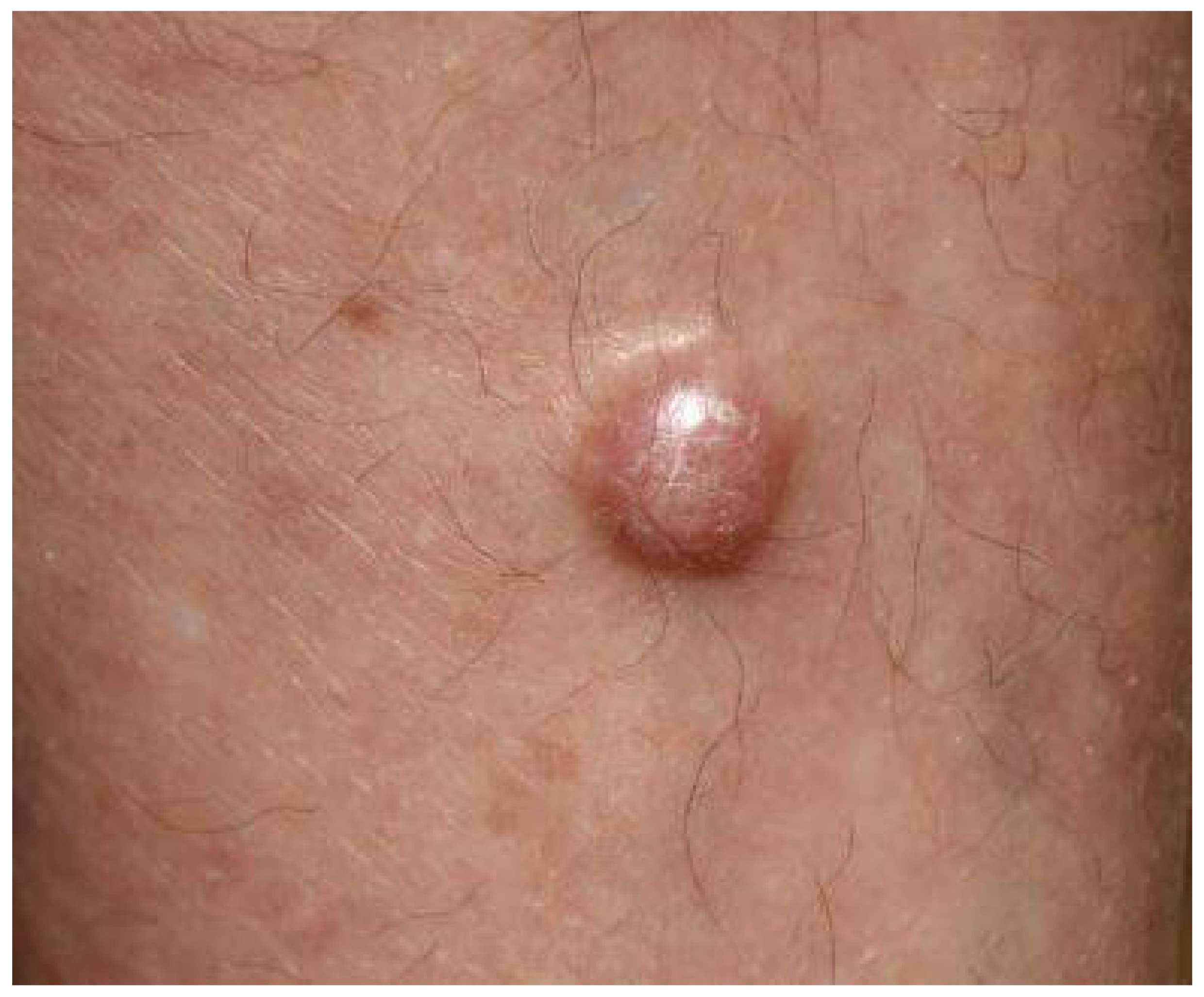
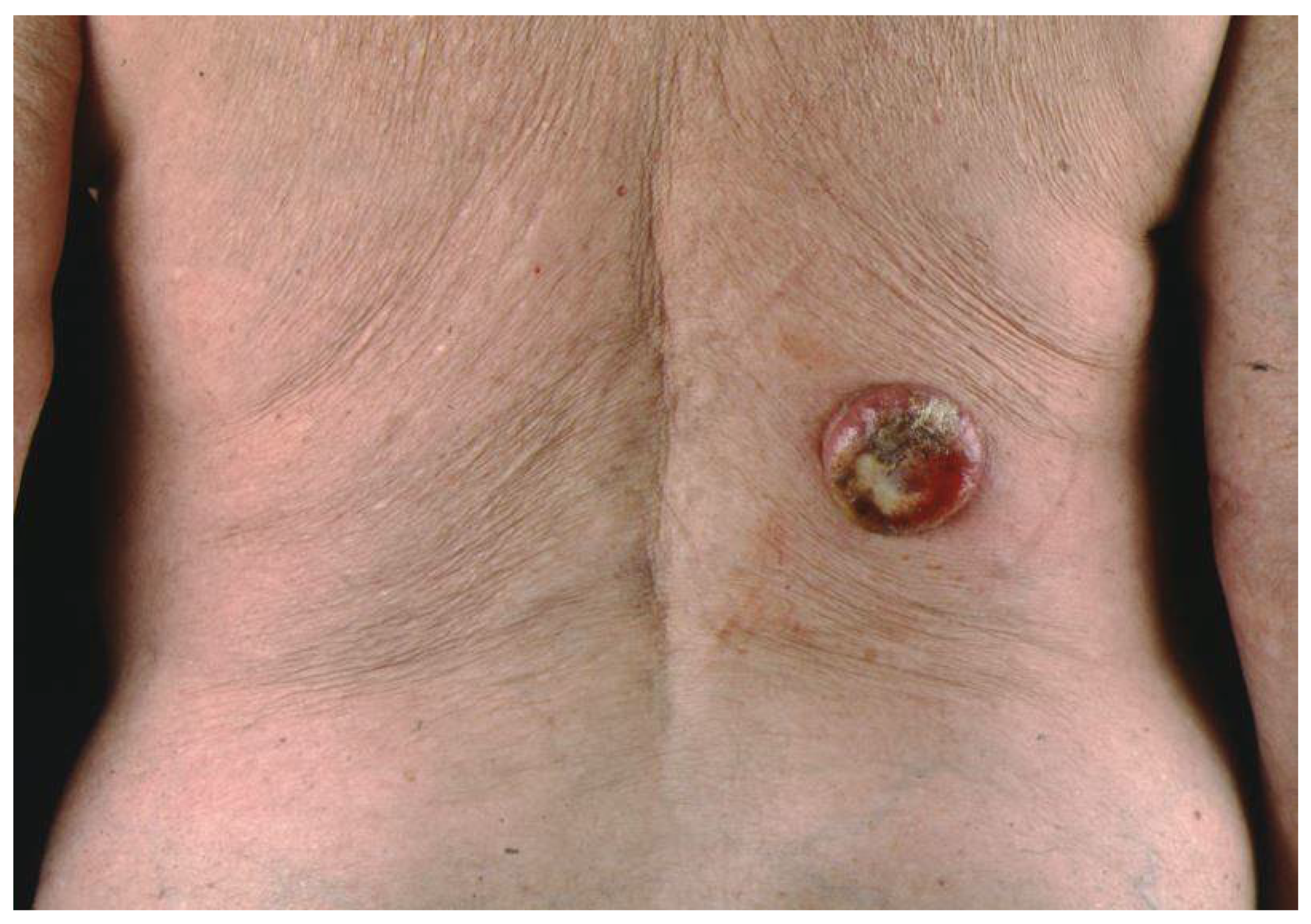
3. Clinical Presentation
4. Etiopathogenesis
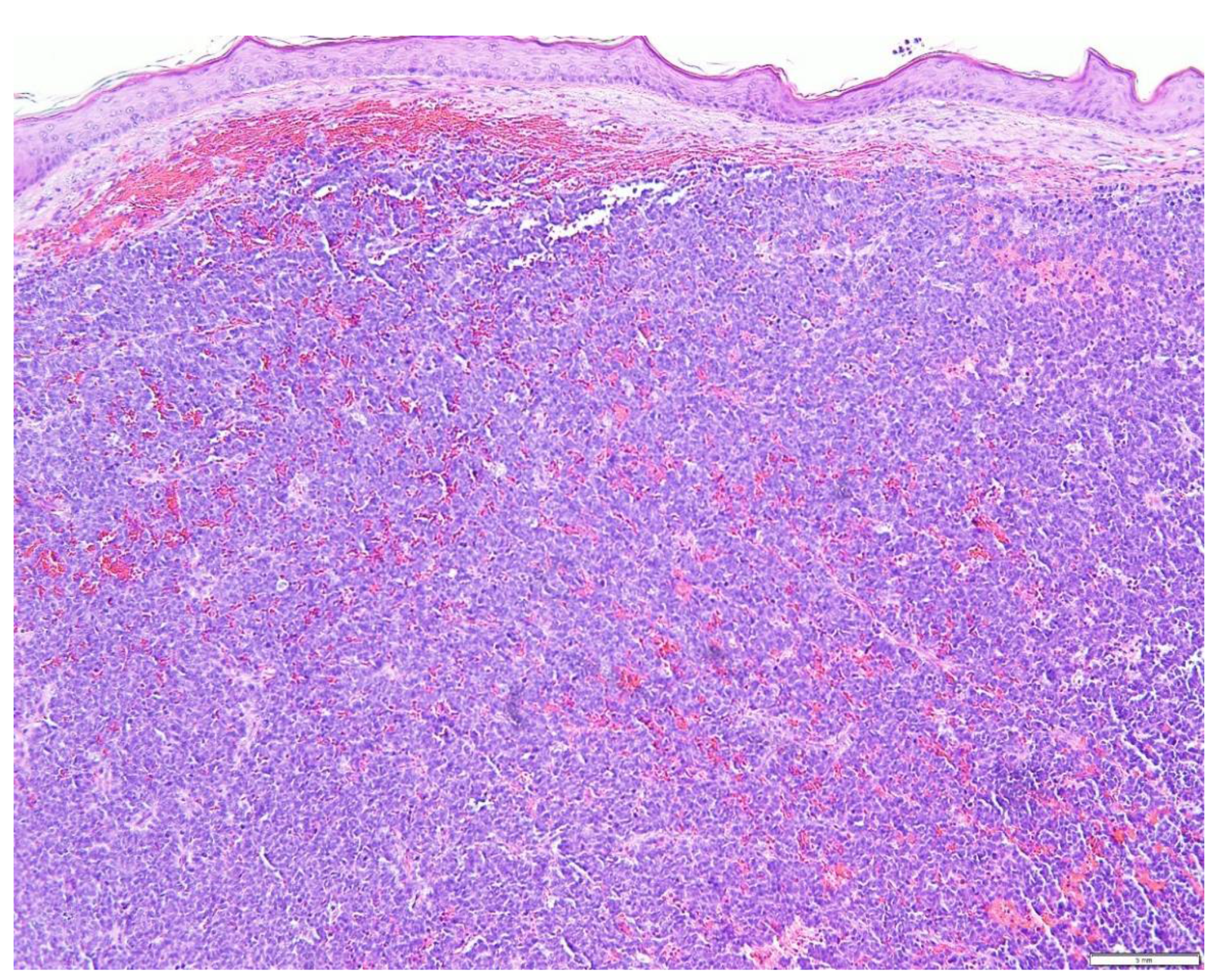
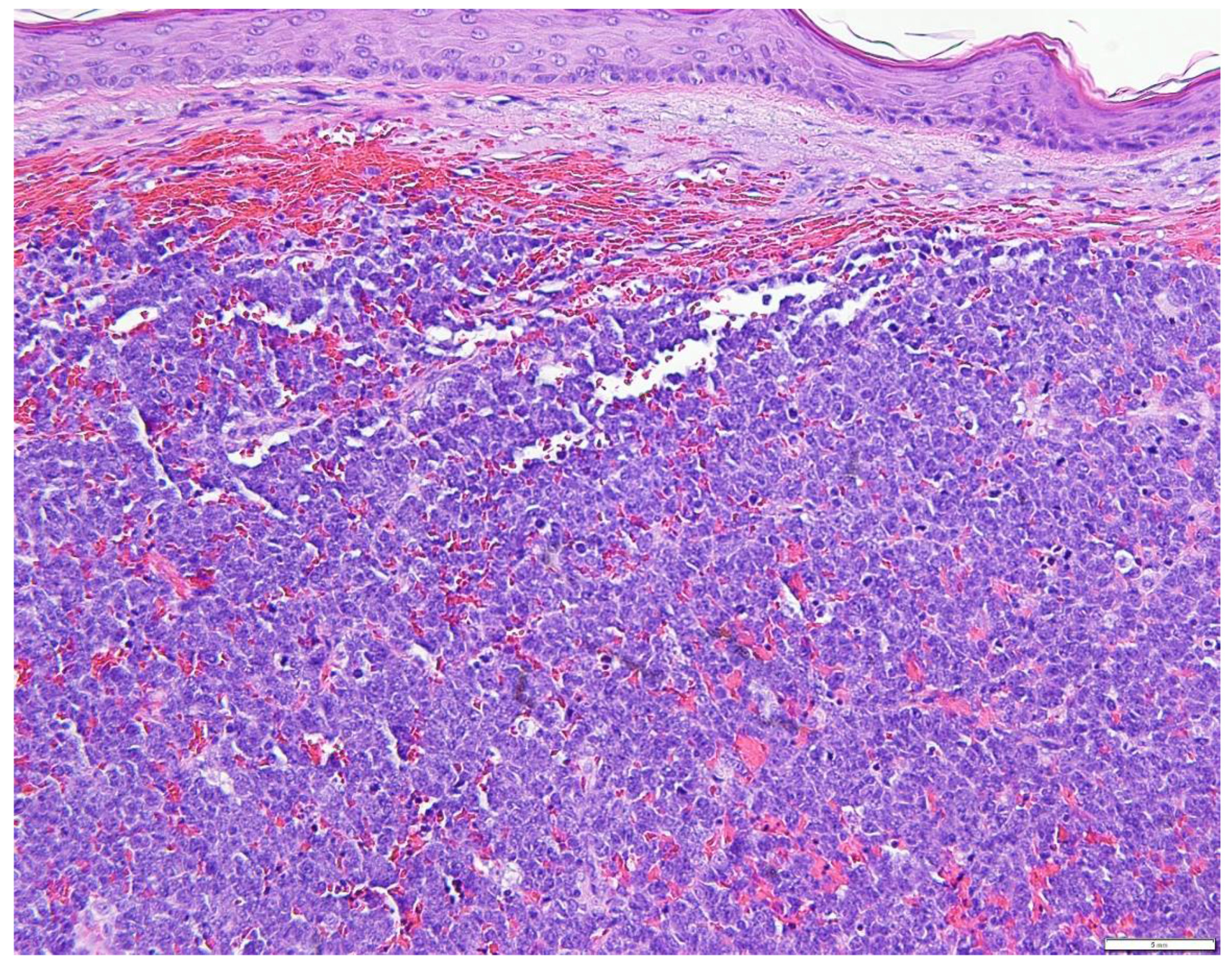
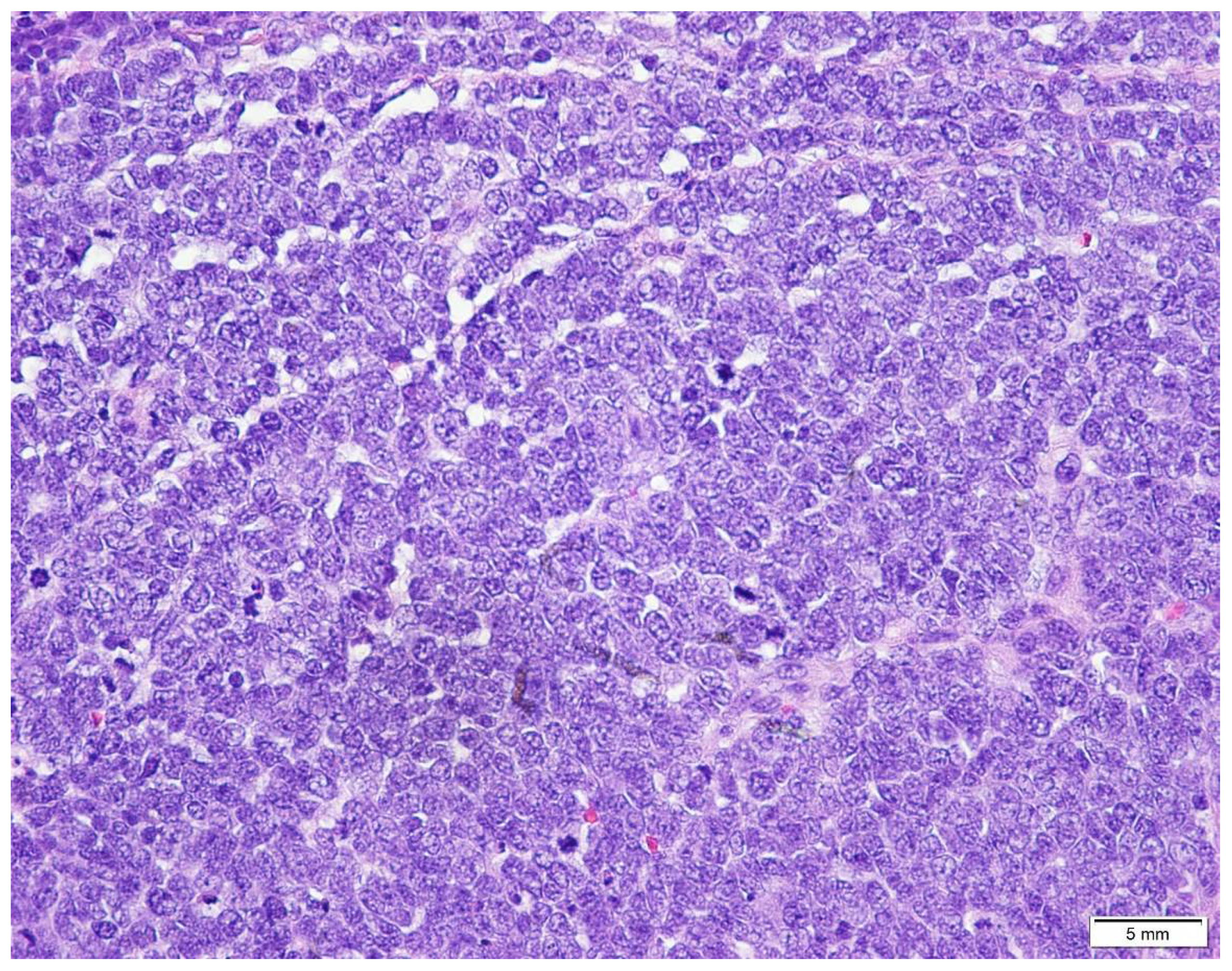
5. Diagnostic Evaluation/Staging
6. Prognostic Factors and Disease Progression
7. Treatment Options
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pan, D.; Narayan, D.; Ariyan, S. Merkel cell carcinoma: Five case reports using sentinel lymph node biopsy and a review of 110 new cases. Plast. Reconstr. Surg. 2002, 110, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Leech, S.N.; Kolar, A.J.O.; Barrett, P.D.; Sinclair, S.A.; Leonard, N. Merkel cell carcinoma can be distinguished from metastatic small cell carcinoma using antibodies to cytokeratin 20 and thyroid transcription factor 1. J. Clin. Pathol. 2001, 54, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Erovic, I.; Erovic, B.M. Merkel cell carcinoma: The past, the present, and the future. J. Ski. Cancer 2013, 2013, 929364. [Google Scholar] [CrossRef] [PubMed]
- Touzé, A.; Gaitan, J.; Maruani, A.; Le Bidre, E.; Doussinaud, A.; Clavel, C.; Durlach, A.; Aubin, F.; Guyétant, S.; Lorette, G.; et al. Merkel Cell Polyoma virus Strains in Patients with Merkel Cell Carcinoma. Emerg. Infect. Dis. 2009, 15, 960. [Google Scholar] [CrossRef]
- Wang, T.S.; Byrne, P.J.; Jacobs, L.K.; Taube, J.M. Merkel cell carcinoma: Update and review. Semin. Cutan. Med. Surg. 2011, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Hussain, K. Merkel Cell Carcinoma. Clin. Exp. Dermatol. 2021, 46, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Koljonen, V. Merkel cell carcinoma. World J. Surg. Oncol. 2006, 4, 7. [Google Scholar] [CrossRef]
- Becker, J.C.; Eigentler, T.; Frerich, B.; Gambichler, T.; Grabbe, S.; Höller, U.; Klumpp, B.; Loquai, C.; Krause-Bergmann, A.; Müller-Richter, U.; et al. S2k guidelines for merkel cell carcinoma (MCC, neuroendocrine carcinoma of the skin)—Update 2018. J. Dtsch. Dermatol. Ges. 2019, 17, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, C.; Juliano Carlos, S.; Balchiero, J.C.; Neto, B.R.C.; Graziosi, G.B.; de Paiva Dumaresq, F. Merkel cell carcinoma: Clinical presentation, prognostic factors, treatment and survival in 32 patients. Rev. Bras. Cir. Plástica 2013, 28, 196–200. [Google Scholar] [CrossRef]
- Schadendorf, D.; Lebbe, C.; Hausen, A.; Avril, M.-F.; Hariharan, S.; Bharmal, M.; Becker, J.C. Merkel cell carcinoma: Epidemiology, prognosis, therapy, and unmet medical needs. Eur. J. Cancer 2017, 71, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Senchenkov, A.; Steven, M.D.; Moran, L. Merkel cell carcinoma: Diagnosis, management, and outcomes. Plast. Reconstr. Surg. 2013, 131, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Tirumani, S.H.; Shinagare, A.B.; Sakellis, C.; Saboo, S.S.; Jagannathan, J.P.; Krajewski, K.M.; Ramaiya, N.H. Merkel cell carcinoma: A primer for the radiologist. Am. J. Roentgenol. 2013, 200, 1186–1196. [Google Scholar] [CrossRef]
- Coggshall, K.; Tello, T.L.; Jeffrey, P.; North, J.P.; Yu, S.S. Merkel cell carcinoma: An update and review: Pathogenesis, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, C.; Becker, J.C.; Grob, J.J.; Malvehy, J.; del Marmol, V.; Pehamberger, H.; Peris, K.; Saiag, P.; Middleton, M.R.; Bastholt, L.; et al. Diagnosis and treatment of Merkel Cell Carcinoma. European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Merkel Cell Carcinoma. Chapter 30. In AJCC Cancer Staging Manual; Springer: Berlin/Heidelberg, Germany, 2009.
- Bichakjian, C.K.; Lowe, L.; Lao, C.D.; Sandler, H.M.; Bradford, C.R.; Johnson, T.M.; Wong, S.L. Merkel cell carcinoma: Critical review with guidelines for multidisciplinary management. Cancer 2007, 110, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Panqueva, R.D.P.L.; Suarez-Zamora, D.A.; Barrera-Herrera, L.E.; Cadena, M.R. Merkel cell carcinoma and diagnostic experience in a reference hospital: A case series. Case Rep. Med. 2020, 2020, 8391510. [Google Scholar] [CrossRef]
- Ramahi, E.; Choi, J.; Fuller, C.D.; Eng, T.Y. Merkel cell carcinoma. Am. J. Clin. Oncol. 2013, 36, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C. Merkel cell carcinoma. Ann. Oncol. 2010, 21, 81–85. [Google Scholar] [CrossRef]
- Cassler, N.M.; Merrill, E.; Bichakjian, C.K.; Brownell, I. Merkel cell carcinoma therapeutic update. Curr. Treat. Options Oncol. 2016, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Pulitzer, M. Merkel cell carcinoma. Surg. Pathol. Clin. 2017, 10, 399–408. [Google Scholar] [CrossRef]
- Monteiro, A.; Gouveia, E.; Garcez, D.; Donato, S.; Martins-Branco, D.; Marques, J.; Nunes, H.; Passos, M.J.; Clara, A.I.; Moreira, A. Challenges of new approaches in metastatic merkel cell carcinoma. Case Rep. Oncol. 2020, 13, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Beebe, V. A review of merkel cell carcinoma for dermatology nurses. J. Dermatol. Nurses’ Assoc. 2009, 1, 173–179. [Google Scholar] [CrossRef]
- Gambichler, T.; Wieland, U.; Silling, S.; Dreißigacker, M.; Schaller, J.; Schulze, H.-J.; Oellig, F.; Kreuter, A.; Stücker, M.; Bechara, F.G.; et al. Left-Sided laterality of Merkel cell carcinoma in a German population: More than just sun exposure. J. Cancer Res. Clin. Oncol. 2016, 143, 347–350. [Google Scholar] [CrossRef]
- Ferringer, T.; Rogers, H.C.; Metcalf, J.S. Merkel cell carcinoma In Situ. J. Cutan. Pathol. 2005, 32, 162–165. [Google Scholar] [CrossRef]
- Bayrou, O.; Avril, M.F.; Charpentier, P.; Caillou, B.; Guillaume, J.C.; Prade, M. Primary neuroendocrine carcinoma of the skin. Clinicopathologic study of 18 cases. J. Am. Acad. Dermatol. 1991, 24, 198–207. [Google Scholar] [CrossRef]
- Sidhu, G.S.; Feiner, H.; Flotte, T.J.; Mullins, J.D.; Schaefler, K.; Schultenhover, S.J. Merkel cell neoplasms. Histology, electron microscopy, biology, and histogenesis. Am. J. Dermatopathol. 1980, 2, 101–119. [Google Scholar] [CrossRef]
- Wick, M.R.; Kaye, V.N.; Sibley, R.K.; Tyler, R.; Frizzera, G. Primary neuroendocrine carcinoma and small-cell malignant lymphoma of the skin. A discriminant immunohistochemical comparison. J. CutanPathol. 1986, 13, 347–358. [Google Scholar] [CrossRef]
- Vazmitel, M.; Michal, M.; Shelekhova, K.V.; Sima, R.; Mukensnabl, P.; Kazakov, D.V. Vascular changes in Merkel cell carcinoma based on a histopathological study of 92 cases. Am. J. Dermatopathol. 2008, 30, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M. Keratin 20: Immunohistochemical marker for gastrointestinal, urothelial, and merkel cell carcinomas. Mod. Pathol. 1995, 8, 384–388. [Google Scholar] [PubMed]
- Scott, M.P.; Helm, K.F. Cytokeratin 20: A marker for diagnosing merkel cell carcinoma. Am. J. Dermatopathol. 1999, 21, 16–20. [Google Scholar] [CrossRef]
- Bobos, M.; Hytiroglou, P.; Kostopoulos, I.; Karkavelas, G.; Papadimitriou, C.S. Immunohistochemical distinction between merkel cell carcinoma and small cell carcinoma of the lung. Am. J. Dermatopathol. 2006, 28, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Gloster, A.L.; Khoor, A.; Glass, L.F.; Messina, J.L.; Whitsett, J.A.; Livingstone, S.K.; Cagle, P.T. Differential expression of thyroid transcription factor 1 in small cell lung carcinoma and Merkel cell tumor. Hum. Pathol. 2000, 31, 58–62. [Google Scholar] [CrossRef]
- Chan, J.K.; Suster, S.; Wenig, B.M.; Tsang, W.Y.; Chan, J.B.; Lau, A.L. Cytokeratin 20 immunoreactivity distinguishes Merkel cell (primary cutaneous neuroendocrine) carcinomas and salivary gland small cell carcinomas from small cell carcinomas of various sites. Am. J. Surg. Pathol. 1997, 21, 226–234. [Google Scholar] [CrossRef]
- Calder, K.B.; Coplowitz, S.; Schlauder, S.; Morgan, M.B. A case series and immunophenotypic analysis of CK20–/CK7+ primary neuroendocrine carcinoma of the skin. J. CutanPathol. 2007, 34, 918–923. [Google Scholar] [CrossRef]
- Jaeger, T.; Ring, J.; Andres, C. Histological, immunohistological, and clinical features of merkel cell carcinoma in correlation to merkel cell polyomavirus status. J. Ski. Cancer 2012, 2012, 983421. [Google Scholar] [CrossRef]
- Chauhan, P.; Gupta, N.; Mardi, K.; Sharma, S.K.; Negi, A. Cytological, histopathological and immunohistochemical features of merkel cell carcinoma—A case report. Arch. Med. Health Sci. 2018, 6, 122. [Google Scholar] [CrossRef]
- Schrama, D.; Peitsch, W.K.; Zapatka, M.; Kneitz, H.; Houben, R.; Eib, S.; Haferkamp, S.; Moore, P.; Shuda, M.; Thompson, J.; et al. Merkel cell polyomavirus status is not associated with clinical course of merkel cell carcinoma. J. Investig. Dermatol. 2011, 131, 1631–1638. [Google Scholar] [CrossRef]
- Becker, J.C.; Kauczok, C.S.; Ugurel, S.; Eib, S.; Bröcker, E.B.; Houben, R. Merkel cell carcinoma: Molecular pathogenesis, clinical features, and therapy. J. Ger. Soc. Dermatol. 2008, 6, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.Y.; Liu, W.; Cohen, P.; Mahle, C.E.; Zhang, W. B-Cell-Specific activation protein encoded by the PAX-5 gene is commonly expressed in merkel cell carcinoma and small cell carcinomas. Am. J. Surg. Pathol. 2005, 29, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Buresh, C.J.; Oliai, B.R.; Miller, R.T. Reactivity with TdT in merkel cell carcinoma: A potential diagnostic pitfall. Am. J. Clin. Pathol. 2008, 129, 894–898. [Google Scholar] [CrossRef]
- Sur, M.; AlArdati, H.; Ross, C.; Alowami, S. TdT expression in merkel cell carcinoma: Potential diagnostic pitfall with blastic hematological malignancies and expanded immunohistochemical analysis. Mod. Pathol. 2007, 20, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Fang, W.; Liu, X.; Weiss, L.M.; Chu, P.G. Frequent expression of glypican-3 in Merkel cell carcinoma: An immunohistochemical study of 55 cases. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Brinkschmidt, C.; Stolze, P.; Fahrenkamp, A.G.; Hundeiker, M.; Fischer-Colbrie, R.; Zelger, B.; Bocker, W.; Schmid, K.W. Immunohistochemical demonstration of chromogranin A, chromogranin B, and secretoneurin in merkel cell carcinoma of the skin. An immunohistochemical study on 18 cases suggesting two types of Merkel cell carcinoma. Appl. Immunohistochem. 1995, 3, 37–44. [Google Scholar]
- Sibley, R.K.; Dahl, D. Primary neuroendocrine (merkel cell?) carcinoma of the skin. II. An immunocytochemical study of 21 cases. Am. J. Surg. Pathol. 1985, 9, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable tumor regression and overall survival in patients with advanced merkel cell carcinoma receiving pembrolizumab as first-line therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbé, C.; Lewis, K.D.; et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: Long-Term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J. Immunother. Cancer 2019, 8, e000674. [Google Scholar] [CrossRef]
- Reichgelt, B.; Visser, O. Epidemiology and survival of merkel cell carcinoma in the Netherlands. A population-based study of 808 cases in 1993–2007. Eur. J. Cancer 2011, 47, 579–585. [Google Scholar] [CrossRef]
- Tam, M.; Luu, M.; Barker, C.A.; Gharavi, N.M.; Hamid, O.; Shiao, S.L.; Nguyen, A.T.; Lu, D.J.; Ho, A.S.; Zumsteg, Z.S. Improved survival in women versus men with merkel cell carcinoma. J. Am. Acad. Dermatol. 2020, 84, 321–329. [Google Scholar] [CrossRef]
- Frohm, M.L.; Griffith, K.A.; Harms, K.L.; Hayman, J.A.; Fullen, D.R.; Nelson, C.C.; Wong, S.L.; Schwartz, J.L.; Bichakjian, C.K. Recurrence and survival in patients with merkel cell carcinoma undergoing surgery without adjuvant radiation therapy to the primary site. JAMA Dermatol. 2016, 152, 1001–1007. [Google Scholar] [CrossRef]
- Samimi, M.; Molet, L.; Fleury, M.J.J.; Laude, H.; Carlotti, A.; Gardair, C.; Baudin, M.; Gouguet, L.; Maubec, E.; Avenel-Audran, M.; et al. Prognostic value of antibodies to merkel cell polyomavirus T antigens and VP1 protein in patients with merkel cell carcinoma. Br. J. Dermatol. 2015, 174, 813–822. [Google Scholar] [CrossRef]
- Feldmeyer, L.; Hudgens, C.W.; Lyons, G.R.; Nagarajan, P.; Aung, P.P.; Curry, J.L.; Torres-Cabala, C.A.; Mino, B.; Rodriguez-Canales, J.; Reuben, A.; et al. Density, distribution, and composition of immune infiltrates correlate with survival in Merkel cell carcinoma. Clin. Cancer Res. 2016, 22, 5553–5563. [Google Scholar] [CrossRef] [PubMed]
- Husein-El Ahmed, H.; Ramos-Pleguezuelos, F.; Ruiz-Molina, I.; Civico-Amat, V.; Solis-García, E.; Galán-Gutierrez, M.; Ruiz-Villaverde, R. Histological features, p53, c-Kit, and poliomavirus status and impact on survival in merkel cell carcinoma patients. Am. J. Dermatopathol. 2016, 38, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.W.; Ng, L.B.; Murray, K. Mast cells have prognostic value in merkel cell carcinoma. Am. J. Dermatopathol. 2008, 30, 27–30. [Google Scholar] [CrossRef]
- Ng, L.; Beer, T.W.; Murray, K. Vascular density has prognostic value in merkel cell carcinoma. Am. J. Dermatopathol. 2008, 30, 442–445. [Google Scholar] [CrossRef]
- Llombart, B.; Monteagudo, C.; López-Guerrero, J.A.; Carda, C.; Jorda, E.; Sanmartin, O.; Almenar, S.; Molina, I.; Martín, J.M.; Llombart-Bosch, A. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005, 46, 622–634. [Google Scholar] [CrossRef]
- Bickle, K.; Glass, L.F.; Messina, J.L.; Fenske, N.A.; Siegrist, K. Merkel cell carcinoma: A clinical, histopathologic, and immunohistochemical review. Semin. Cutan. Med. Surg. 2003, 23, 46–53. [Google Scholar] [CrossRef]
- Stetsenko, G.Y.; Malekirad, J.; Paulson, K.; Iyer, J.G.; Thibodeau, R.M.; Nagase, K.; Schmidt, M.; Storer, B.E.; Argenyi, Z.B.; Nghiem, P. P63 expression in merkel cell carcinoma predicts poorer survival yet may have limited clinical utility. Am. J. Clin. Pathol. 2013, 140, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Green, M.D.; Hayman, J.A. Radiotherapy in the multidisciplinary management of merkel cell carcinoma. J. Natl. Compr. Cancer Netw. 2018, 16, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Ghidini, A.; Torchio, M.; Prinzi, N.; Trevisan, F.; Dallera, P.; De Stefani, A.; Russo, A.; Vitali, E.; Bruschieri, L.; et al. Adjuvant radiotherapy for merkel cell carcinoma: A systematic review and meta-analysis. Radiother. Oncol. 2019, 134, 211–219. [Google Scholar] [CrossRef]
- Mortier, L.; Mirabel, X.; Fournier, C.; Piette, F.; Lartigau, E. Radiotherapy alone for primary merkel cell carcinoma. Arch. Dermatol. 2003, 139, 1587–1590. [Google Scholar] [CrossRef]
- Lawenda, B.D.; Arnold, M.G.; Tokarz, V.A.; Silverstein, J.R.; Busse, P.M.; McIntyre, J.F.; Deschler, D.G.; Baldini, E.H.; Kachnic, L.A. Analysis of radiation therapy for the control of merkel cell carcinoma of the head and neck based on 36 cases and a literature review. Ear Nose Throat J. 2008, 87, 11. [Google Scholar] [CrossRef]
- Maclean, M.; Cook, M.M.; Stephanie, K.; Schaub, S.K.; Peter, H.; Goff, P.H.; Alex, F.A.; Park, S.Y.; Hippe, D.S.; Liao, J.J.; et al. Postoperative, single-fraction radiation therapy in merkel cell carcinoma of the head and neck. Adv. Radiat. Oncol. 2020, 5, 1248–1254. [Google Scholar]
- Schmults, C.D.; Donigan, J.M.; Park, S.; Blitzblau, R.; Farma, J.M.; Patel, T.; Aasi, S.Z.; Ghosh, K.; Puzanov, I.; Alam, M.; et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Merkel Cell Carcinoma Version 1.2021—February 18, 2021; National Comprehensive Cancer Network: Plymouth Meeting, PA, USA, 2021. [Google Scholar]
- Bhatia, S.; Storer, B.E.; Iyer, J.G.; Moshiri, A.; Parvathaneni, U.; Byrd, D.; Sober, A.J.; Sondak, V.K.; Gershenwald, J.E.; Nghiem, P. Adjuvant radiation therapy and chemotherapy in merkel cell carcinoma: Survival analyses of 6908 cases from the national cancer data base. J. Natl. Cancer Inst. 2016, 108, djw042. [Google Scholar] [CrossRef] [PubMed]
- Servy, A.; Maubec, E.; Sugier, P.; Grange, F.; Mansard, S.; Lesimple, T.; Marinho, E.; Couturaud, B.; Girod, A.; Albert, S.; et al. Merkel cell carcinoma: Value of sentinel lymph-node status and adjuvant radiation therapy. Ann. Oncol. 2016, 27, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.J.; Tanabe, K.K.; Gadd, M.A.; Stark, P.; Smith, B.L.; Finkelstein, D.M.; Souba, W.W. multimodality management of merkel cell carcinoma. Arch. Surg. 1999, 134, 388–393. [Google Scholar] [CrossRef]
- Chan, I.; Bhatia, S.; Kaufman, H.L.; Lipson, E.J. Immunotherapy for merkel cell carcinoma: A turning point in patient care. J. Immunother. Cancer 2018, 6, 23. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer.org|1.800.227.2345. Available online: https://www.cancer.org/content/dam/CRC/PDF/Public/8893.00.pdf (accessed on 16 September 2021).
- Colunga, A.; Pulliam, T.; Nghiem, P. Merkel cell carcinoma in the age of immunotherapy: Facts and hopes. Clin. Cancer Res. 2017, 24, 2035–2043. [Google Scholar] [CrossRef]
- Stege, H.M.; Bradfisch, F.; Fleischer, M.I.; Mohr, P.; Ugurel, S.; Terheyden, P.; Thiem, A.; Kiecker, F.; Leiter, U.; Becker, J.C.; et al. Retrospective multicentre analysis of the outcome of a re-induction with immune checkpoint inhibitors in advanced merkel cell carcinoma. SN Compr. Clin. Med. 2020, 2, 2202–2207. [Google Scholar] [CrossRef]
- Paulson, K.G.; Bhatia, S. Advances in immunotherapy for metastatic merkel cell carcinoma: A clinician’s guide. J. Natl. Compr. Cancer Netw. 2018, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | MCC | Lymphoma | Melanoma | SCLC |
|---|---|---|---|---|
| Cytokeratin 20 (CK 20) | + | − | − | − |
| Cytokeratin7 (CK 7) | − | − | − | + |
| Chromogranin A | +/− | − | − | +/− |
| HBM45 | − | − | + | − |
| Huntingtin-interacting protein 1 (HIP1) | + | +/− | − | − |
| Melan-A/MART-1 | − | − | + | − |
| Leucocyte common antigen (LCA) | − | + | − | |
| S100B | − | − | + | − |
| Thyroid transcription factor 1 (TTF-1) | − | − | − | + |
| Neuron-specific enolase | + | − | − | +/− |
| Vimentin | − | + | + | − |
| Stage | Primary Tumor | Lymph Node Status | Distant Metastasis |
|---|---|---|---|
| 0 | In situ | No nodes | No |
| I Clinical | Tumor ≤ 2 cm in maximum dimension | No nodes on clinical examination (histopathological examination not performed) | No |
| I Pathological | Tumor ≤ 2 cm in maximum dimension | Nodes negative by histopathologic examination | No |
| IIA Clinical | Tumor > 2 cm | No nodes on clinical examination (histopathological examination not performed) | No |
| IIA Pathological | Tumor > 2 cm | Nodes negative by histopathologic examination | No |
| IIB Clinical | Primary tumor involves fascia, muscle, bone, or cartilage | Clinically absent nodes (histopathology not performed) | No |
| IIB Pathological | Primary tumor involves fascia, muscle, bone, or cartilage | Histopathology demonstrates negative nodes | No |
| III Clinical | Tumor of any size/depth | Clinically nodes are present (histopathology not performed) | No |
| IIIA Pathological | Tumor of any size/depth | No nodes present on clinical examination, but nodes positive by pathological examination | No |
| Unknown primary | Nodes present on clinical examination and confirmed by histopathological examination | No | |
| IIIB Pathological | Tumor of any size/depth | Clinically nodes are present which are confirmed by histopathology or in-transit lesions | No |
| IV Clinical | Any | Present/absent | Clinically metastasis is present |
| IV Pathological | Any | Present/absent | Histopathological confirmation of metastasis |
| Site | Clinical Scenario | Recommended Dose |
|---|---|---|
| Primary | Surgical resection with wide margins (e.g., 1–2 cm) Small tumor size (<1–2 cm) | Consider observation |
| Surgical resection with negative margins | 50–56 Gy | |
| Surgical resection with histologically positive margins | 56–60 Gy | |
| Surgical resection with gross positive margins | 60–66 Gy | |
| No resection performed; as definitive therapy | 60–66 Gy | |
| Nodal basin | Node-negative (clinical examination); SLNB negative | Observation |
| Node-negative (clinical examination); SLNB/lymph node dissection not performed | 46–50 Gy | |
| Node-negative (clinical examination) SLNB positive, lymph node dissection not performed | 50–56 Gy | |
| Lymph node dissection with extracapsular extension; multiple positive Lymph nodes | 50–60 Gy | |
| Node positive (clinical examination); SLNB or lymph node dissection not performed | 60–66 Gy |
| Avelumab | Pembrolizumab | Nivolumab | Ipilimumab | |
|---|---|---|---|---|
| Mechanism of action | Anti-PD-L1 | Anti-PD-1 | Anti-PD-1 | Anti-CTLA-4 |
| Dose and schedule of administration | 10 mg/kg IV q2 week | 2 mg/kg q3 week | 240 mg IV q2 week | 3 mg/kg IV q3 weeks × 4 doses |
| ORR in chemotherapy naïve MCC | 69% (n = 16) | 56% (n = 26) | 71% (n = 14) | 40% (n = 5) |
| ORR in chemotherapy treated/second line MCC | 33% (n = 88) | N/A | 63% (n = 8) | |
| The median time of response | 6.1 weeks | 12 weeks | 2 months |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farooq Baba, P.U.; Rasool, Z.; Younas Khan, I.; Cockerell, C.J.; Wang, R.; Kassir, M.; Stege, H.; Grabbe, S.; Goldust, M. Merkel Cell Carcinoma: From Pathobiology to Clinical Management. Biology 2021, 10, 1293. https://doi.org/10.3390/biology10121293
Farooq Baba PU, Rasool Z, Younas Khan I, Cockerell CJ, Wang R, Kassir M, Stege H, Grabbe S, Goldust M. Merkel Cell Carcinoma: From Pathobiology to Clinical Management. Biology. 2021; 10(12):1293. https://doi.org/10.3390/biology10121293
Chicago/Turabian StyleFarooq Baba, Peerzada Umar, Zubaida Rasool, Ishrat Younas Khan, Clay J. Cockerell, Richard Wang, Martin Kassir, Henner Stege, Stephan Grabbe, and Mohamad Goldust. 2021. "Merkel Cell Carcinoma: From Pathobiology to Clinical Management" Biology 10, no. 12: 1293. https://doi.org/10.3390/biology10121293
APA StyleFarooq Baba, P. U., Rasool, Z., Younas Khan, I., Cockerell, C. J., Wang, R., Kassir, M., Stege, H., Grabbe, S., & Goldust, M. (2021). Merkel Cell Carcinoma: From Pathobiology to Clinical Management. Biology, 10(12), 1293. https://doi.org/10.3390/biology10121293