
Transcriptome Analysis Reveals Different Responsive Patterns to Nitrogen Deficiency in Two Wheat Near-Isogenic Lines Contrasting for Nitrogen Use Efficiency


 ,
, 
 ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Pot Experiment and Sampling
2.2. RNA Extraction, Library Construction, and Transcript Profiling
2.3. Confirmation of RNA-Seq Data by qRT-PCR Analysis
2.4. Statistical Analysis
3. Results
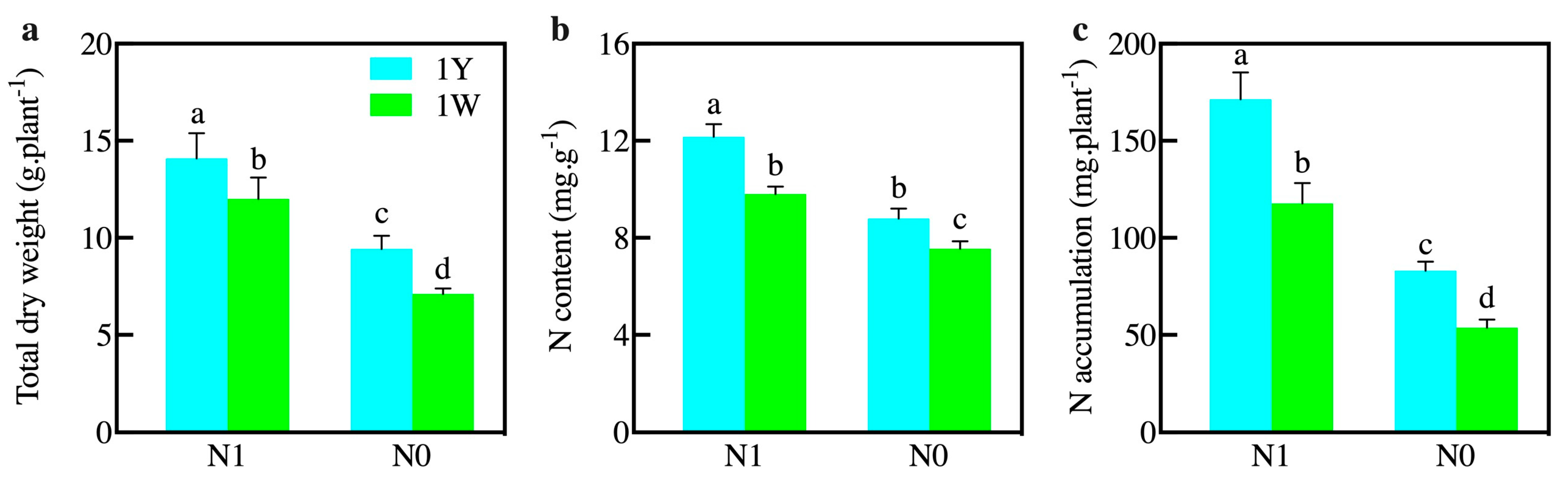
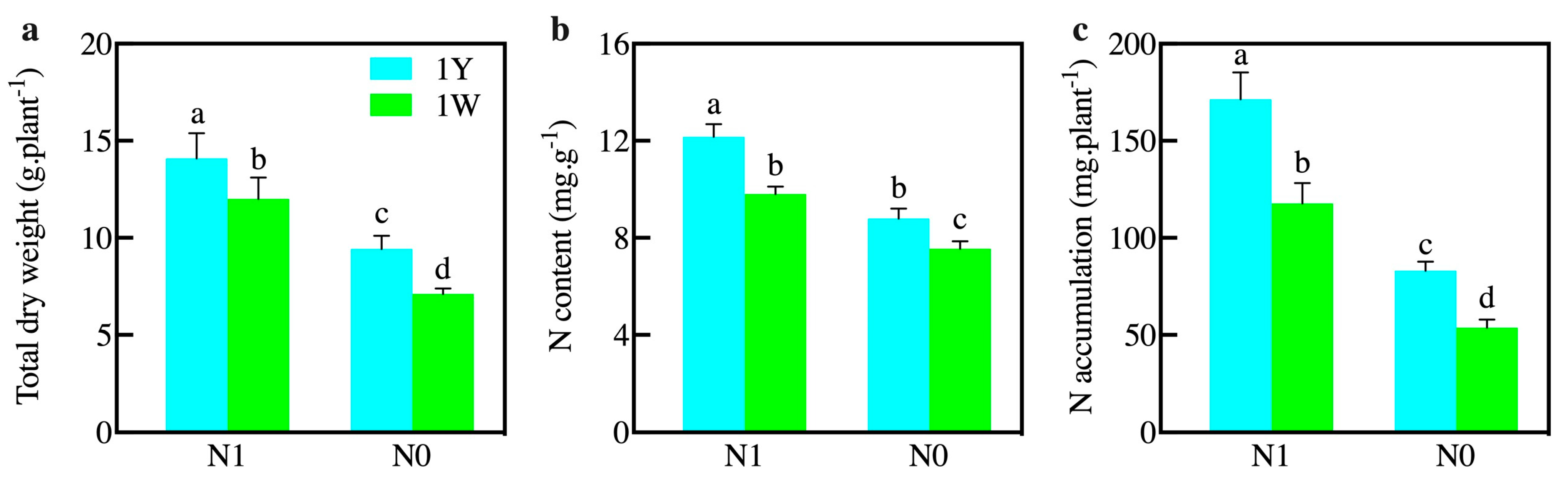
3.1. Influences of N Level on the Growth Parameters and Enzyme Activities of Wheat NILs
3.2. Analysis of Transcriptome Sequencing Data
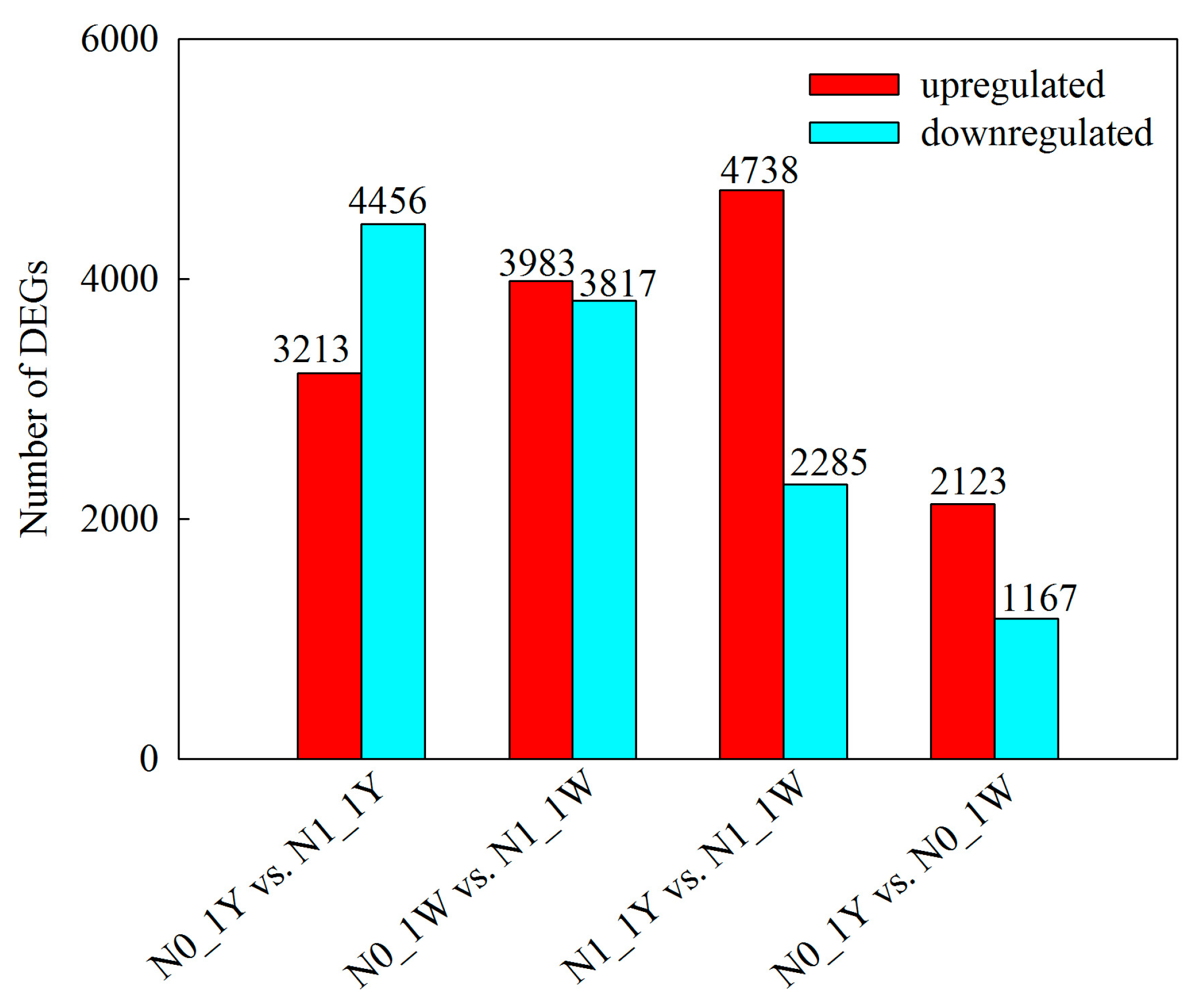
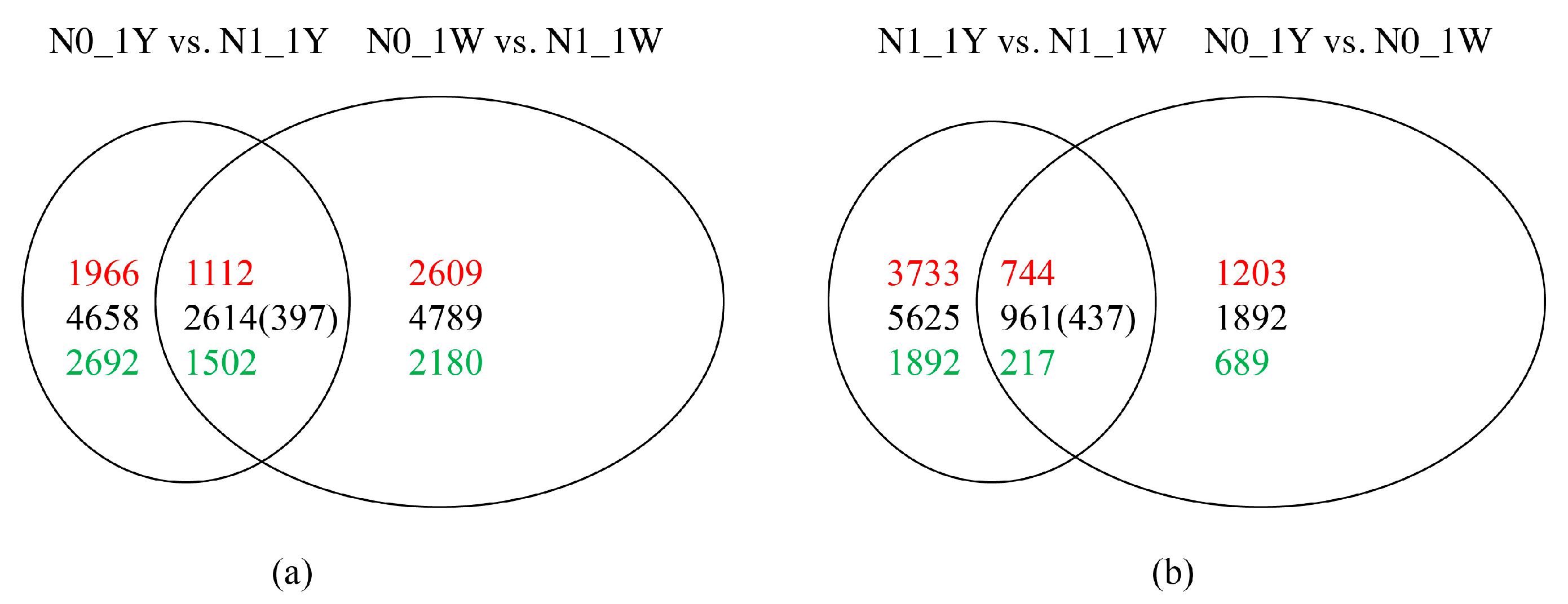
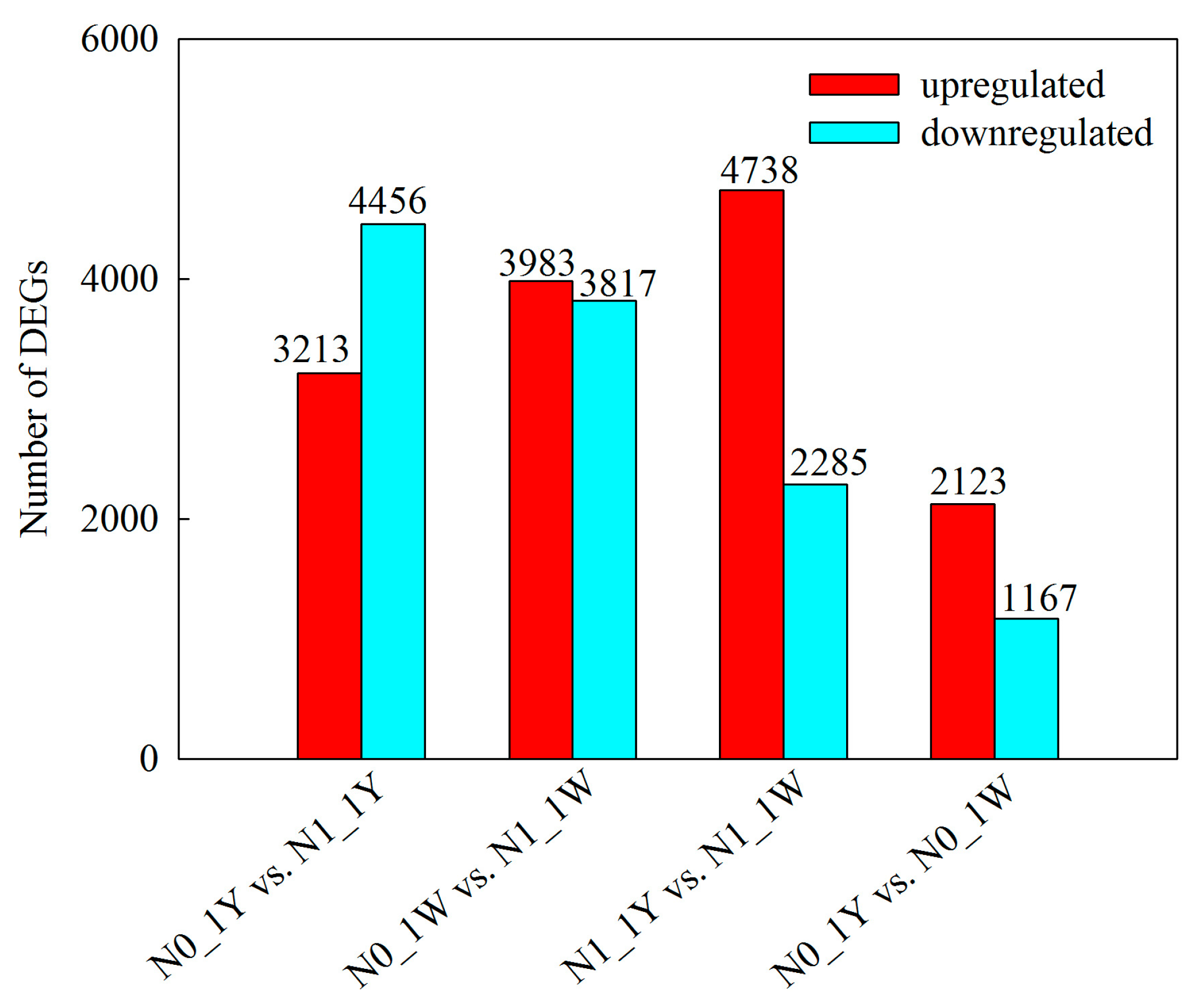
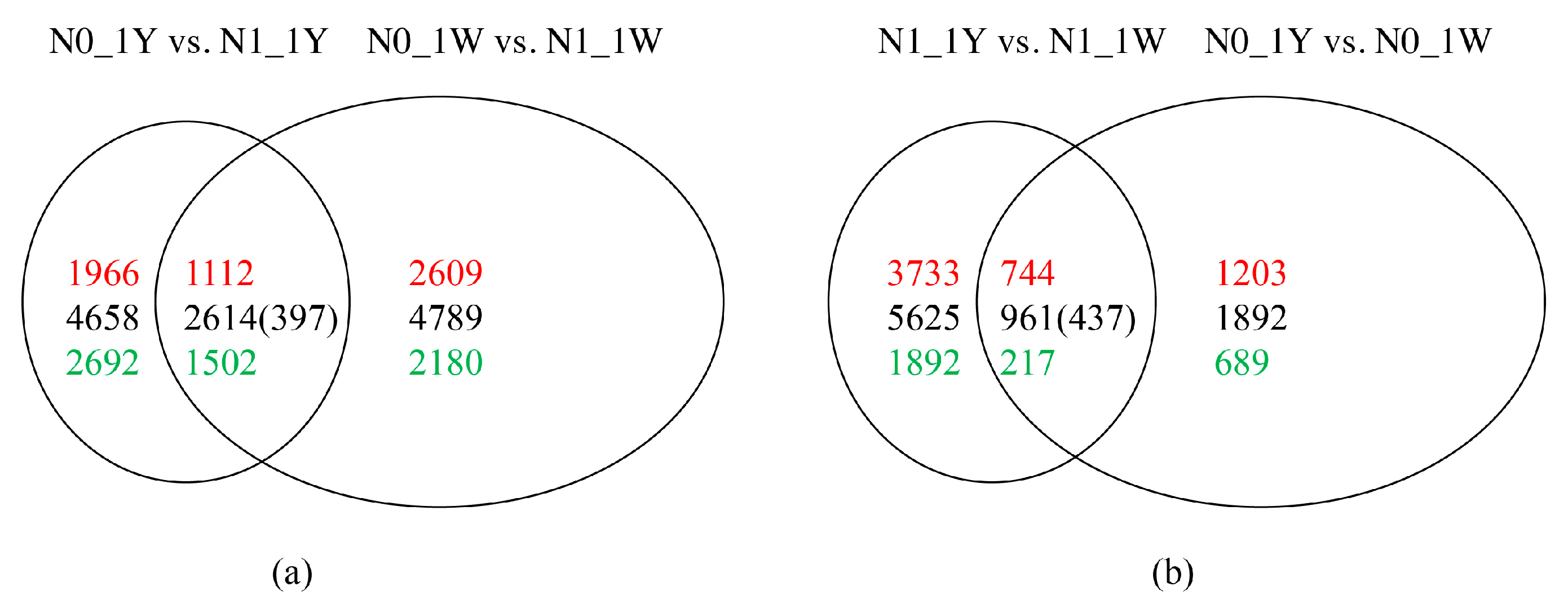
3.3. Identification and Validation of DEGs
3.4. Functional Characterization of the DEGs by GO and KEGG Pathway Analysis
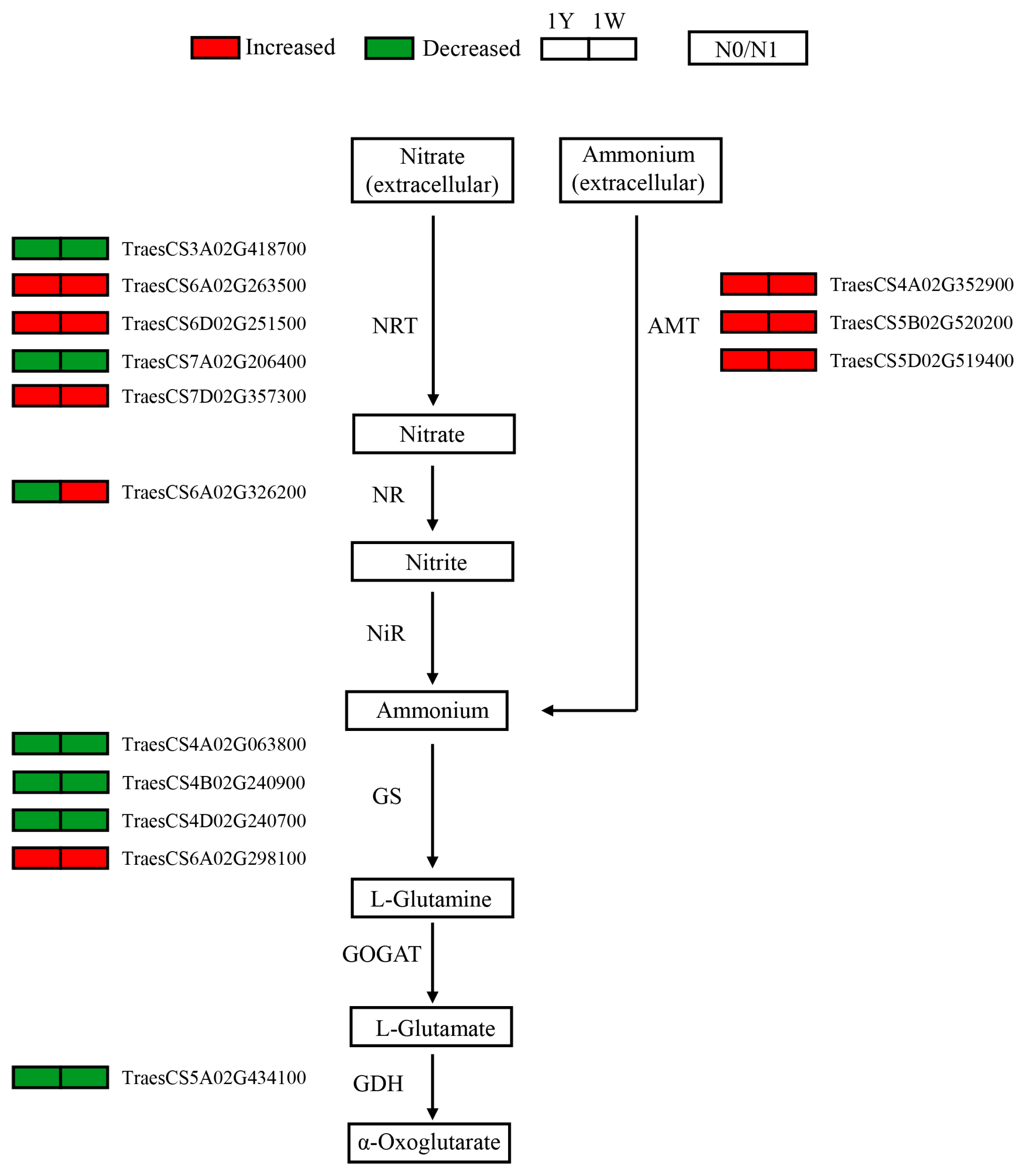
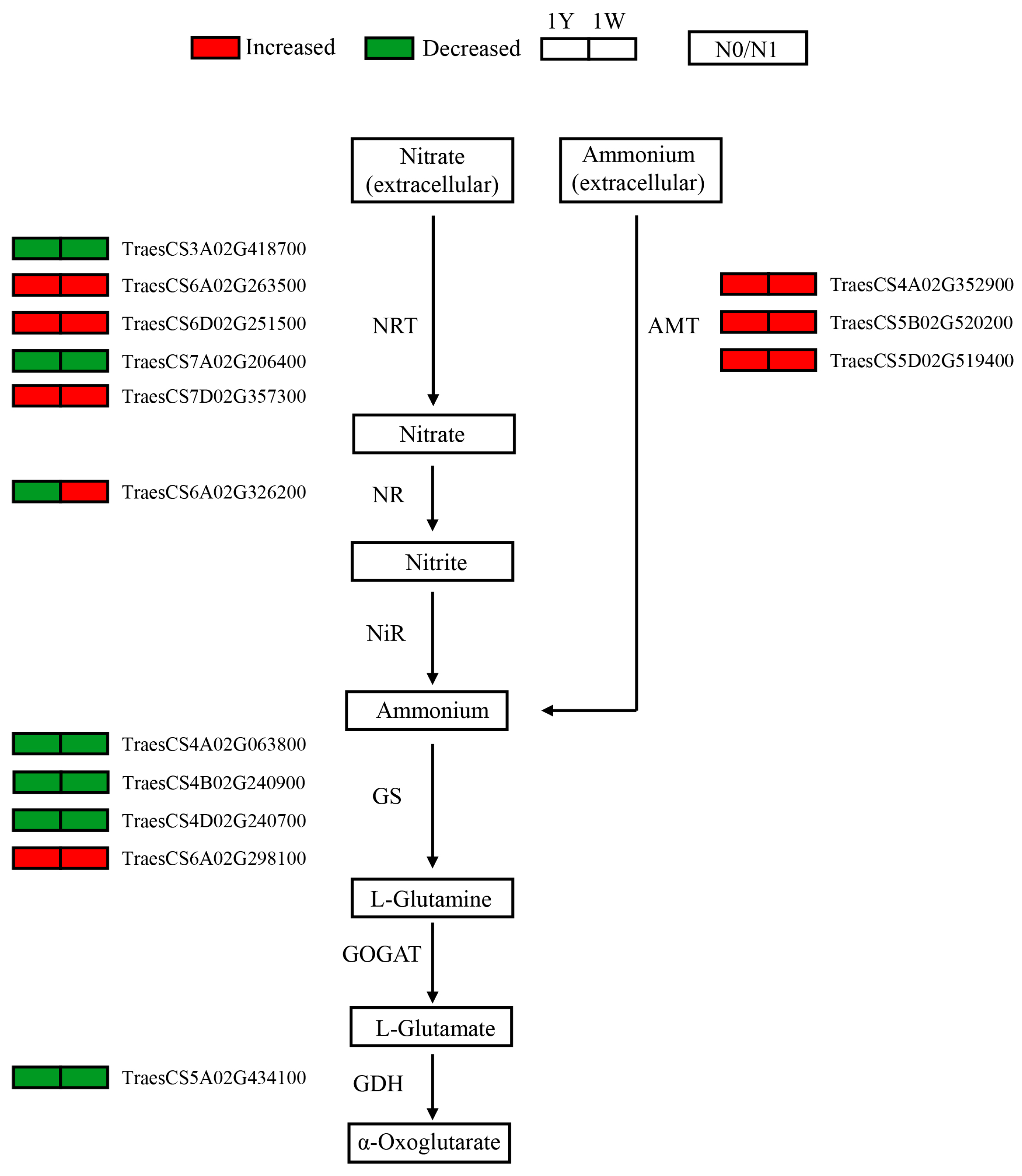
3.5. Responses of Genes Involved in Nitrogen Metabolism to N-Deficit Conditions
3.6. Specific Responses of the High-NUE Wheat to N-Deficit Conditions
3.7. Specific Responses of the Low-NUE Wheat to N-Deficit Conditions
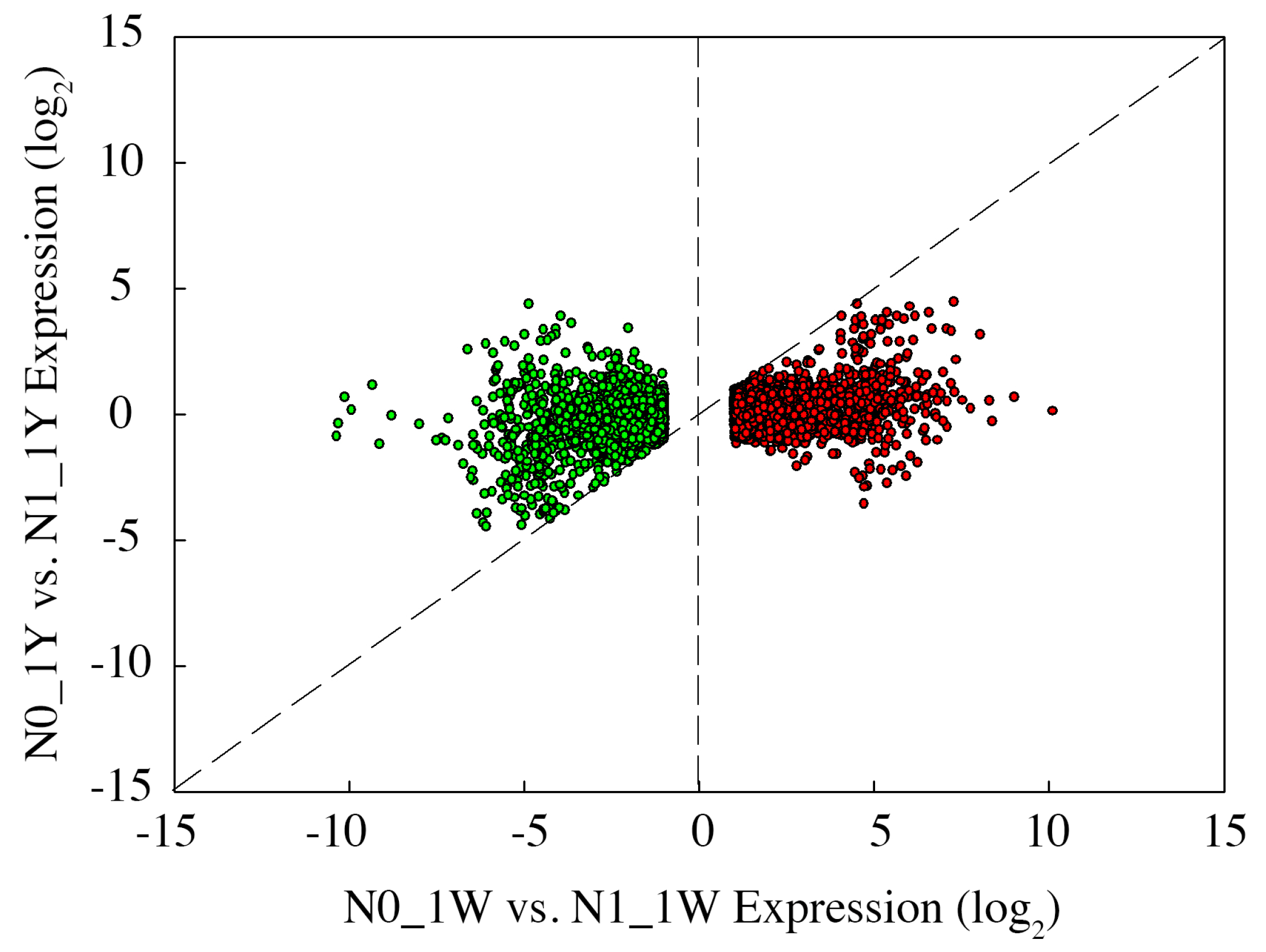
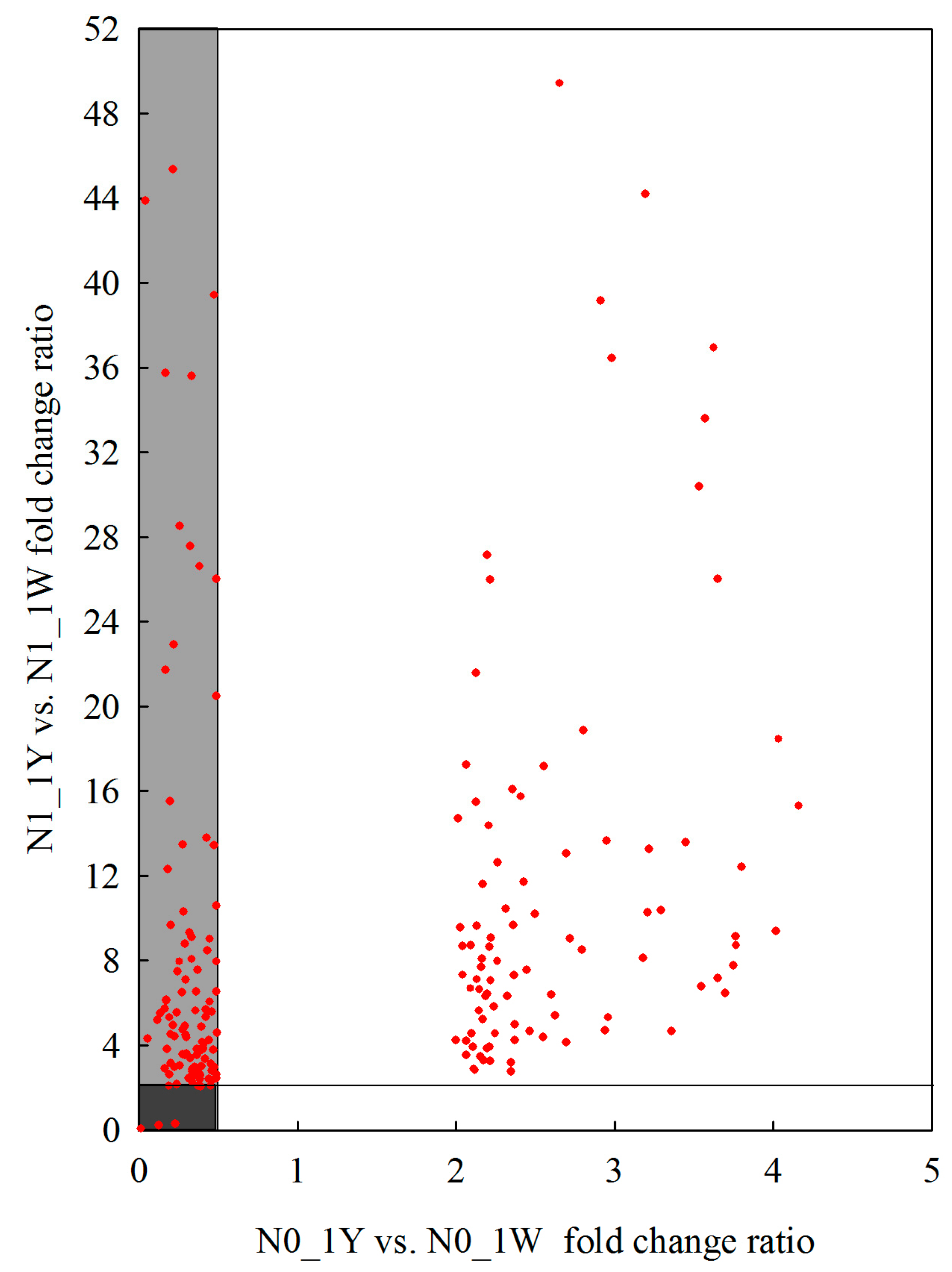
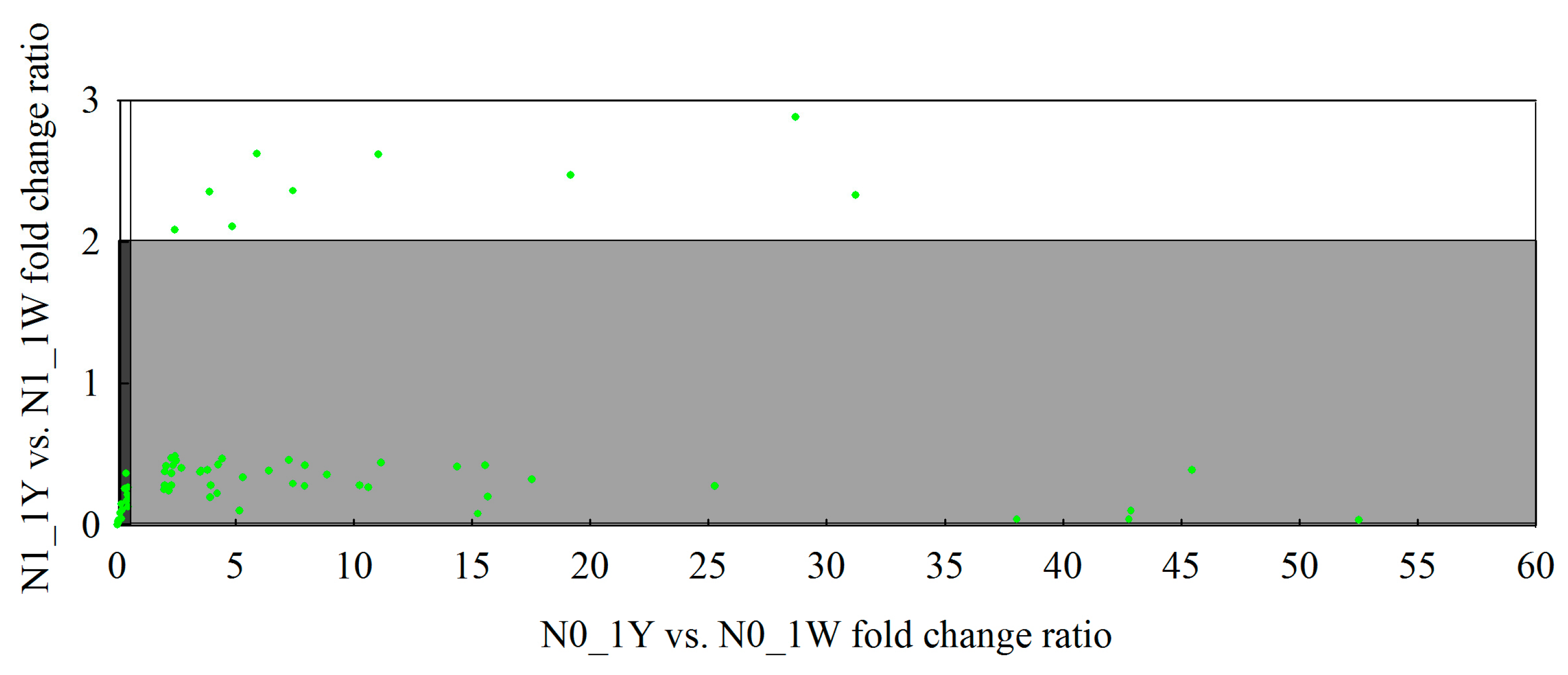
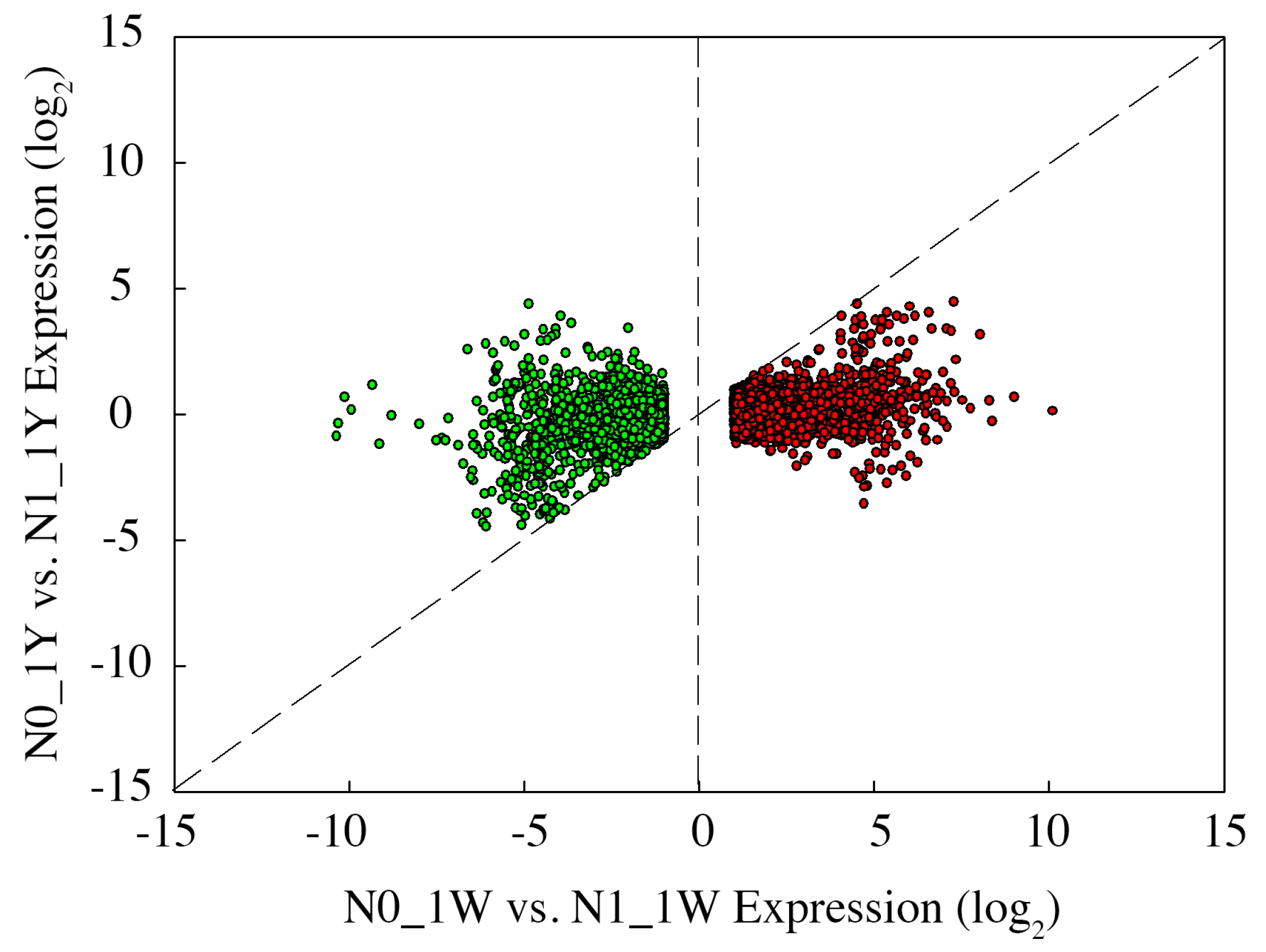
3.8. Classification of N-Responsive Genes in Wheat
4. Discussion
4.1. Nitrogen Metabolism Genes Responsive to N-Deficient Conditions
4.2. Some Potential Genes for High-NUE Wheat Response to N-Deficient Conditions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, Z.; Yu, Z.; Zhao, J. Theory and application for the promotion of wheat production in China: Past, present and future. J. Sci. Food Agric. 2013, 93, 2339–2350. [Google Scholar] [CrossRef]
- Tegeder, M.; Masclaux-Daubresse, C. Source and sink mechanisms of nitrogen transport and use. New Phytol. 2018, 217, 35–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Song, K.; Sun, L.; Qin, Q.; Sun, Y.; Pan, J.; Xue, Y. Morphological and Transcriptome Analysis of Wheat Seedlings Response to Low Nitrogen Stress. Plants 2019, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Good, A.G.; Beatty, P.H. Fertilizing nature: A tragedy of excess in the commons. PLoS Biol. 2011, 9, 1001124. [Google Scholar] [CrossRef] [PubMed]
- Gaju, O.; Allard, V.; Martre, P.; Snape, J.W.; Heumez, E.; LeGouis, J. Identification of traits to improve the nitrogen-use efficiency of wheat genotypes. Field Crops Res. 2011, 123, 139–152. [Google Scholar] [CrossRef]
- Ahmed, M.; Rauf, M.; Mukhtar, Z.; Saeed, N.A. Excessive use of nitrogenous fertilizers: An unawareness causing serious threats to environment and human health. Environ. Sci. Pollut. Res. Int. 2017, 24, 26983–26987. [Google Scholar] [CrossRef] [PubMed]
- Randall, C.W. The environmental, economic and societal consequences of inadequate nitrogen pollution controls. Water Sci. Technol. 2004, 49, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Motesharezadeh, B.; Etesami, H.; Bagheri-Novair, S.; Amirmokri, H. Fertilizer consumption trend in developing countries vs. developed countries. Environ. Monit. Assess. 2017, 189, 103. [Google Scholar] [CrossRef]
- Wu, Z.; Luo, J.; Han, Y.; Hua, Y.; Guan, C.; Zhang, Z. Low Nitrogen Enhances Nitrogen Use Efficiency by Triggering NO3− Uptake and Its Long-Distance Translocation. J. Agric. Food Chem. 2019, 67, 6736–6747. [Google Scholar] [CrossRef]
- Iqbal, A.; Qiang, D.; Alamzeb, M.; Xiangru, W.; Huiping, G.; Hengheng, Z.; Nianchang, P.; Xiling, Z.; Meizhen, S. Untangling the molecular mechanisms and functions of nitrate to improve nitrogen use efficiency. J. Sci. Food Agric. 2020, 100, 904–914. [Google Scholar] [CrossRef]
- Han, M.; Okamoto, M.; Beatty, P.H.; Rothstein, S.J.; Good, A.G. The Genetics of Nitrogen Use Efficiency in Crop Plants. Annu. Rev. Genet. 2015, 49, 269–289. [Google Scholar] [CrossRef]
- Vidal, E.A.; Alvarez, J.M.; Araus, V.; Riveras, E.; Brooks, M.D.; Krouk, G.; Ruffel, S.; Lejay, L.; Crawford, N.M.; Coruzzi, G.M.; et al. Nitrate in 2020: Thirty Years from Transport to Signaling Networks. Plant Cell 2020, 32, 2094–2119. [Google Scholar] [CrossRef] [PubMed]
- Baslam, M.; Mitsui, T.; Sueyoshi, K.; Ohyama, T. Recent Advances in Carbon and Nitrogen Metabolism in C3 Plants. Int. J. Mol. Sci. 2020, 22, 318. [Google Scholar] [CrossRef]
- Swarbreck, S.M.; Wang, M.; Wang, Y.; Kindred, D.; Sylvester-Bradley, R.; Shi, W.; Varinderpal-Singh; Bentley, A.R.; Griffiths, H. A Roadmap for Lowering Crop Nitrogen Requirement. Trends Plant Sci. 2019, 24, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Hawkesford, M.J. Reducing the reliance on nitrogen fertilizer for wheat production. J. Cereal Sci. 2014, 59, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, J.M.; Muntaha, S.T.; Ali, E.; Khan, A.A.; Zaidi, S.H.R.; Shahzad, A.N.; Hassan, Z.; Nawaz, A.; Rashid, M.; Bukhari, S.A.H. Comparative study of the genetic basis of nitrogen use efficiency in wild and cultivated barley. Physiol. Mol. Biol. Plants 2019, 25, 1435–1444. [Google Scholar] [CrossRef]
- Wei, Y.; Shi, A.; Jia, X.; Zhang, Z.; Ma, X.; Gu, M.; Meng, X.; Wang, X. Nitrogen Supply and Leaf Age Affect the Expression of TaGS1 or TaGS2 Driven by a Constitutive Promoter in Transgenic Tobacco. Genes 2018, 9, 406. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Bi, Y.M.; Rothstein, S.J. Understanding plant response to nitrogen limitation for the improvement of crop nitrogen use efficiency. J. Exp. Bot. 2011, 62, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Chen, X.; Wu, K.; Fu, X. Nitrogen signaling and use efficiency in plants: What’s new? Curr. Opin. Plant Biol. 2015, 27, 192–198. [Google Scholar] [CrossRef]
- Goel, P.; Sharma, N.K.; Bhuria, M.; Sharma, V.; Chauhan, R.; Pathania, S.; Swarnkar, M.K.; Chawla, V.; Acharya, V.; Shankar, R.; et al. Transcriptome and Co-Expression Network Analyses Identify Key Genes Regulating Nitrogen Use Efficiency in Brassica juncea L. Sci. Rep. 2018, 8, 7451. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Yang, X.; Yu, H.; Jiang, W.; Sun, N.; Liu, X.; Liu, X.; Zhang, X.; Wang, Y.; Gu, X. RNA-Seq-based transcriptome profiling of early nitrogen deficiency response in cucumber seedlings provides new insight into the putative nitrogen regulatory network. Plant Cell Physiol. 2015, 56, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yin, C.; Xiang, L.; Jiang, W.; Xu, S.; Mao, Z. Transcription strategies related to photosynthesis and nitrogen metabolism of wheat in response to nitrogen deficiency. BMC Plant Biol. 2020, 20, 448. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Li, Z.; Zhao, Y.; Peng, C.; Zhao, C. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlüter, U.; Mascher, M.; Colmsee, C.; Scholz, U.; Bräutigam, A.; Fahnenstich, H.; Sonnewald, U. Maize source leaf adaptation to nitrogen deficiency affects not only nitrogen and carbon metabolism but also control of phosphate homeostasis. Plant Physiol. 2012, 160, 1384–1406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, F.; Ding, Y.; Ma, Q.; Yi, Y.; Zhu, M.; Ding, J.; Li, C.; Guo, W.; Zhu, X. Transcriptome Analysis of Two Near-Isogenic Lines with Different NUE under Normal Nitrogen Conditions in Wheat. Biology 2021, 10, 787. [Google Scholar] [CrossRef]
- Munoz-Huerta, R.F.; Guevara-Gonzalez, R.G.; Contreras-Medina, L.M.; Torres-Pacheco, I.; Prado-Olivarez, J.; Ocampo-Velazquez, R.V. A review of methods for sensing the nitrogen status in plants: Advantages, disadvantages and recent advances. Sensors 2013, 13, 10823–10843. [Google Scholar] [CrossRef]
- Siren, J.; Valimaki, N.; Makinen, V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.; Zeng, J.; Chen, G.; Zhang, G. Transcriptomic analysis reveals adaptive strategies to chronic low nitrogen in Tibetan wild barley. BMC Plant Biol. 2019, 19, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2^(−delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]
- Zörb, C.; Ludewig, U.; Hawkesford, M.J. Perspective on Wheat Yield and Quality with Reduced Nitrogen Supply. Trends Plant Sci. 2018, 23, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Cormier, F.; Faure, S.; Dubreuil, P.; Heumez, E.; Beauchêne, K.; Lafarge, S.; Praud, S.; Le Gouis, J. A multi-environmental study of recent breeding progress on nitrogen use efficiency in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2013, 126, 3035–3048. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Yadav, B.; Raj, U.; Freilich, S.; Varadwaj, P.K. Transcriptomic Analysis of Soil Grown T. aestivum cv. Root to Reveal the Changes in Expression of Genes in Response to Multiple Nutrients Deficiency. Front. Plant Sci. 2017, 8, 1025. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.Y.; Jeong, J.S.; Lim, J.Y.; Kim, T.; Park, J.H.; Kim, J.K.; Shin, C. Transcriptomic analyses of rice (Oryza sativa) genes and non-coding RNAs under nitrogen starvation using multiple omics technologies. BMC Genom. 2018, 19, 532. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.; Zeng, J.; Ye, L.; Chen, G.; Han, Z.; Shah, J.M.; Zhang, G. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016, 16, 30. [Google Scholar] [CrossRef] [Green Version]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef]
- Balotf, S.; Kavoosi, G.; Kholdebarin, B. Nitrate reductase, nitrite reductase, glutamine synthetase, and glutamate synthase expression and activity in response to different nitrogen sources in nitrogen-starved wheat seedlings. Biotechnol. Appl. Biochem. 2016, 63, 220–229. [Google Scholar] [CrossRef]
- Tian, R.; Yang, Y.; Chen, M. Genome-wide survey of the amino acid transporter gene family in wheat (Triticum aestivum L.): Identification, expression analysis and response to abiotic stress. Int. J. Biol. Macromol. 2020, 162, 1372–1387. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; King, R.; Mitchell, R.A.C.; Hassani-Pak, K.; Hawkesford, M.J. Spatiotemporal expression patterns of wheat amino acid transporters reveal their putative roles in nitrogen transport and responses to abiotic stress. Sci. Rep. 2017, 7, 5461. [Google Scholar] [CrossRef] [PubMed]
- Del Río, L.A. ROS and RNS in plant physiology: An overview. J. Exp. Bot. 2015, 66, 2827–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef]
- Nadarajah, K.K. ROS Homeostasis in Abiotic Stress Tolerance in Plants. Int. J. Mol. Sci. 2020, 21, 5208. [Google Scholar] [CrossRef]
- Pandian, B.A.; Sathishraj, R.; Djanaguiraman, M.; Prasad, P.V.V.; Jugulam, M. Role of Cytochrome P450 Enzymes in Plant Stress Response. Antioxidants 2020, 9, 454. [Google Scholar] [CrossRef] [PubMed]
- Curci, P.L.; Cigliano, R.A.; Zuluaga, D.L.; Janni, M.; Sanseverino, W.; Sonnante, G. Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 2017, 7, 1176. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Wang, Z.; Ma, Z.; Song, W.; Lu, W.; Xiao, K. Characterization on TaMPK14, an MAPK family gene of wheat, in modulating N-starvation response through regulating N uptake and ROS homeostasis. Plant Cell Rep. 2020, 39, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ding, Y.; Jiang, Q.; Wang, F.; Sun, J.; Zhu, C. The role of receptor-like protein kinases (RLKs) in abiotic stress response in plants. Plant Cell Rep. 2017, 36, 235–242. [Google Scholar] [CrossRef]
- Khan, M.U.; Li, P.; Amjad, H.; Khan, A.Q.; Arafat, Y.; Waqas, M.; Li, Z.; Noman, A.; Islam, W.; Wu, L.; et al. Exploring the Potential of Overexpressed OsCIPK2 Rice as a Nitrogen Utilization Efficient Crop and Analysis of Its Associated Rhizo-Compartmental Microbial Communities. Int. J. Mol. Sci. 2019, 20, 3636. [Google Scholar] [CrossRef] [Green Version]
- Gaudinier, A.; Rodriguez-Medina, J.; Zhang, L.; Olson, A.; Liseron-Monfils, C.; Bågman, A.M.; Foret, J.; Abbitt, S.; Tang, M.; Li, B.; et al. Transcriptional regulation of nitrogen-associated metabolism and growth. Nature 2018, 563, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Heerah, S.; Katari, M.; Penjor, R.; Coruzzi, G.; Marshall-Colon, A. WRKY1 Mediates Transcriptional Regulation of Light and Nitrogen Signaling Pathways. Plant Physiol. 2019, 181, 1371–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, D.; Tao, Z.; Yang, Y.; Gao, Z.; Zhao, G.; Chang, X. Impacts of Nitrogen Deficiency on Wheat (Triticum aestivum L.) Grain during the Medium Filling Stage: Transcriptomic and Metabolomic Comparisons. Front Plant Sci. 2021, 12, 674433. [Google Scholar] [CrossRef]
- Yang, T.; Hao, L.; Yao, S.; Zhao, Y.; Lu, W.; Xiao, K. TabHLH1, a bHLH-type transcription factor gene in wheat, improves plant tolerance to Pi and N deprivation via regulation of nutrient transporter gene transcription and ROS homeostasis. Plant Physiol. Biochem. 2016, 104, 99–113. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
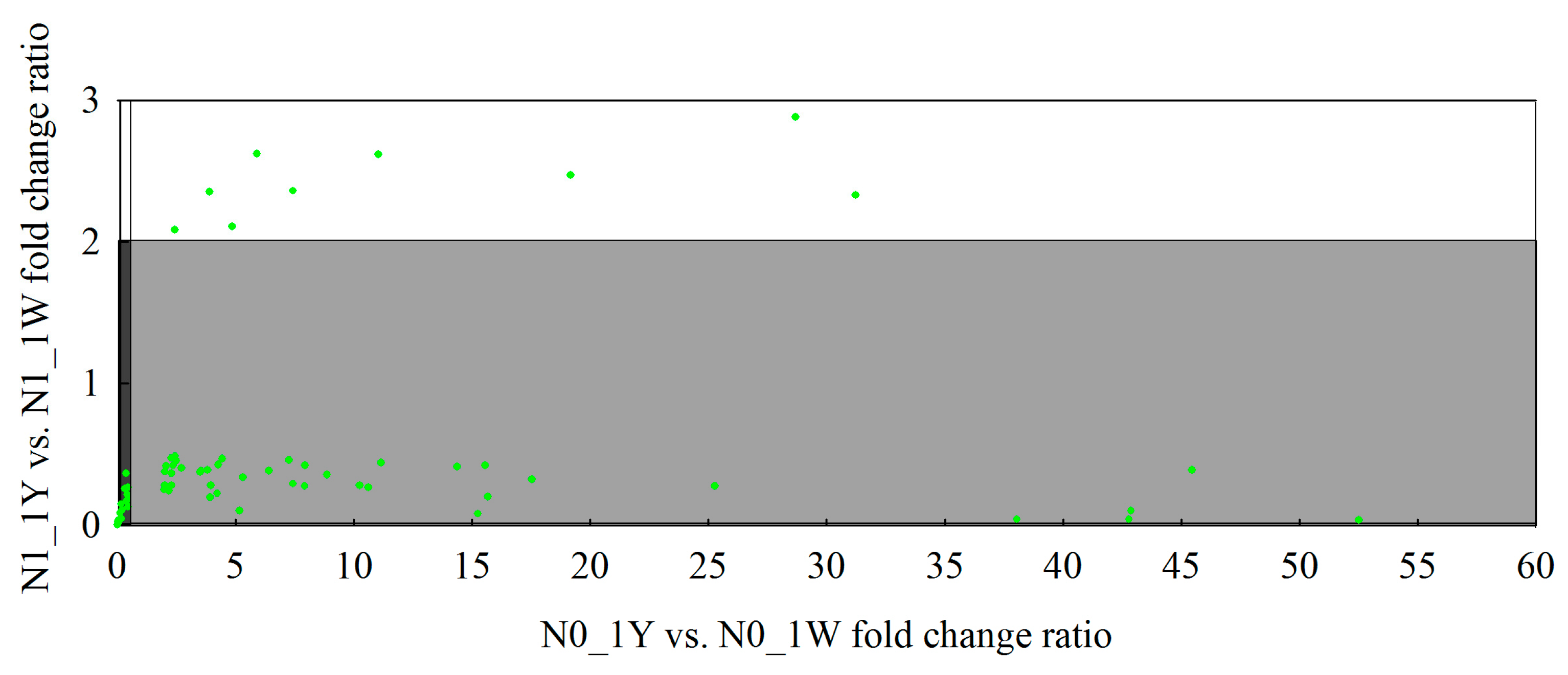{kind=link}
| Samples | Raw Reads | Q30 (%) | Clean Reads | Clean Reads % | Total Mapped | Uniquely Mapped |
|---|---|---|---|---|---|---|
| N0_1W | 42,598,731 | 94.81 | 42,364,637 | 99.45 | 40,389,624 (95.34%) | 38,025,789 (94.15%) |
| N0_1Y | 46,724,559 | 94.49 | 46,447,429 | 99.41 | 44,065,654 (94.87%) | 41,507,838 (94.20%) |
| NI_1W | 41,396,203 | 93.84 | 41,144,746 | 99.39 | 39,078,506 (94.98%) | 37,029,109 (94.76%) |
| NI_1Y | 45,007,941 | 94.62 | 44,758,766 | 99.44 | 42,670,214 (95.33%) | 40,297,380 (94.44%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Ma, Q.; Li, F.; Ding, Y.; Yi, Y.; Zhu, M.; Ding, J.; Li, C.; Guo, W.; Zhu, X. Transcriptome Analysis Reveals Different Responsive Patterns to Nitrogen Deficiency in Two Wheat Near-Isogenic Lines Contrasting for Nitrogen Use Efficiency. Biology 2021, 10, 1126. https://doi.org/10.3390/biology10111126
Zhang X, Ma Q, Li F, Ding Y, Yi Y, Zhu M, Ding J, Li C, Guo W, Zhu X. Transcriptome Analysis Reveals Different Responsive Patterns to Nitrogen Deficiency in Two Wheat Near-Isogenic Lines Contrasting for Nitrogen Use Efficiency. Biology. 2021; 10(11):1126. https://doi.org/10.3390/biology10111126
Chicago/Turabian StyleZhang, Xinbo, Quan Ma, Fujian Li, Yonggang Ding, Yuan Yi, Min Zhu, Jinfeng Ding, Chunyan Li, Wenshan Guo, and Xinkai Zhu. 2021. "Transcriptome Analysis Reveals Different Responsive Patterns to Nitrogen Deficiency in Two Wheat Near-Isogenic Lines Contrasting for Nitrogen Use Efficiency" Biology 10, no. 11: 1126. https://doi.org/10.3390/biology10111126
APA StyleZhang, X., Ma, Q., Li, F., Ding, Y., Yi, Y., Zhu, M., Ding, J., Li, C., Guo, W., & Zhu, X. (2021). Transcriptome Analysis Reveals Different Responsive Patterns to Nitrogen Deficiency in Two Wheat Near-Isogenic Lines Contrasting for Nitrogen Use Efficiency. Biology, 10(11), 1126. https://doi.org/10.3390/biology10111126